
Graph Pangenomes Track Genetic Variants for Crop Improvement

Abstract
:1. Introduction
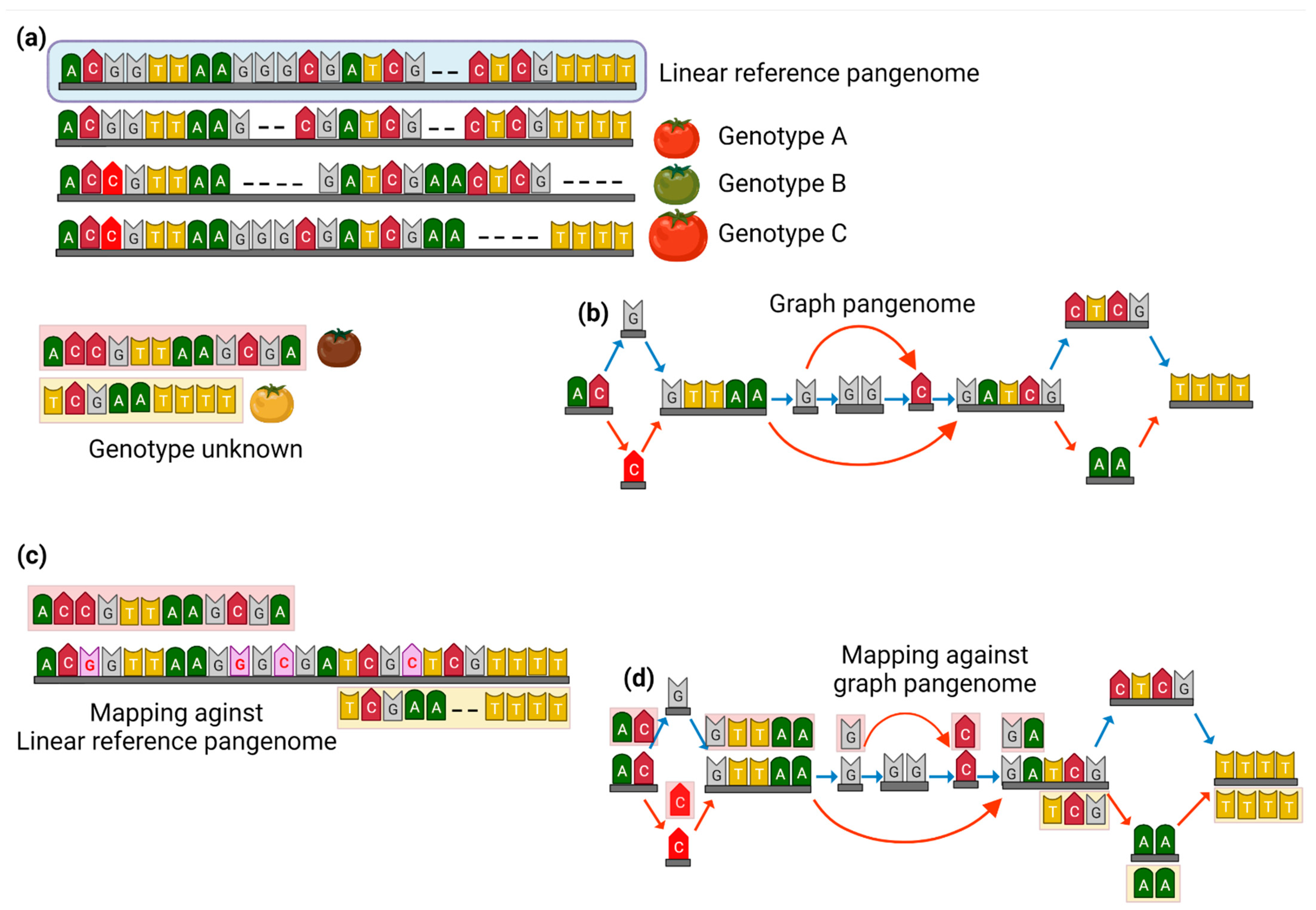
2. The Transition from Linear High-Quality Reference Genome to Graph Pangenome
3. Graph Pangenomes to Track Genetic Variability within Crop Plants
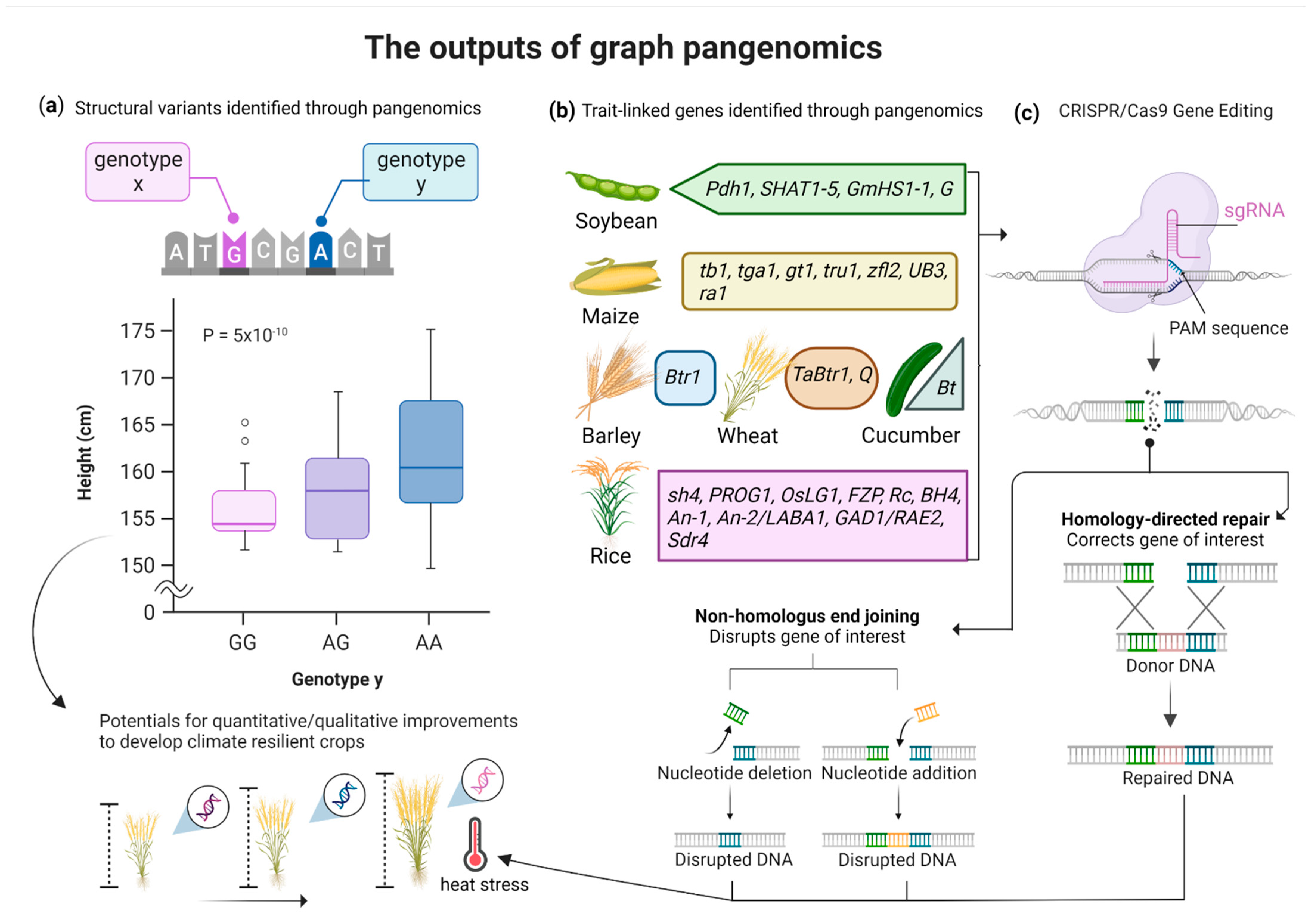
4. Graph Pangenomics for Crops Improvement
4.1. Pangenomics to Identify the Disease Resistance Potential of Crops
4.2. Pangenomics to Identify the Quantitative Yield Potential of Crops
4.3. Pangenomics to Develop Climate-Resilient Germplasms
5. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, W.; Pijl, A.; Tarolli, P. Future climate-zone shifts are threatening steep-slope agriculture. Nat. Food 2022, 3, 193–196. [Google Scholar] [CrossRef]
- Erenstein, O.; Poole, N.; Donovan, J. Role of staple cereals in human nutrition: Separating the wheat from the chaff in the infodemics age. Trends Food Sci. Technol. 2022, 119, 508–513. [Google Scholar] [CrossRef]
- Huang, X.; Huang, S.; Han, B.; Li, J. The integrated genomics of crop domestication and breeding. Cell 2022, 185, 2828–2839. [Google Scholar] [CrossRef]
- Kaul, S.; Koo, H.L.; Jenkins, J.; Rizzo, M.; Rooney, T.; Tallon, L.J.; Feldblyum, T.; Nierman, W.; Benito, M.I.; Lin, X.; et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar]
- Kersey, P.J. Plant genome sequences: Past, present, future. Curr. Opin. Plant Biol. 2019, 48, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Danilevicz, M.F.; Fernandez, C.G.T.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Golicz, A.A.; Bayer, P.E.; Bhalla, P.L.; Batley, J.; Edwards, D. Pangenomics comes of age: From bacteria to plant and animal applications. Trends Genet. 2020, 36, 132–145. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Zhang, H.; Liu, Z.; Wang, Y.; Xing, L.; He, Q.; Du, H. Plant pan-genomics: Recent advances, new challenges, and roads ahead. J. Genet. Genom. 2022, 49, 833–846. [Google Scholar] [CrossRef]
- Zanini, S.F.; Bayer, P.E.; Wells, R.; Snowdon, R.J.; Batley, J.; Varshney, R.K.; Nguyen, H.T.; Edwards, D.; Golicz, A.A. Pangenomics in crop improvement—From coding structural variations to finding regulatory variants with pangenome graphs. Plant Genome 2022, 15, e20177. [Google Scholar] [CrossRef]
