
SDG102, a H3K36-Methyltransferase-Encoding Gene, Plays Pleiotropic Roles in Growth and Development of Maize (Zea mays L.)


 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. SDG102 Phylogenetics and Sequence Alignment
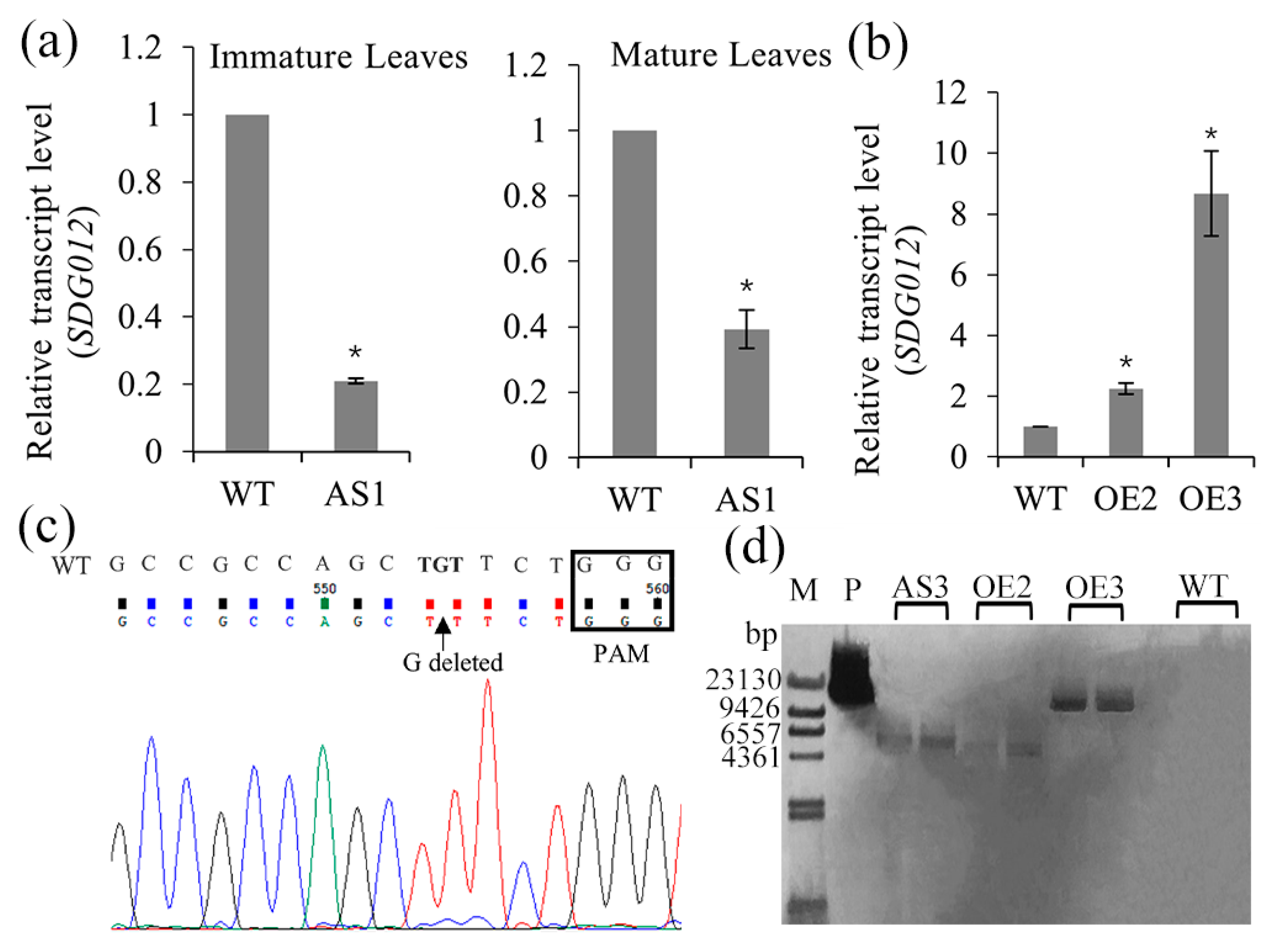
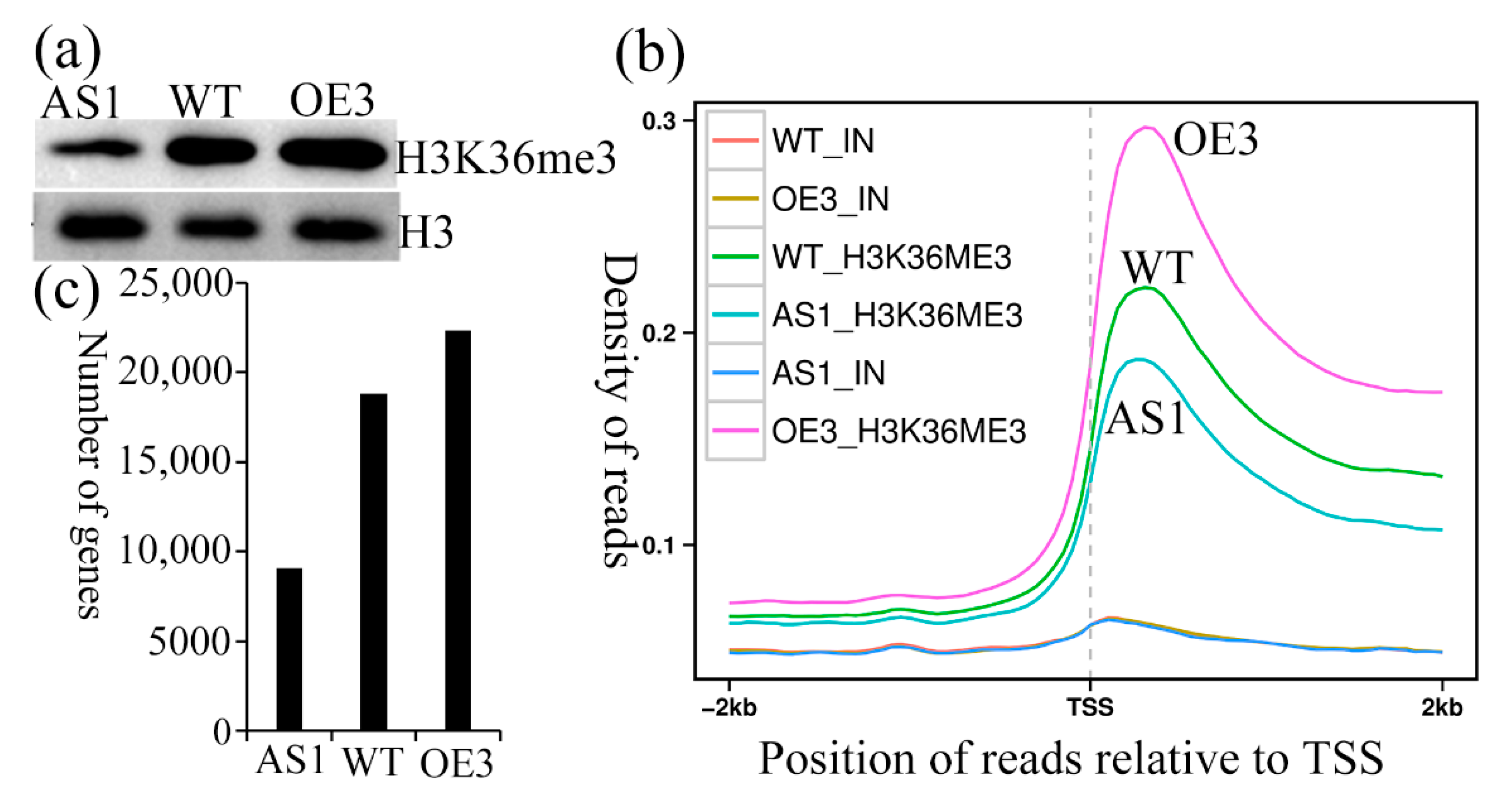
2.2. Generation of SDG102 Downregulation and Overexpression Lines
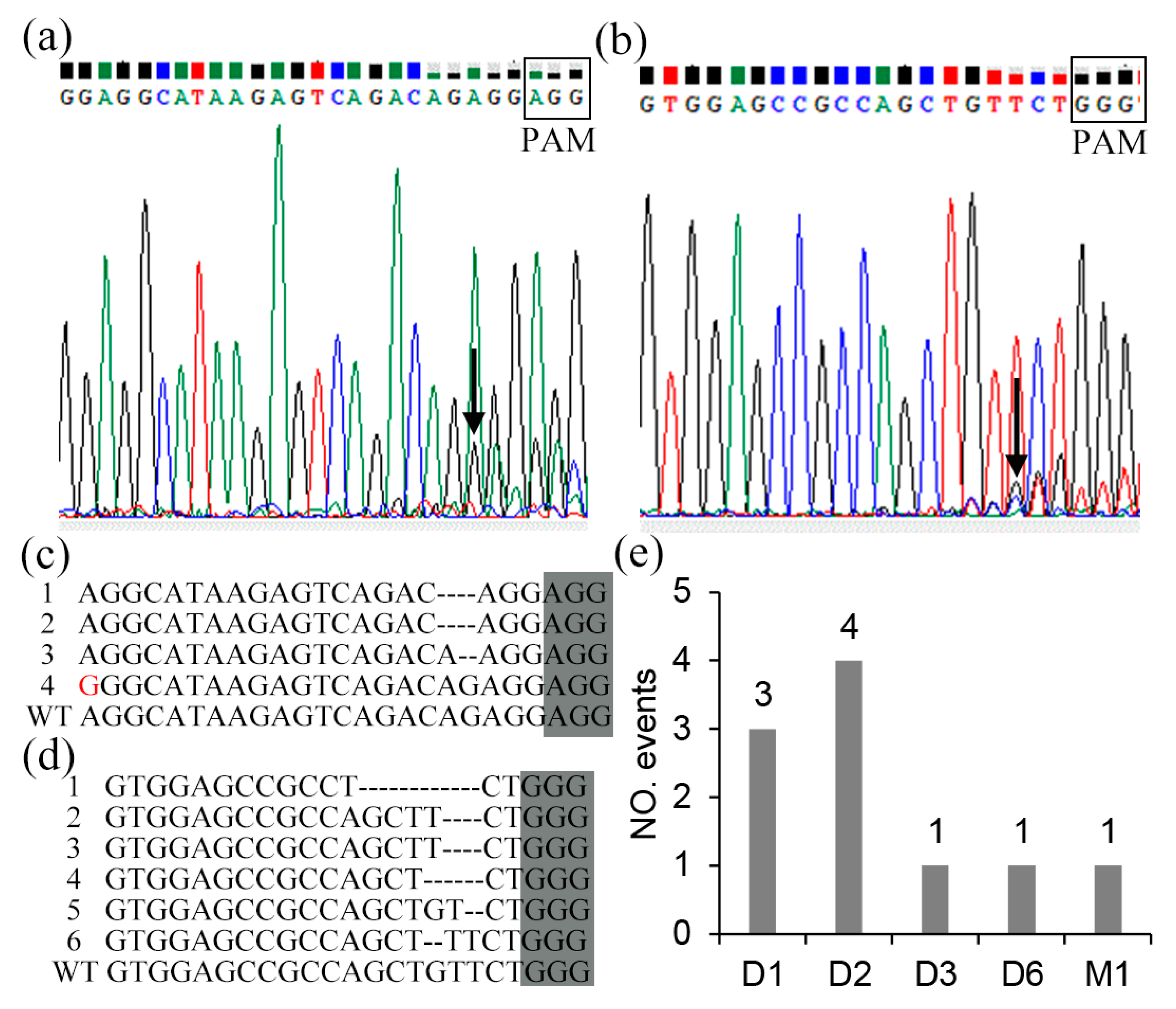
2.3. Mutation Types and Editing Rate of CRISPR/Cas9
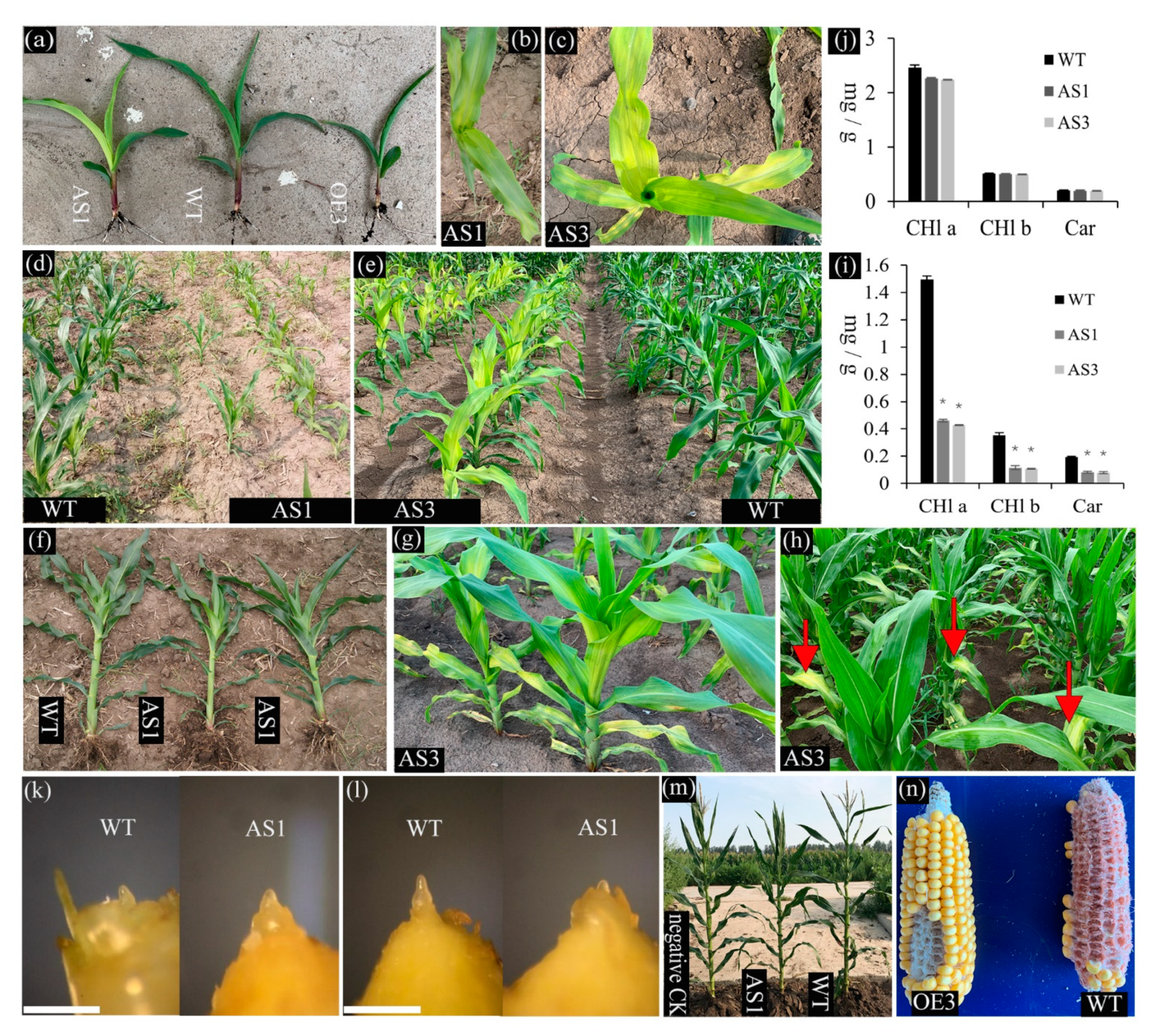
2.4. Phenotypic Alteration in the Down- and Upregulation Lines of SDG102 Expression
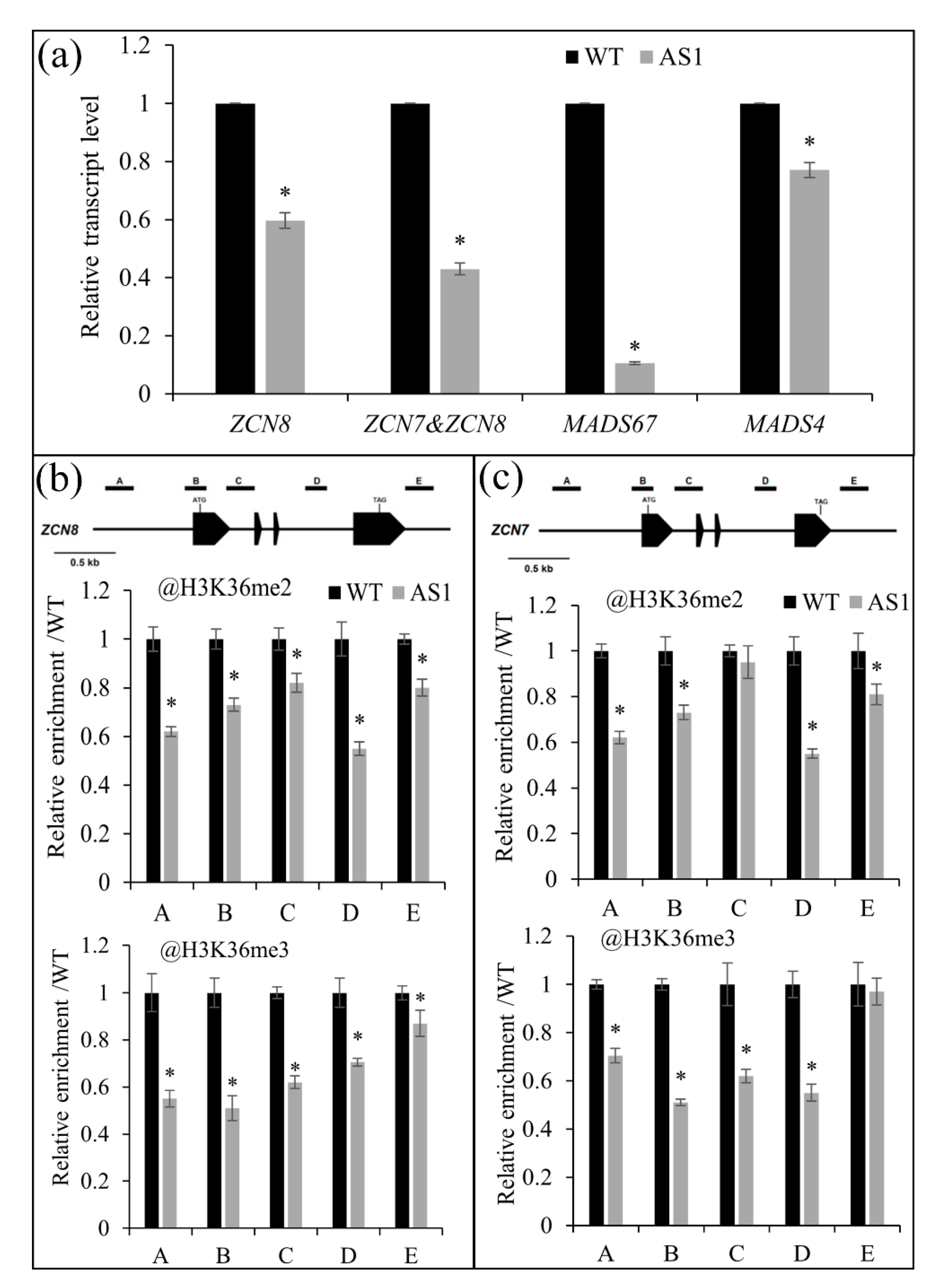
2.5. SDG102 Is Required for Normal Expression of a Set of Genes Involved in Various Biological Processes
2.6. SDG102 Promotes Flowering by Regulating H3K36 Methylation of Flowering Genes
2.7. SDG102 Is a Broad Trimethyl Transferase on H3K36
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Genetic Transformation
4.2. Transient Expression in Maize Protoplast
4.3. Mutation Detection
4.4. Phenotypic and Morphological Analysis
4.5. Measurement of Photosynthetic Pigments
4.6. Gene Expression Analysis
4.7. RNA Sequencing
4.8. ChIP Assays
4.9. ChIP-Seq
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephenson, E.; Estrada, S.; Meng, X.; Ourada, J.; Muszynski, M.G.; Habben, J.E.; Danilevskaya, O.N. Over-expression of the photoperiod response regulator ZmCCT10 modifies plant architecture, flowering time and inflorescence morphology in maize. PLoS ONE 2019, 14, e0203728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McSteen, P.; Laudencia-Chingcuanco, D.; Colasanti, J. A floret by any other name: Control of meristem identity in maize. Trends Plant Sci. 2000, 5, 61–66. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Wigge, P.A. FT protein acts as a long-range signal in Arabidopsis. Curr. Biol. 2007, 17, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, C. Long-distance regulation of flowering time. J. Exp. Bot. 2011, 62, 4399–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbesier, L.; Vincent, C.; Jang, S.; Fornara, F.; Fan, Q.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigge, P.A.; Kim, M.C.; Jaeger, K.E.; Busch, W.; Schmid, M.; Lohmann, J.U.; Weigel, D. Integration of spatial and temporal information during floral induction in Arabidopsis. Science 2005, 309, 1056–1059. [Google Scholar] [CrossRef]
