
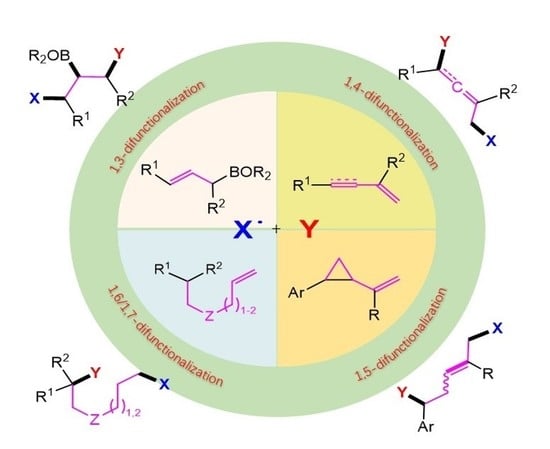
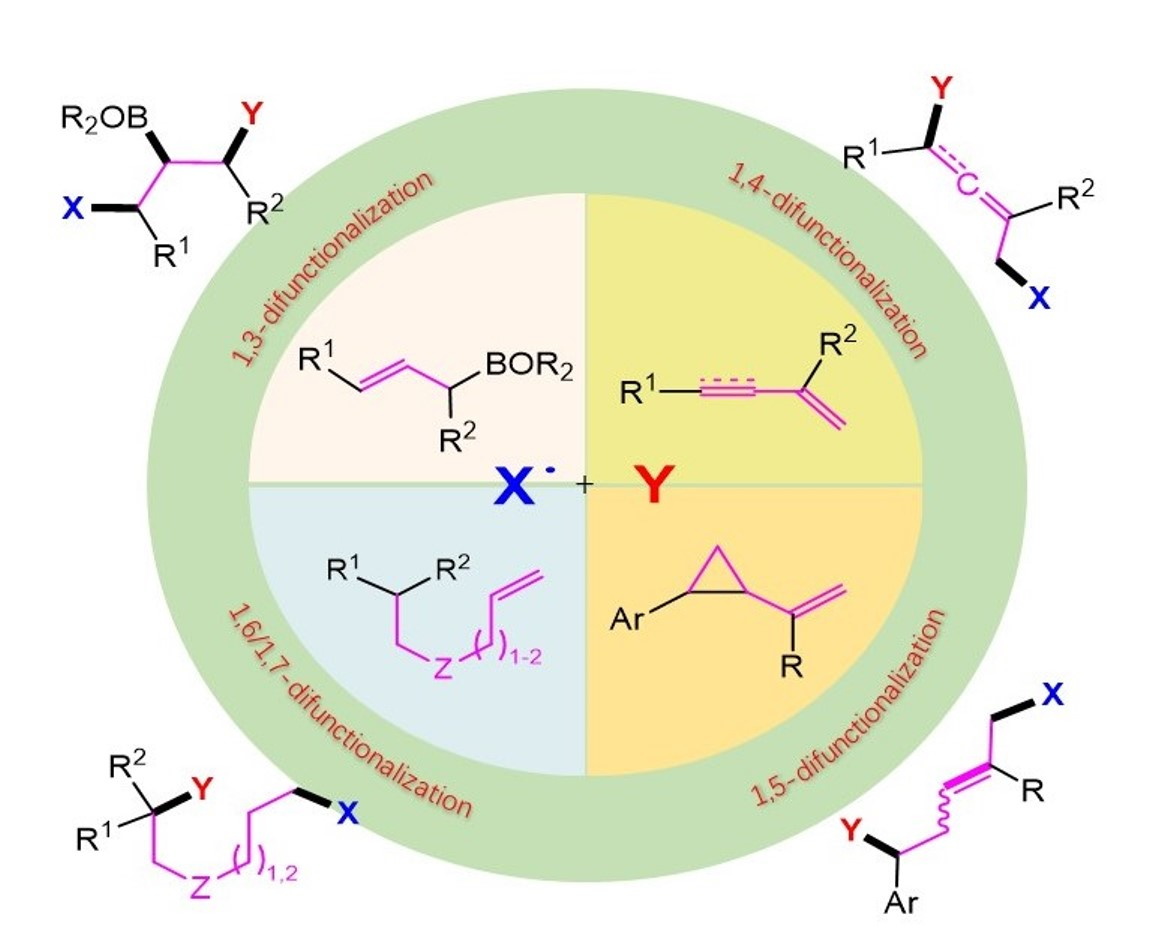
Remote Radical 1,3-, 1,4-, 1,5-, 1,6- and 