- Golicz, A.A.; Batley, J.; Edwards, D. Towards plant pangenomics. Plant Biotechnol. J. 2016, 14, 1099–1105. [Google Scholar] [CrossRef]
- Hübner, S. Are we there yet? Driving the road to evolutionary graph-pangenomics. Curr. Opin. Plant Biol. 2022, 66, 102195. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.; Batley, J. Graph pangenomes find missing heritability. Nat. Genet. 2022, 54, 919–920. [Google Scholar] [CrossRef] [PubMed]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Shang, L.; Li, X.; He, H.; Yuan, Q.; Song, Y.; Wei, Z.; Lin, H.; Hu, M.; Zhao, F.; Zhang, C. A super pan-genomic landscape of rice. Cell Res. 2022, 32, 878–896. [Google Scholar] [CrossRef]
- Jayakodi, M.; Schreiber, M.; Stein, N.; Mascher, M. Building pan-genome infrastructures for crop plants and their use in association genetics. DNA Res. 2021, 28, dsaa030. [Google Scholar] [CrossRef]
- Baaijens, J.A.; Bonizzoni, P.; Boucher, C.; Della Vedova, G.; Pirola, Y.; Rizzi, R.; Sirén, J. Computational graph pangenomics: A tutorial on data structures and their applications. Nat. Comput. 2022, 21, 81–108. [Google Scholar] [CrossRef]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Li, H.; Feng, X.; Chu, C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 2020, 21, 265. [Google Scholar] [CrossRef]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 2020, 182, 145–161.e23. [Google Scholar] [CrossRef]
- Tang, D.; Jia, Y.; Zhang, J.; Li, H.; Cheng, L.; Wang, P.; Bao, Z.; Liu, Z.; Feng, S.; Zhu, X. Genome evolution and diversity of wild and cultivated potatoes. Nature 2022, 606, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Z.; Bao, Z.; Li, H.; Lyu, Y.; Zan, Y.; Wu, Y.; Cheng, L.; Fang, Y.; Wu, K. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 2022, 606, 527–534. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 2022, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.; Lenderts, B.; Feigenbutz, L.; Barone, P.; Llaca, V.; Fengler, K.; Svitashev, S. CRISPR–Cas9-mediated 75.5-Mb inversion in maize. Nat. Plants 2020, 6, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J. Molecular markers in plant improvement: Manipulation of genes affecting quantitative traits. Crop Sci. 1993, 33, 660–668. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; De Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.G.; Redenbaugh, K. Commercialization of a tomato with an antisense polygalacturonase gene: The FLAVR SAVR™ tomato story. Euphytica 1994, 79, 293–297. [Google Scholar] [CrossRef]