- Borner, R.; Kampmann, G.; Chandler, J.; Gleissner, R.; Wisman, E.; Apel, K.; Melzer, S. A MADS domain gene involved in the transition to flowering in Arabidopsis. Plant J. 2000, 24, 591–599. [Google Scholar] [CrossRef]
- Danilevskaya, O.N.; Meng, X.; Hou, Z.; Ananiev, E.V.; Simmons, C.R. A genomic and expression compendium of the expanded PEBP gene family from maize. Plant Physiol. 2008, 146, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Lazakis, C.M.; Coneva, V.; Colasanti, J. ZCN8 encodes a potential orthologue of Arabidopsis FT florigen that integrates both endogenous and photoperiod flowering signals in maize. J. Exp. Bot. 2011, 62, 4833–4842. [Google Scholar] [CrossRef]
- Meng, X.; Muszynski, M.G.; Danilevskaya, O.N. The FT-like ZCN8 Gene Functions as a Floral Activator and Is Involved in Photoperiod Sensitivity in Maize. Plant Cell 2011, 23, 942–960. [Google Scholar] [CrossRef] [Green Version]
- Mascheretti, I.; Turner, K.; Brivio, R.S.; Hand, A.; Colasanti, J.; Rossi, V. Florigen-Encoding Genes of Day-Neutral and Photoperiod-Sensitive Maize Are Regulated by Different Chromatin Modifications at the Floral Transition. Plant Physiol. 2015, 168, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelletti, S.; Coupel-Ledru, A.; Granato, I.; Palaffre, C.; Cabrera-Bosquet, L.; Tonelli, C.; Nicolas, S.D.; Tardieu, F.; Welcker, C.; Conti, L. Maize adaptation across temperate climates was obtained via expression of two florigen genes. PLoS Genet. 2020, 16, e1008882. [Google Scholar] [CrossRef] [PubMed]
- Muszynski, M.G.; Dam, T.; Li, B.; Shirbroun, D.M.; Hou, Z.; Bruggemann, E.; Archibald, R.; Ananiev, E.V.; Danilevskaya, O.N. Delayed flowering1 Encodes a basic leucine zipper protein that mediates floral inductive signals at the shoot apex in maize. Plant Physiol. 2006, 142, 1523–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilevskaya, O.N.; Meng, X.; Selinger, D.A.; Deschamps, S.; Hermon, P.; Vansant, G.; Gupta, R.; Ananiev, E.V.; Muszynski, M.G. Involvement of the MADS-box gene ZMM4 in floral induction and inflorescence development in maize. Plant Physiol. 