1,7-Difunctionalization Reactions †

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
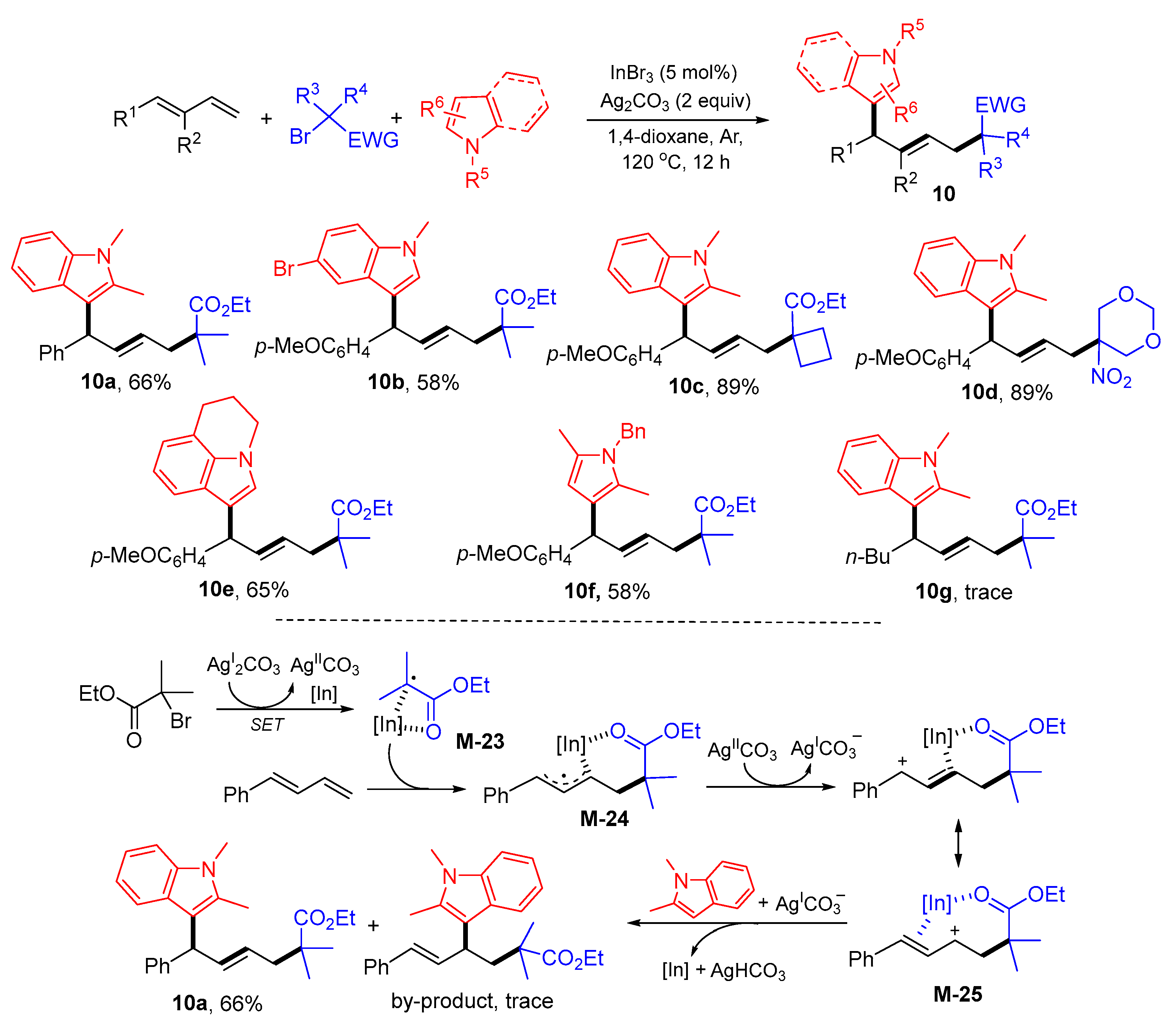{kind=link}