- Jones, D.M.; Wells, R.; Pullen, N.; Trick, M.; Irwin, J.A.; Morris, R.J. Spatio-temporal expression dynamics differ between homologues of flowering time genes in the allopolyploid Brassica napus. Plant J. 2018, 96, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.-A.; Zhang, H.; Liu, Z.; Shi, M. Pan-genome of wild and cultivated soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef]
- Li, J.; Yuan, D.; Wang, P.; Wang, Q.; Sun, M.; Liu, Z.; Si, H.; Xu, Z.; Ma, Y.; Zhang, B. Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 2021, 22, 119. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.B.; Seetharam, A.S.; Woodhouse, M.R.; Chougule, K.M.; Ou, S.; Liu, J.; Ricci, W.A.; Guo, T.; Olson, A.; Qiu, Y. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 2021, 373, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, L.; Yang, J.; He, H.; Jin, H.; Li, X.; Ren, T.; Ren, Z.; Li, F.; Han, X. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat. Genet. 2021, 53, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Hübner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Odenheimer, J.; Ziegler, E.; Lee, J.S.; Baute, G.J.; Owens, G.L.; Grassa, C.J. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 2019, 5, 54–62. [Google Scholar] [CrossRef]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolatabadian, A.; Bayer, P.E.; Tirnaz, S.; Hurgobin, B.; Edwards, D.; Batley, J. Characterization of disease resistance genes in the Brassica napus pangenome reveals significant structural variation. Plant Biotechnol. J. 2020, 18, 969–982. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.; Alderman, S.; Garvin, D.F.; Pfender, W.F. Infection of Brachypodium distachyon by formae speciales of Puccinia graminis: Early infection events and host-pathogen incompatibility. PLoS ONE 2013, 8, e56857. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Petereit, J.; Durant, É.; Monat, C.; Rouard, M.; Hu, H.; Chapman, B.; Li, C.; Cheng, S.; Batley, J. Wheat Panache: A pangenome graph database representing presence–absence variation across sixteen bread wheat genomes. Plant Genome 2022, 15, e20221. [Google Scholar] [CrossRef]