2008, 147, 2054–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Wang, C.; Chen, X.; Liu, H.; Huang, Y.; Li, S.; Dong, Z.; Zhao, X.; Tian, F.; Jin, W. dlf1 promotes floral transition by directly activating ZmMADS4 and ZmMADS67 in the maize shoot apex. New Phytol. 2020, 228, 1386–1400. [Google Scholar] [CrossRef] [PubMed]
- Alter, P.; Bircheneder, S.; Zhou, L.Z.; Schlüter, U.; Gahrtz, M.; Sonnewald, U.; Dresselhaus, T. Flowering Time-Regulated Genes in Maize Include the Transcription Factor ZmMADS1. Plant Physiol. 2016, 172, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Wang, X.; Zhao, M.; Huang, C.; Li, C.; Li, D.; Yang, C.J.; York, A.M.; Xue, W.; Xu, G.; et al. Stepwise cis-Regulatory Changes in ZCN8 Contribute to Maize Flowering-Time Adaptation. Curr. Biol. 2018, 28, 3005–3015.e3004. [Google Scholar] [CrossRef] [Green Version]
- Colasanti, J.; Yuan, Z.; Sundaresan, V. The indeterminate gene encodes a zinc finger protein and regulates a leaf-generated signal required for the transition to flowering in maize. Cell 1998, 93, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.Y.; Colasanti, J. Maize floral regulator protein INDETERMINATE1 is localized to developing leaves and is not altered by light or the sink/source transition. J. Exp. Bot. 2007, 58, 403–414. [Google Scholar] [CrossRef]
- Salvi, S.; Sponza, G.; Morgante, M.; Tomes, D.; Niu, X.; Fengler, K.A.; Meeley, R.; Ananiev, E.V.; Svitashev, S.; Bruggemann, E. Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc. Natl. Acad. Sci. USA 2007, 104, 11376–11381. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone methylation in higher plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Y.; Liang, Y.; Zhou, D.; Li, S.; Lin, S.; Dong, H.; Huang, L. The function of histone lysine methylation related SET domain group proteins in plants. Protein Sci. 2020, 29, 1120–1137. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.W.K.; Wang, T.; Chanbrasekaran, M.B.; Aramayo, R.; Kertbundit, S.; Hall, T. Plant SET domain-containing proteins: Structure, function and regulation. Biochim. Biophys. Acta 2017, 2007, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Huang, X.; Ouyang, Y.; Yao, J. Genome-Wide Identification, Phylogenetic and Co-Expression Analysis of OsSET Gene Family in Rice. PLoS ONE 2013, 8, e65426. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhao, Z.; Dong, A.; Soubigou-Taconnat, L.; Renou, J.P.; Steinmetz, A.; Shen, W.H. Di- and tri- but not monomethylation on histone H3 lysine 36 marks active transcription of genes involved in flowering time regulation and other processes in Arabidopsis thaliana. Mol. Cell. Biol. 2008, 28, 1348–1360. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; He, Y.; Jacob, Y.; Noh, Y.S.; Michaels, S.; Amasino, R. Establishment of the vernalization-responsive, winter-annual habit in Arabidopsis requires a putative histone H3 methyl transferase. Plant Cell 2005, 17, 3301–3310. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Yu, Y.; Meyer, D.; Wu, C.; Shen, W.H. Prevention of early flowering by expression of FLOWERING LOCUS C requires methylation of histone H3 K36. Nat. Cell Biol. 2005, 7, 1256–1260. [Google Scholar] [CrossRef]
- Berr, A.; McCallum, E.J.; Alioua, A.; Heintz, D.; Heitz, T.; Shen, W.H. Arabidopsis histone methyltransferase SET DOMAIN GROUP8 mediates induction of the jasmonate/ethylene pathway genes in plant defense response to necrotrophic fungi. Plant Physiol. 2010, 154, 1403–1414. [Google Scholar] [CrossRef] [Green Version]
- Cazzonelli, C.I.; Cuttriss, A.J.; Cossetto, S.B.; Pye, W.; Crisp, P.; Whelan, J.; Finnegan, E.J.; Turnbull, C.; Pogson, B.J. Regulation of carotenoid composition and shoot branching in Arabidopsis by a chromatin modifying histone methyltransferase, SDG8. Plant Cell 2009, 21, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Ma, D.P.; Li, J. The histone methyltransferase SDG8 regulates shoot branching in Arabidopsis. Biochem. Biophys. Res. Commun. 2008, 373, 659–664. [Google Scholar] [CrossRef]