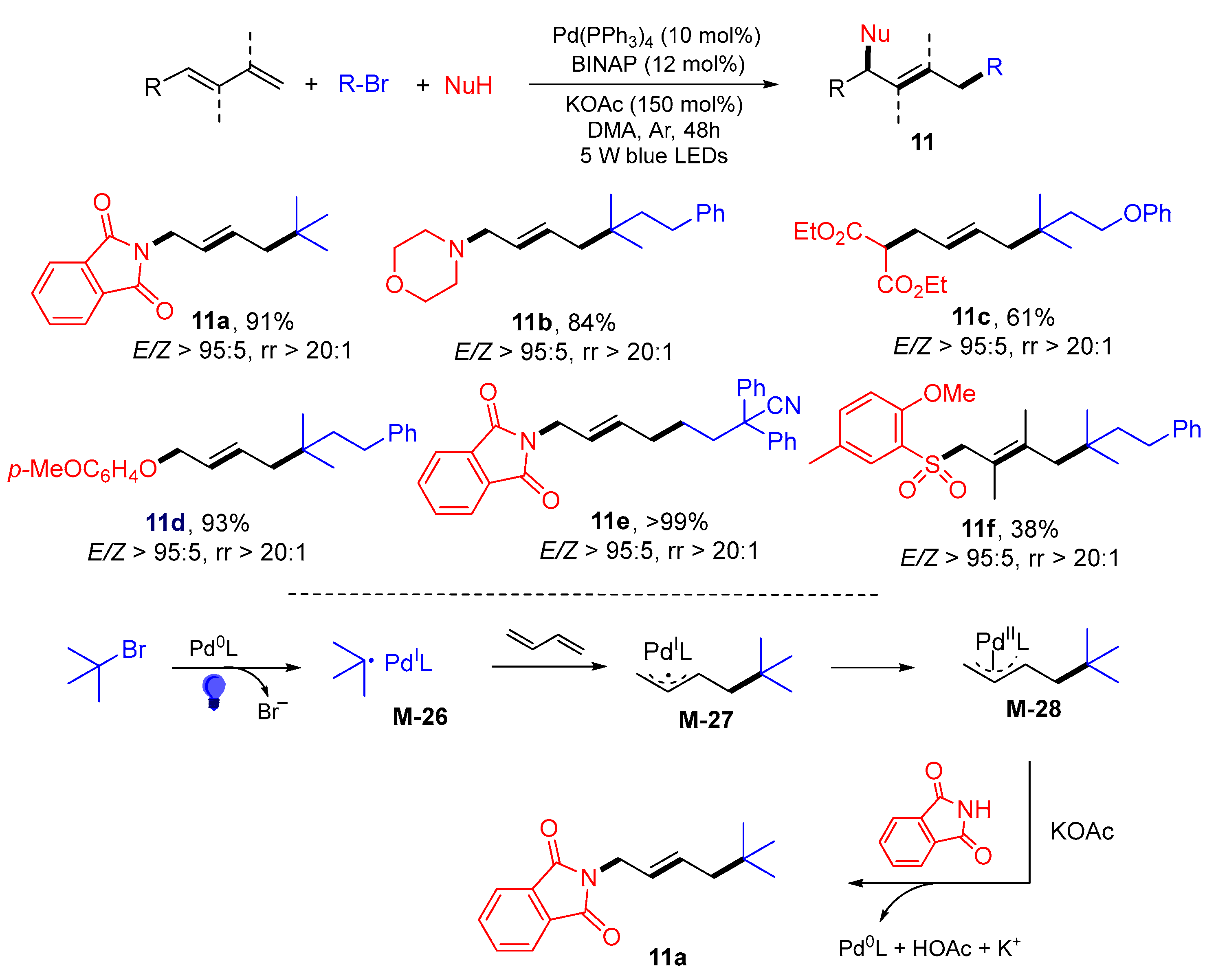{kind=link}
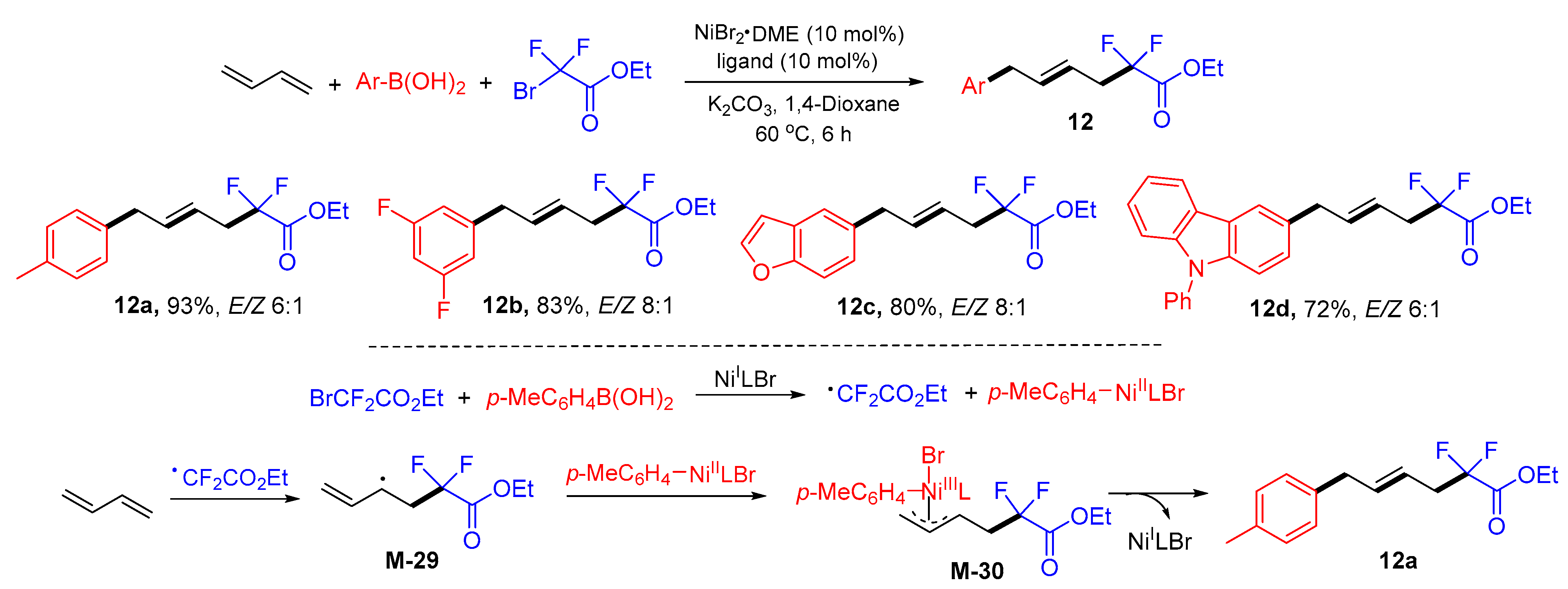{kind=link}
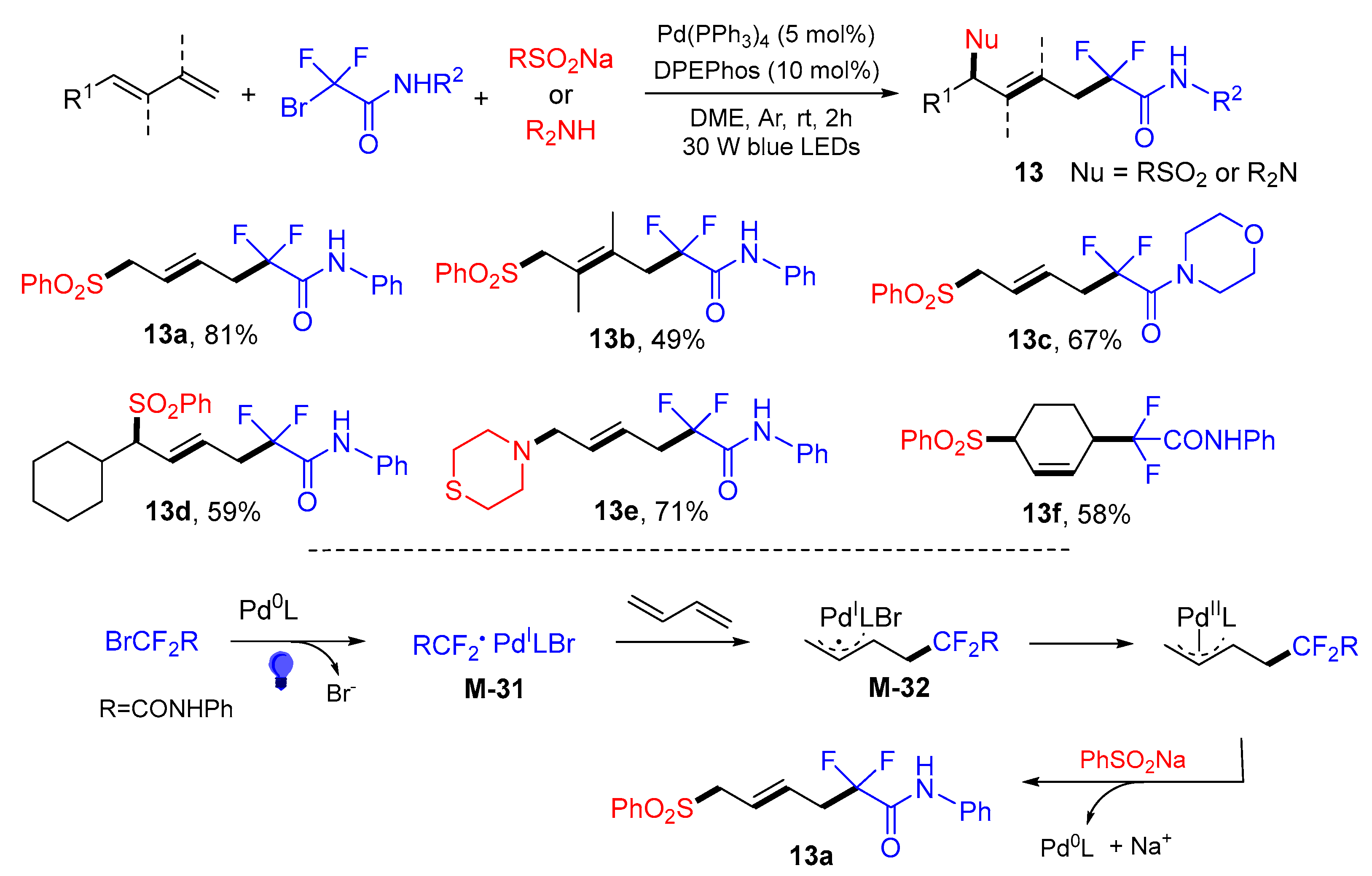