- Ou, L.; Li, D.; Lv, J.; Chen, W.; Zhang, Z.; Li, X.; Yang, B.; Zhou, S.; Yang, S.; Li, W. Pan-genome of cultivated pepper (Capsicum) and its use in gene presence–absence variation analyses. New Phytol. 2018, 220, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Petereit, J.; Bayer, P.E.; Thomas, W.J.; Tay Fernandez, C.G.; Amas, J.; Zhang, Y.; Batley, J.; Edwards, D. Pangenomics and Crop Genome Adaptation in a Changing Climate. Plants 2022, 11, 1949. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E.; Scheben, A.; Golicz, A.A.; Yuan, Y.; Faure, S.; Lee, H.; Chawla, H.S.; Anderson, R.; Bancroft, I.; Raman, H. Modelling of gene loss propensity in the pangenomes of three Brassica species suggests different mechanisms between polyploids and diploids. Plant Biotechnol. J. 2021, 19, 2488–2500. [Google Scholar] [CrossRef] [PubMed]
- Khush, G.S. Green revolution: The way forward. Nat. Rev. Genet. 2001, 2, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.; Atkin, O.K.; Bennett, M.; Cooper, M.; Dodd, I.C.; Foulkes, M.J.; Frohberg, C.; Hammer, G.; Henderson, I.R.; Huang, B. Addressing research bottlenecks to crop productivity. Trends Plant Sci. 2021, 26, 607–630. [Google Scholar] [CrossRef] [PubMed]
- Hyten, D.L.; Song, Q.; Zhu, Y.; Choi, I.-Y.; Nelson, R.L.; Costa, J.M.; Specht, J.E.; Shoemaker, R.C.; Cregan, P.B. Impacts of genetic bottlenecks on soybean genome diversity. Proc. Natl. Acad. Sci. USA 2006, 103, 16666–16671. [Google Scholar] [CrossRef] [Green Version]
- Parra-Rondinel, F.; Casas, A.; Begazo, D.; Paco, A.; Márquez, E.; Cruz, A.; Segovia, J.; Torres-García, I.; Zarazúa, M.; Lizárraga, L. Natural and cultural processes influencing gene flow among wild (atoq papa), weedy (araq papa and k’ipa papa), and crop potatoes in the Andean region of southern Peru. Front. Ecol. Evol. 2021, 327. [Google Scholar] [CrossRef]
- Valliyodan, B.; Cannon, S.B.; Bayer, P.E.; Shu, S.; Brown, A.V.; Ren, L.; Jenkins, J.; Chung, C.Y.L.; Chan, T.F.; Daum, C.G. Construction and comparison of three reference-quality genome assemblies for soybean. Plant J. 2019, 100, 1066–1082. [Google Scholar] [CrossRef]