- Grini, P.E.; Thorstensen, T.; Alm, V.; Vizcay-Barrena, G.; Windju, S.S.; Jørstad, T.S.; Wilson, Z.A.; Aalen, R.B. The ASH1 HOMOLOG 2 (ASHH2) histone H3 methyltransferase is required for ovule and anther development in Arabidopsis. PLoS ONE 2009, 4, e7817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Mukherjee, I.; Thum, K.E.; Tanurdzic, M.; Katari, M.S.; Obertello, M.; Edwards, M.B.; McCombie, W.R.; Martienssen, R.A.; Coruzzi, G.M. The histone methyltransferase SDG8 mediates the epigenetic modification of light and carbon responsive genes in plants. Genome Biol. 2015, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, J.; Xie, Z.; Liu, S.; Nolan, T.; Ye, H.; Zhang, M.; Guo, H.; Schnable, P.S.; Li, Z.; et al. Histone lysine methyltransferase SDG8 is involved in brassinosteroid-regulated gene expression in Arabidopsis thaliana. Mol. Plant 2014, 7, 1303–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berr, A.; Shafiq, S.; Pinon, V.; Dong, A.; Shen, W.H. The trxG family histone methyltransferase SET DOMAIN GROUP 26 promotes flowering via a distinctive genetic pathway. Plant J. 2015, 81, 316–328. [Google Scholar] [CrossRef]
- Liu, B.; Berr, A.; Chang, C.; Liu, C.; Shen, W.H.; Ruan, Y. Interplay of the histone methyltransferases SDG8 and SDG26 in the regulation of transcription and plant flowering and development. Biochim. Biophys. Acta 2016, 1859, 581–590. [Google Scholar] [CrossRef]
- Cartagena, J.A.; Matsunaga, S.; Seki, M.; Kurihara, D.; Yokoyama, M.; Shinozaki, K.; Fujimoto, S.; Azumi, Y.; Uchiyama, S.; Fukui, K. The Arabidopsis SDG4 contributes to the regulation of pollen tube growth by methylation of histone H3 lysines 4 and 36 in mature pollen. Dev. Biol. 2008, 315, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Thorstensen, T.; Grini, P.E.; Mercy, I.S.; Alm, V.; Erdal, S.; Aasland, R.; Aalen, R.B. The Arabidopsis SET-domain protein ASHR3 is involved in stamen development and interacts with the bHLH transcription factor ABORTED MICROSPORES (AMS). Plant Mol. Biol. 2008, 66, 47–59. [Google Scholar] [CrossRef]
- Kumpf, R.; Thorstensen, T.; Rahman, M.A.; Heyman, J.; Nenseth, H.Z.; Lammens, T.; Herrmann, U.; Swarup, R.; Veiseth, S.V.; Emberland, G.; et al. The ASH1-RELATED3 SET-Domain Protein Controls Cell Division Competence of the Meristem and the Quiescent Center of the Arabidopsis Primary Root. Plant Physiol. 2014, 166, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Yun, J.Y.; Zhao, W.; Shen, W.H.; Amasino, R.M. A methyltransferase required for proper timing of the vernalization response in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 2269–2274. [Google Scholar] [CrossRef] [Green Version]