{kind=link}
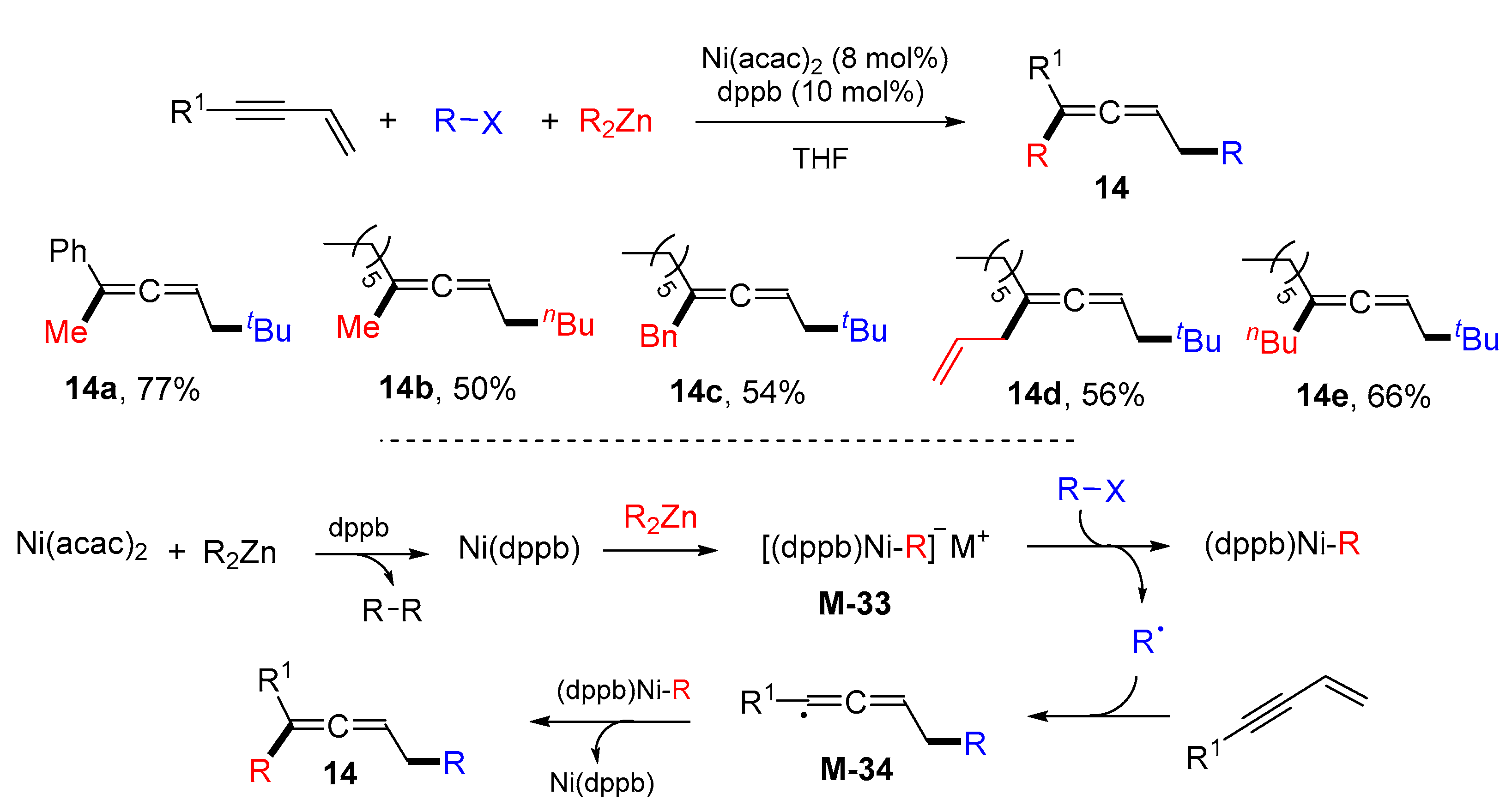{kind=link}
{kind=link}
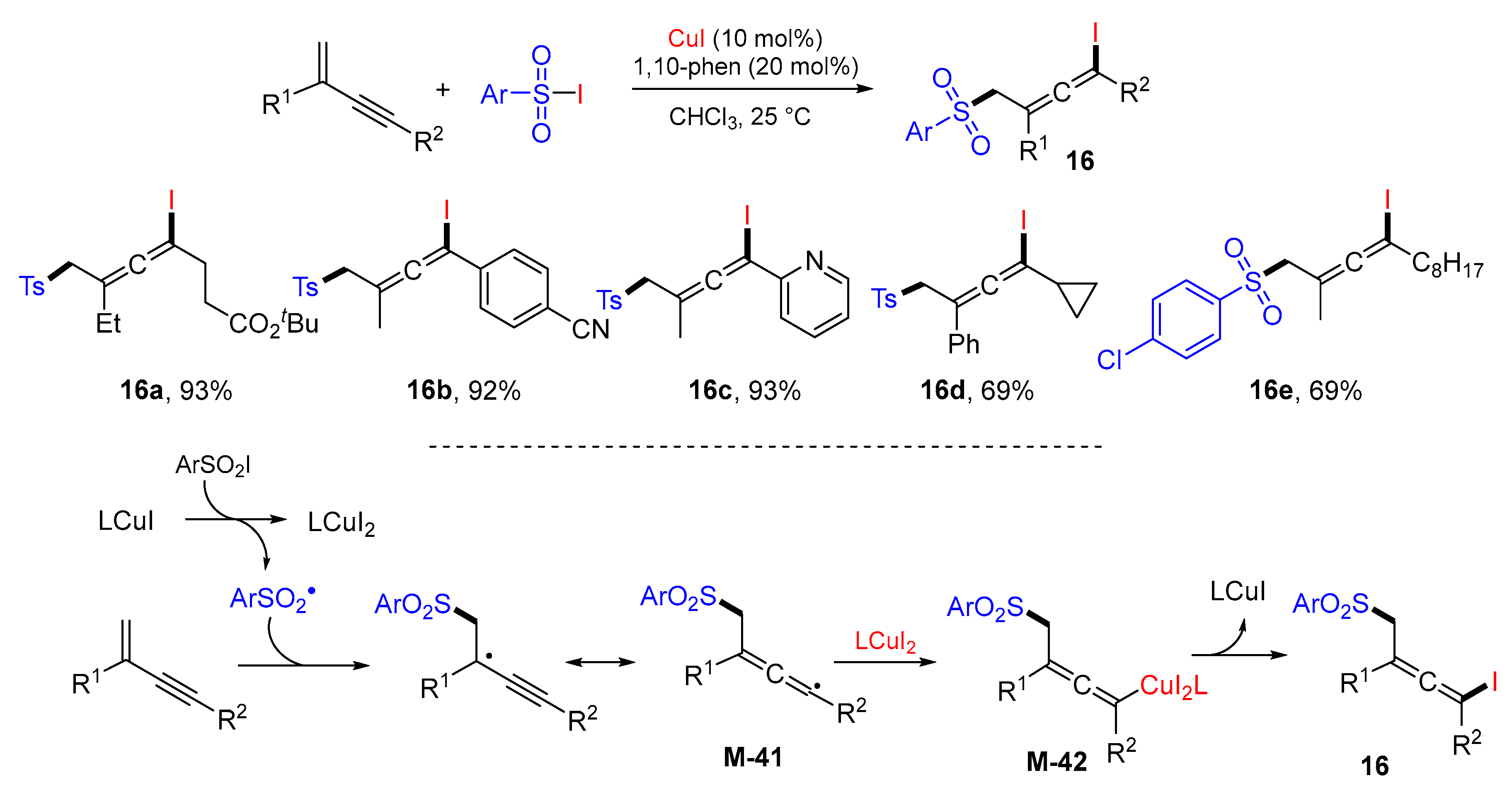{kind=link}