- Hartman, G.; Gardner, M.; Hymowitz, T.; Naidoo, G. Evaluation of perennial Glycine species for resistance to soybean fungal pathogens that cause Sclerotinia stem rot and sudden death syndrome. Crop Sci. 2000, 40, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef]
- Durant, É.; Sabot, F.; Conte, M.; Rouard, M. Panache: A web browser-based viewer for linearized pangenomes. Bioinformatics 2021, 37, 4556–4558. [Google Scholar] [CrossRef] [PubMed]
- Salmen, F.; De Jonghe, J.; Kaminski, T.S.; Alemany, A.; Parada, G.E.; Verity-Legg, J.; Yanagida, A.; Kohler, T.N.; Battich, N.; van den Brekel, F. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Crop | Scientific Name | Pangenome Dataset | Structural Variant | Traits Associated | Number of Accessions | Reference |
|---|---|---|---|---|---|---|
| Rapeseed | Brassica napus | 1.8 Gb; >150,000 genes | InDels, PAV | Seed weight, flowering, silique length | 8 | [29] |
| Soybean | Glycine max; Glycine soja | 57,492 orthologs | PAV | Nutrient uptake | 29 | [30] |
| Cotton | Gossypium hirsutum; Gossypium barbadense | 3.3 Gb; >102,000 genes: 2.5 Gb; >80,000 genes | InDels, PAV, SNPs | Disease resistance, fiber quality, stress resistance | 1581 for G. hirsutum; 226 for G. barbadense | [31] |
| Tomato | Solanum lycopersicum | 1.1 Gb, 40,369 genes | PAV | Fruit flavor, disease resistance | 725 | [32] |
| Maize | Zea mays | >103,000 genes | SNPs, PAV, TE, InDels | Flowering; disease resistance | 26 | [33] |
| Rye | Secale cereale | 7.74 Gb; 86,991 genes | TE, Gene duplications | Starch biosynthesis, disease resistance genes | 295 | [34] |
| Rice | Oryza sativa | 1.52 Gb; 51,359 genes | PAV | Grain weight, improved nitrogen uptake | 251 | [15] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hameed, A.; Poznanski, P.; Nadolska-Orczyk, A.; Orczyk, W. Graph Pangenomes Track Genetic Variants for Crop Improvement. Int. J. Mol. Sci. 2022, 23, 13420. https://doi.org/10.3390/ijms232113420
Hameed A, Poznanski P, Nadolska-Orczyk A, Orczyk W. Graph Pangenomes Track Genetic Variants for Crop Improvement. International Journal of Molecular Sciences. 2022; 23(21):13420. https://doi.org/10.3390/ijms232113420
Chicago/Turabian StyleHameed, Amir, Pawel Poznanski, Anna Nadolska-Orczyk, and Waclaw Orczyk. 2022. "Graph Pangenomes Track Genetic Variants for Crop Improvement" International Journal of Molecular Sciences 23, no. 21: 13420. https://doi.org/10.3390/ijms232113420