- Sui, P.; Jin, J.; Ye, S.; Mu, C.; Gao, J.; Feng, H.; Shen, W.H.; Yu, Y.; Dong, A. H3K36 methylation is critical for brassinosteroid-regulated plant growth and development in rice. Plant J. 2012, 70, 340–347. [Google Scholar] [CrossRef]
- Sui, P.; Shi, J.; Gao, X.; Shen, W.H.; Dong, A. H3K36 methylation is involved in promoting rice flowering. Mol. Plant 2013, 6, 975–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Wei, G.; Shi, J.; Jin, J.; Shen, T.; Ni, T.; Shen, W.H.; Yu, Y.; Dong, A. SET DOMAIN GROUP 708, a histone H3 lysine 36-specific methyltransferase, controls flowering time in rice (Oryza sativa). New Phytol. 2016, 210, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Du, K.; Shi, Y.; Yin, L.; Shen, W.H.; Yu, Y.; Liu, B.; Dong, A. H3K36 methyltransferase SDG708 enhances drought tolerance by promoting abscisic acid biosynthesis in rice. New Phytol. 2021, 230, 1967–1984. [Google Scholar] [CrossRef] [PubMed]
- He, X.H.; Wang, J.B.; Yuan, Y.; Huang, Y. SDG736 Regulating Flowering Timeby Reducing Expression of FLC in Arabidopsis thaliana. Genom. Appl. Biol. 2020, 39, 2118–2126. [Google Scholar]
- Sun, C.; Fang, J.; Zhao, T.; Xu, B.; Zhang, F.; Liu, L.; Tang, J.; Zhang, G.; Deng, X.; Chen, F.; et al. The histone methyltransferase SDG724 mediates H3K36me2/3 deposition at MADS50 and RFT1 and promotes flowering in rice. Plant Cell 2012, 24, 3235–3247. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M.; Napoli, C.A.; Selinger, D.A.; Pandey, R.; Cone, K.C.; Chandler, V.L.; Kaeppler, H.F.; Kaeppler, S.M. Comparative analysis of set domain proteins in maize and Arabidopsis reveals multiple duplications preceding the divergence of monocots and dicots. Plant Physiol. 2003, 132, 907–925. [Google Scholar] [CrossRef] [Green Version]