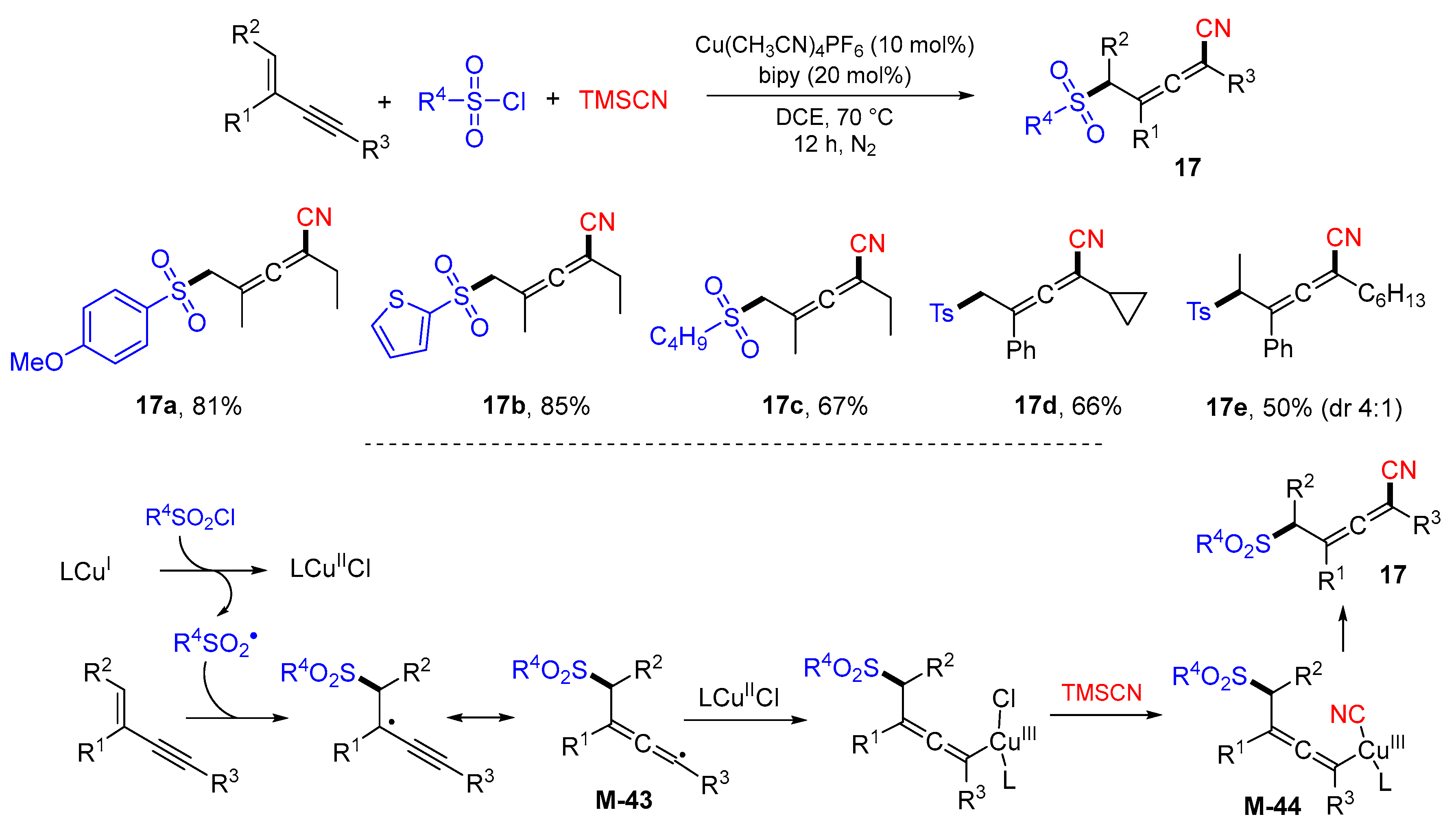{kind=link}
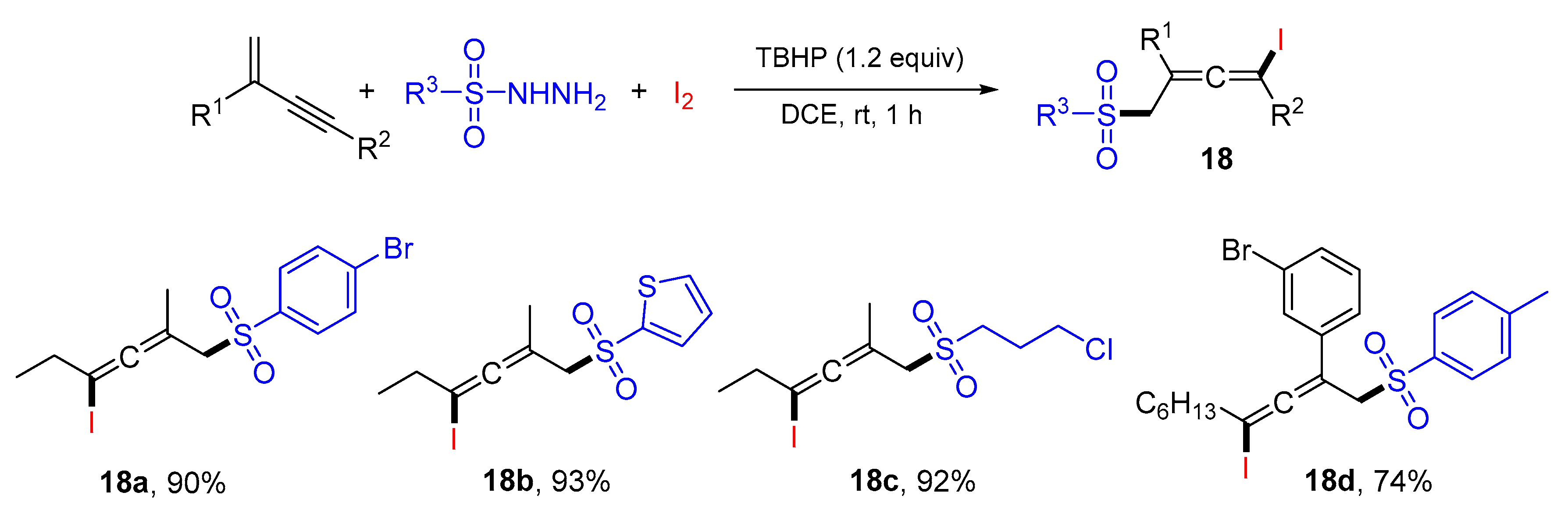{kind=link}