- Campi, M.; D’Andrea, L.; Emiliani, J.; Casati, P. Participation of chromatin-remodeling proteins in the repair of ultraviolet-B-damaged DNA. Plant Physiol. 2012, 158, 981–995. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.M.; Hu, X.J.; Zhao, Y.Z.; Song, W.B.; Zhang, M.; Chen, Z.L.; Chen, W.; Dong, Y.B.; Wang, Z.H.; Lai, J.S. Map-based cloning of zb7 encoding an IPP and DMAPP synthase in the MEP pathway of maize. Mol. Plant 2012, 55, 1100–1112. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, J.; Han, J.J.; Han, M.J.; An, G. Functional analyses of the flowering time gene OsMADS50, the putative SUPPRESSOR OF OVEREXPRESSION OF CO 1/AGAMOUS-LIKE 20 (SOC1/AGL20) ortholog in rice. Plant J. 2004, 38, 754–764. [Google Scholar] [CrossRef]
- Xiao, J.; Lee, U.S.; Wagner, D. Tug of war: Adding and removing histone lysine methylation in Arabidopsis. Curr. Opin. Plant Biol. 2016, 34, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Karimi, M.; Inzé, D.; Depicker, A. GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 2002, 7, 193–195. [Google Scholar] [CrossRef]
- Yu, X.; Meng, X.; Liu, Y.; Wang, X.; Xu, Z.Y. The chromatin remodeler zmchb101 impacts alternative splicing contexts in response to osmotic stress. Plant Cell Rep. 2018, 38, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cui, G.; Wang, Y.; Hao, Y.; Du, J.; Zhang, H.; Wang, C.; Zhang, H.; Wu, S.B.; Sun, Y. Expression of Foreign Genes Demonstrates the Effectiveness of Pollen-Mediated Transformation in Zea mays. Front. Plant Sci. 2017, 8, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Jiang, L.; Wu, R.; Meng, X.; Zhang, A.; Li, N.; Xia, Q.; Qi, X.; Pang, J.; Xu, Z.Y. The Core Subunit of A Chromatin-Remodeling Complex, ZmCHB101, Plays Essential Roles in Maize Growth and Development. Sci. Rep. 2016, 6, 38504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.D.; Cho, Y.H.; Sheen, J. Arabidopsis mesophyll protoplasts: A versatile cell system for transient gene expression analysis. Nat. Protoc. 2007, 2, 1565–1572. [Google Scholar] [CrossRef] [Green Version]
- Boudsocq, M.; Barbier-Brygoo, H.; Laurière, C. Identification of nine sucrose nonfermenting 1-related protein kinases 2 activated by hyperosmotic and saline stresses in Arabidopsis thaliana. J. Biol. Chem. 2004, 279, 41758–41766. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, S.W.; Hanway, J.J.; Benson, G.O. How a Corn Plant Develops; Iowa University of Science and Technology Cooperative Extension Service: Ames, IA, USA, 1997; Volume 48. [Google Scholar]
- Lichtenthaler, H.K. ChlorolShylls and Carotenoids: Pigments of Photosynthetic Biomembranes. Methods Enzymol. 1987, 148, 350–382. [Google Scholar]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [Green Version]
- Haring, M.; Offermann, S.; Danker, T.; Horst, I.; Peterhansel, C.; Stam, M. Chromatin immunoprecipitation: Optimization, quantitative analysis and data normalization. Plant Methods 2007, 24, 3. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Jointing Stage | Flowering Time (Days) | Adult Plant | |||||
|---|---|---|---|---|---|---|---|
| PHT (cm) | Number of Leaves | PHT (cm) | EHT (cm) | Ratio of EHT/PHT | Number of Leaves | ||
| WT | 48.5 ± 4.3 | 6.9 ± 0.2 | 63.3 ± 2.0 | 206.6 ± 9.1 | 84.1 ± 7.7 | 0.41 ± 0.04 | 21.9 ± 0.7 |
| AS1 | 32.1 ± 4.7 * | 5.8 ± 0.4 * | 69.1 ± 2.7 * | 213.6 ± 8.6 * | 95.1 ± 8.2 * | 0.45 ± 0.04 * | 22.6 ± 0.8 * |
| AS3 | 31.1 ± 4.0 * | 5.3 ± 0.5 * | 70.0 ± 2.6 * | 225.3 ± 9.7 * | 100.9 ± 9.5 * | 0.45 ± 0.03 * | 22.8 ± 0.8 * |
| OE3 | 34.5 ± 4.5 * | 6.5 ± 0.5 | 62.3 ± 1.5 | 176.3 ± 15.0 * | 71.3 ± 8.5 * | 0.41 ± 0.03 | 21.5 ± 0.7 |
| OE2 | 35.6 ± 4.3 * | 6.7 ± 0.3 | 63.1 ± 1.3 | 179.6 ± 14.5 * | 72.6 ± 9.2 * | 0.41 ± 0.02 | 21.8 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Sun, W.; Wang, Z.; Wan, C.; Zhang, J.; Qi, X.; Zhang, J. SDG102, a H3K36-Methyltransferase-Encoding Gene, Plays Pleiotropic Roles in Growth and Development of Maize (Zea mays L.). Int. J. Mol. Sci. 2022, 23, 7458. https://doi.org/10.3390/ijms23137458
Li Y, Sun W, Wang Z, Wan C, Zhang J, Qi X, Zhang J. SDG102, a H3K36-Methyltransferase-Encoding Gene, Plays Pleiotropic Roles in Growth and Development of Maize (Zea mays L.). International Journal of Molecular Sciences. 2022; 23(13):7458. https://doi.org/10.3390/ijms23137458
Chicago/Turabian StyleLi, Yongjian, Weifeng Sun, Zhenhui Wang, Chang Wan, Jun Zhang, Xin Qi, and Jian Zhang. 2022. "SDG102, a H3K36-Methyltransferase-Encoding Gene, Plays Pleiotropic Roles in Growth and Development of Maize (Zea mays L.)" International Journal of Molecular Sciences 23, no. 13: 7458. https://doi.org/10.3390/ijms23137458