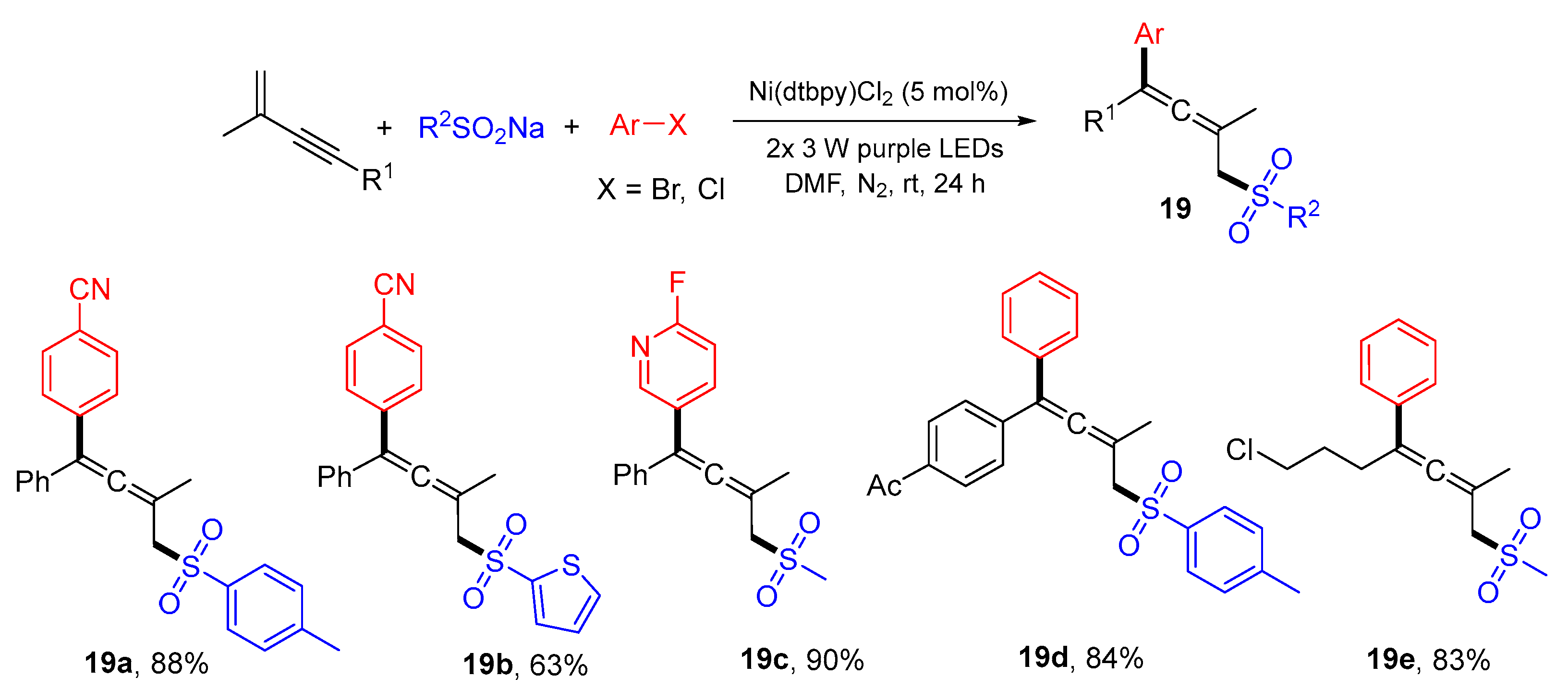{kind=link}
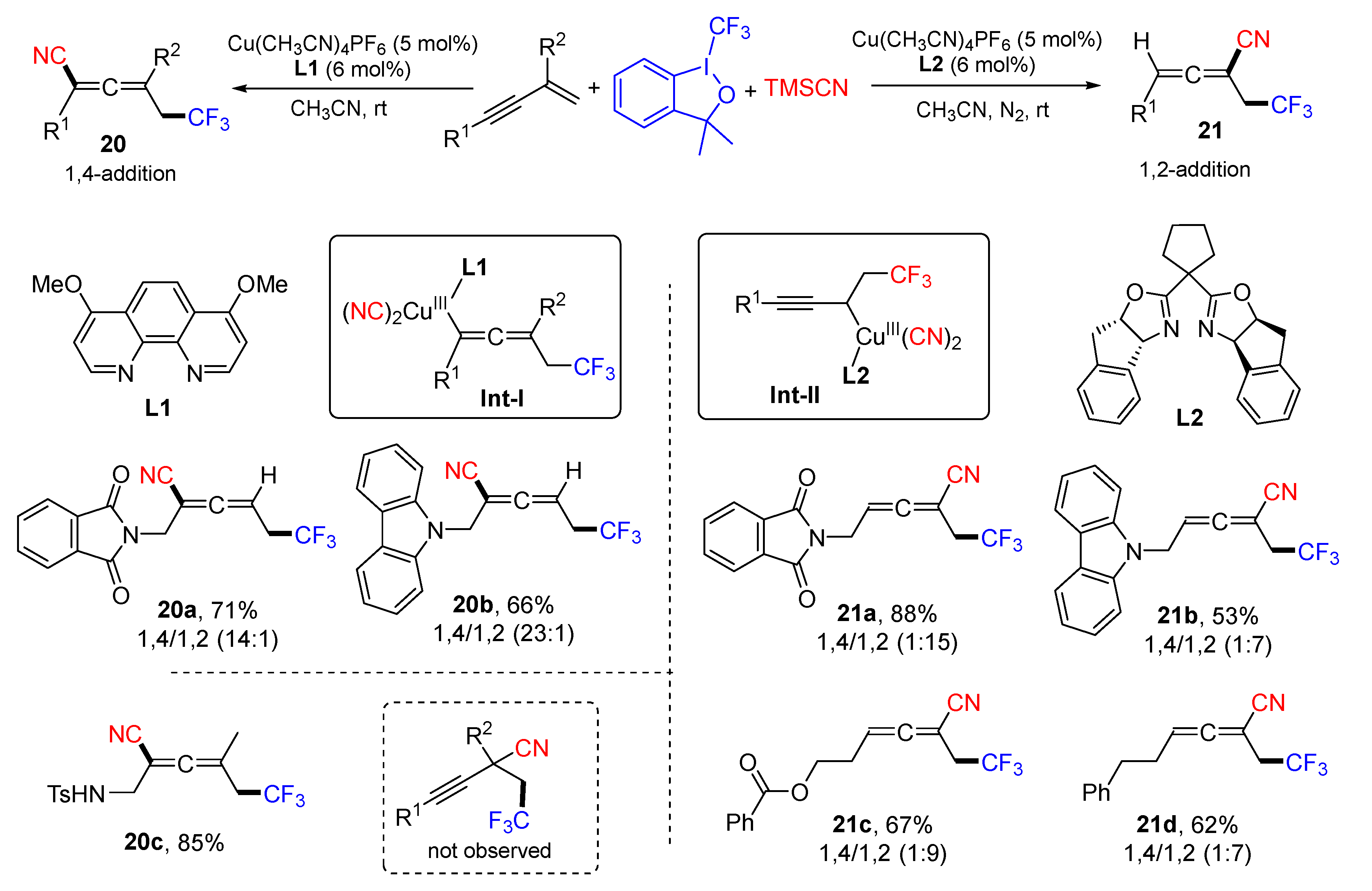{kind=link}
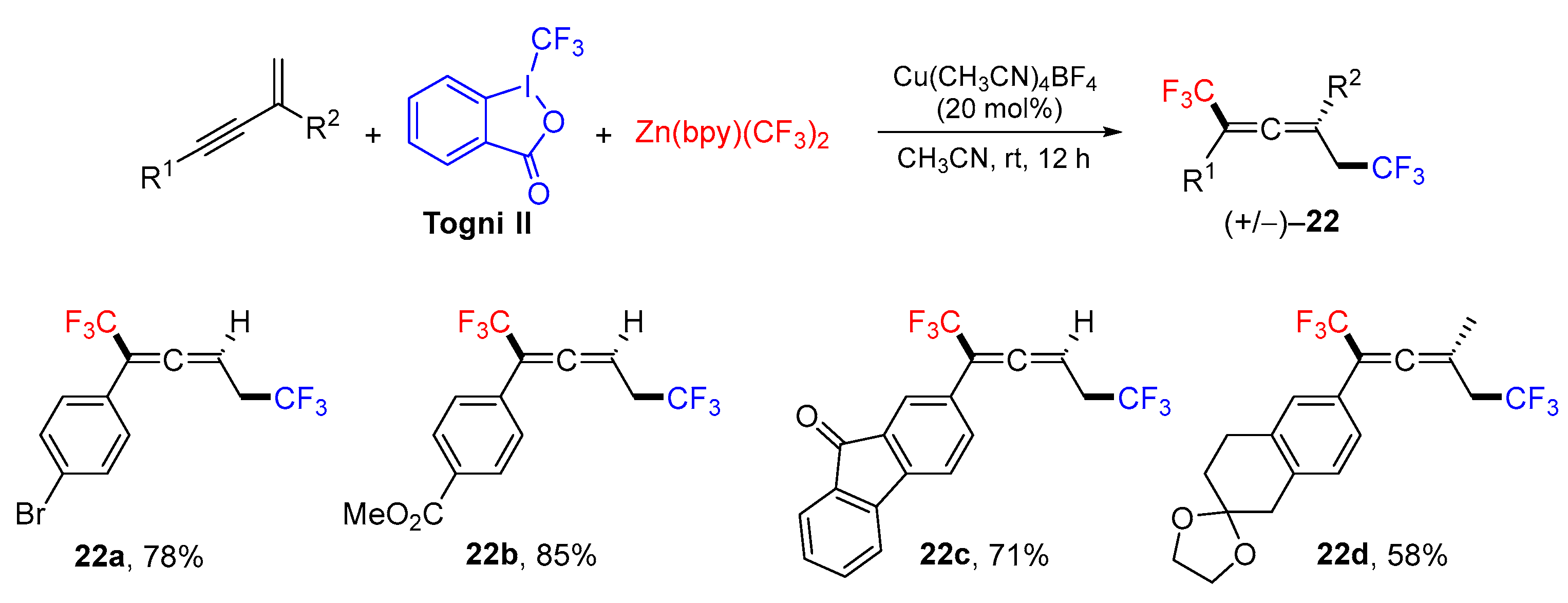{kind=link}
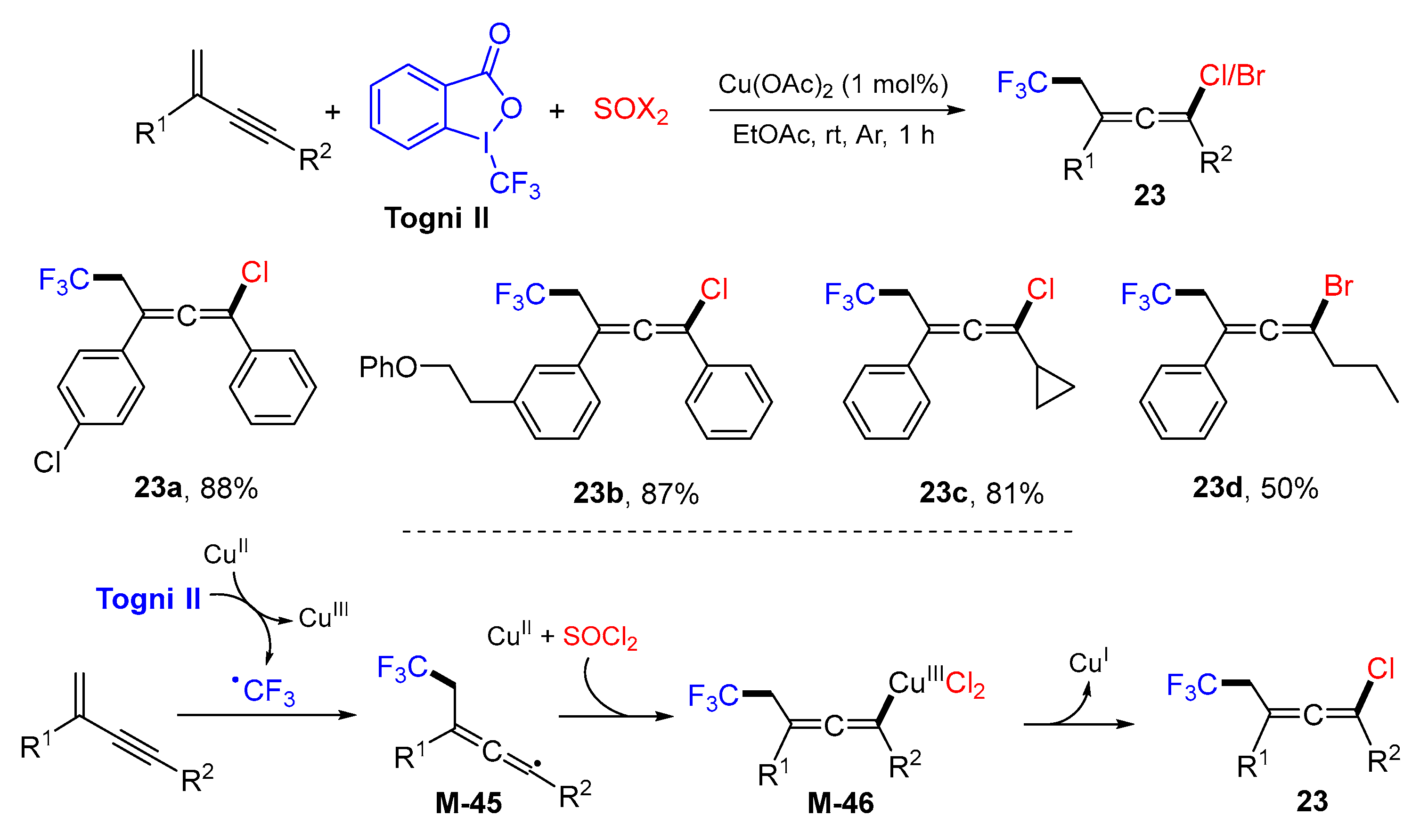{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
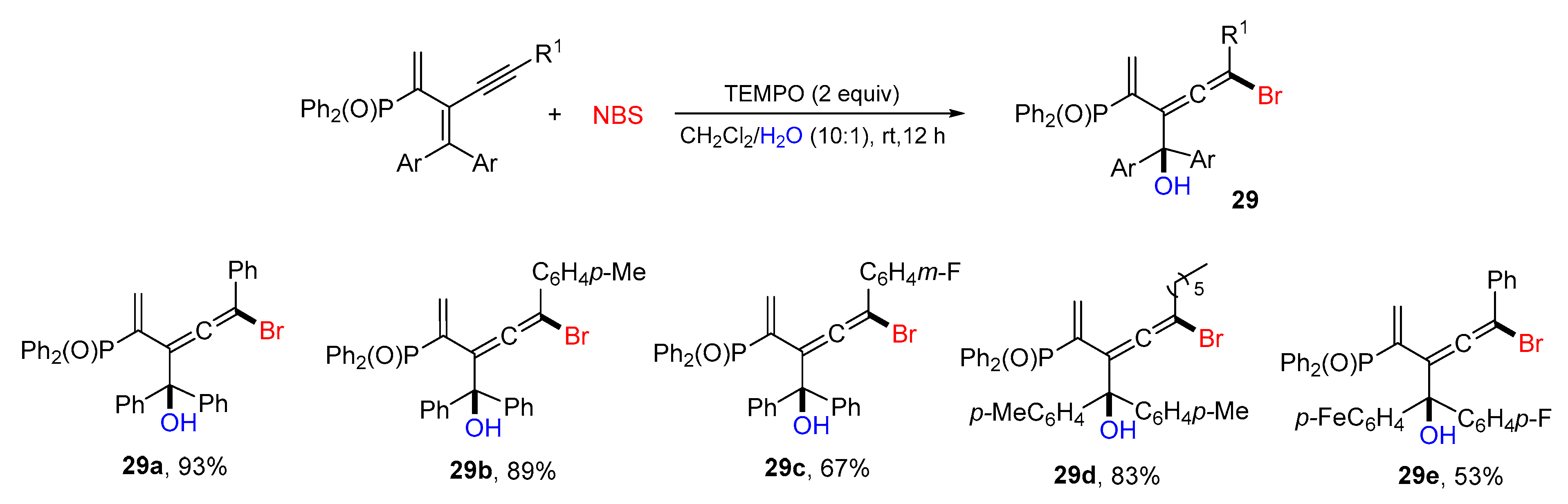{kind=link}
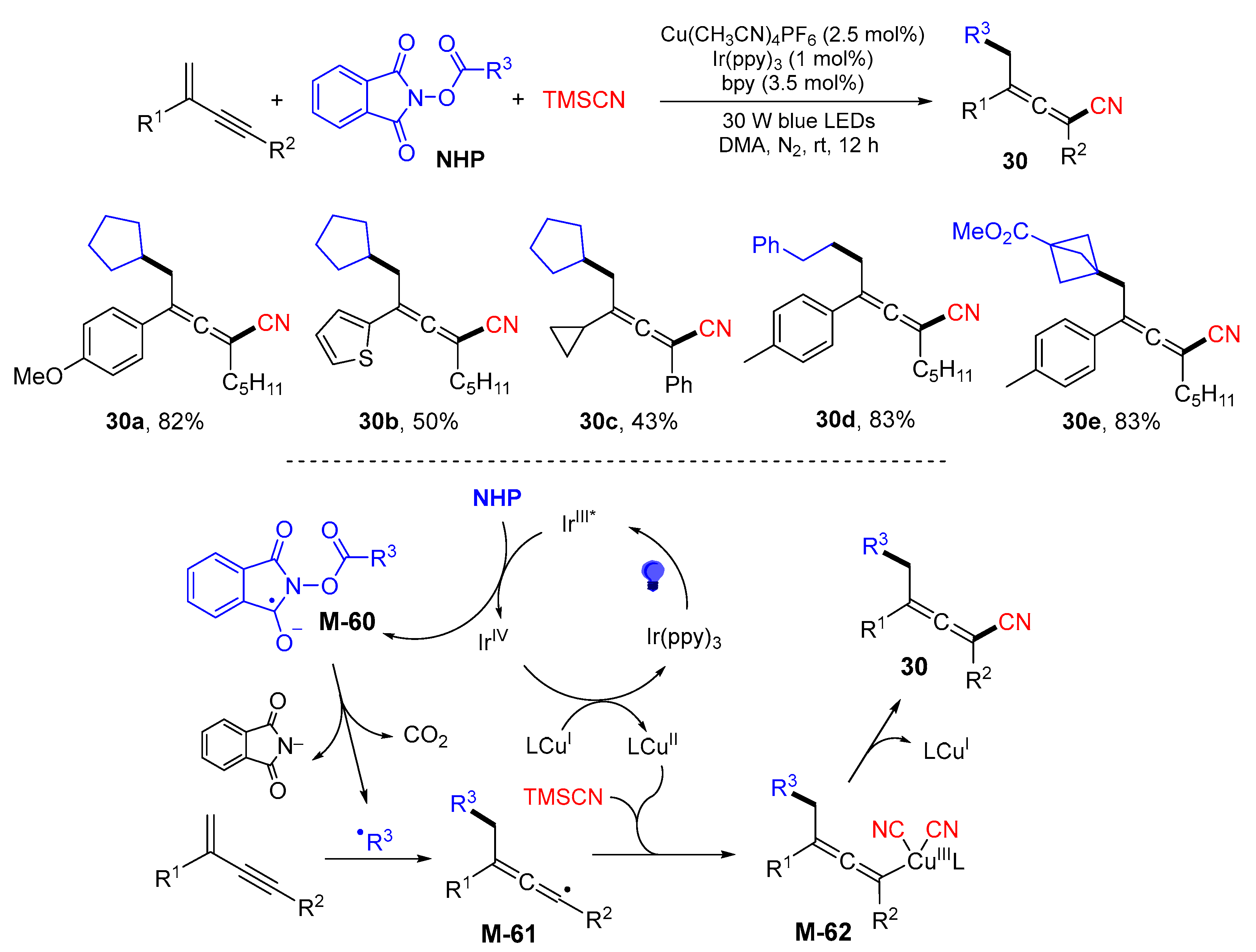{kind=link}
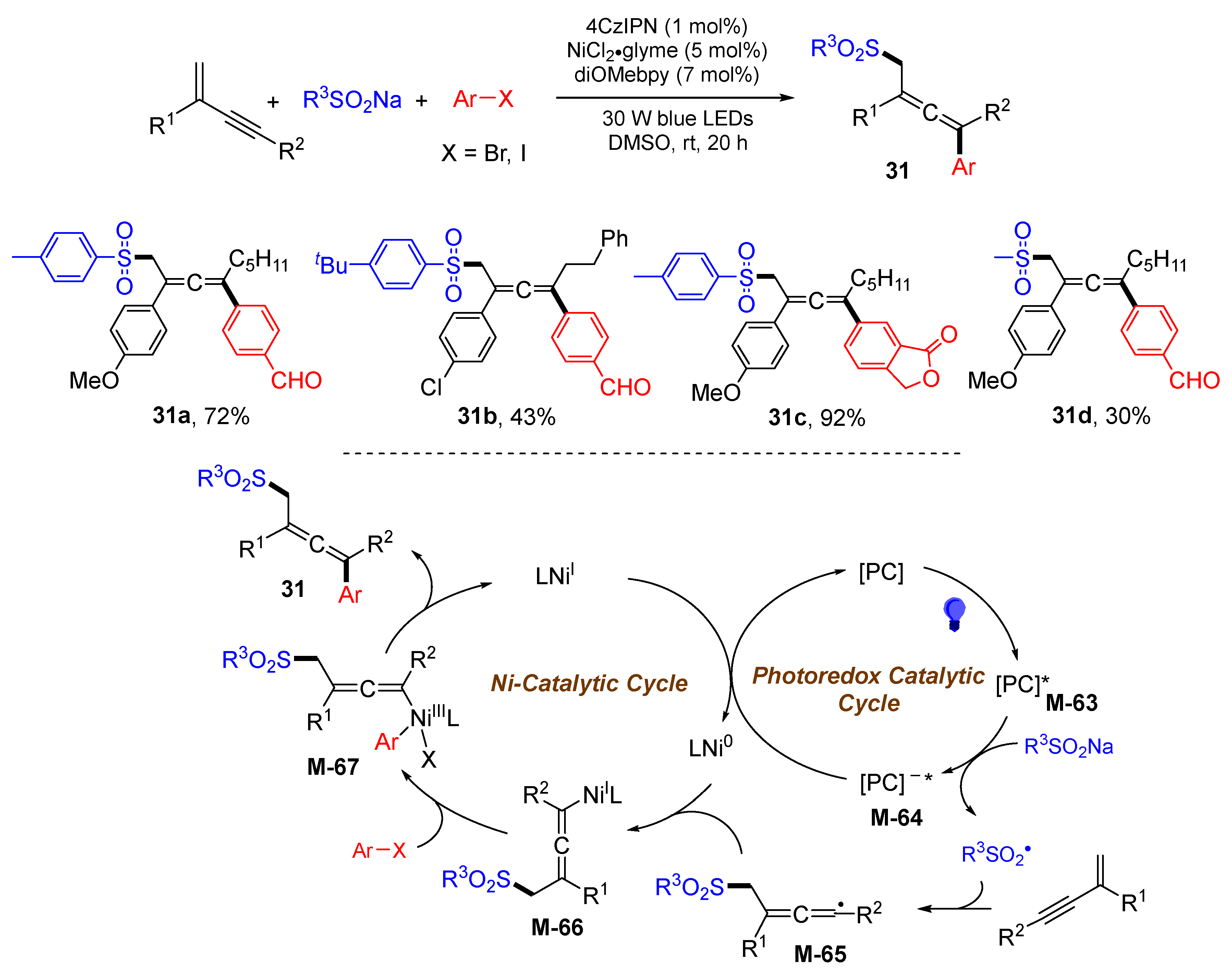{kind=link}
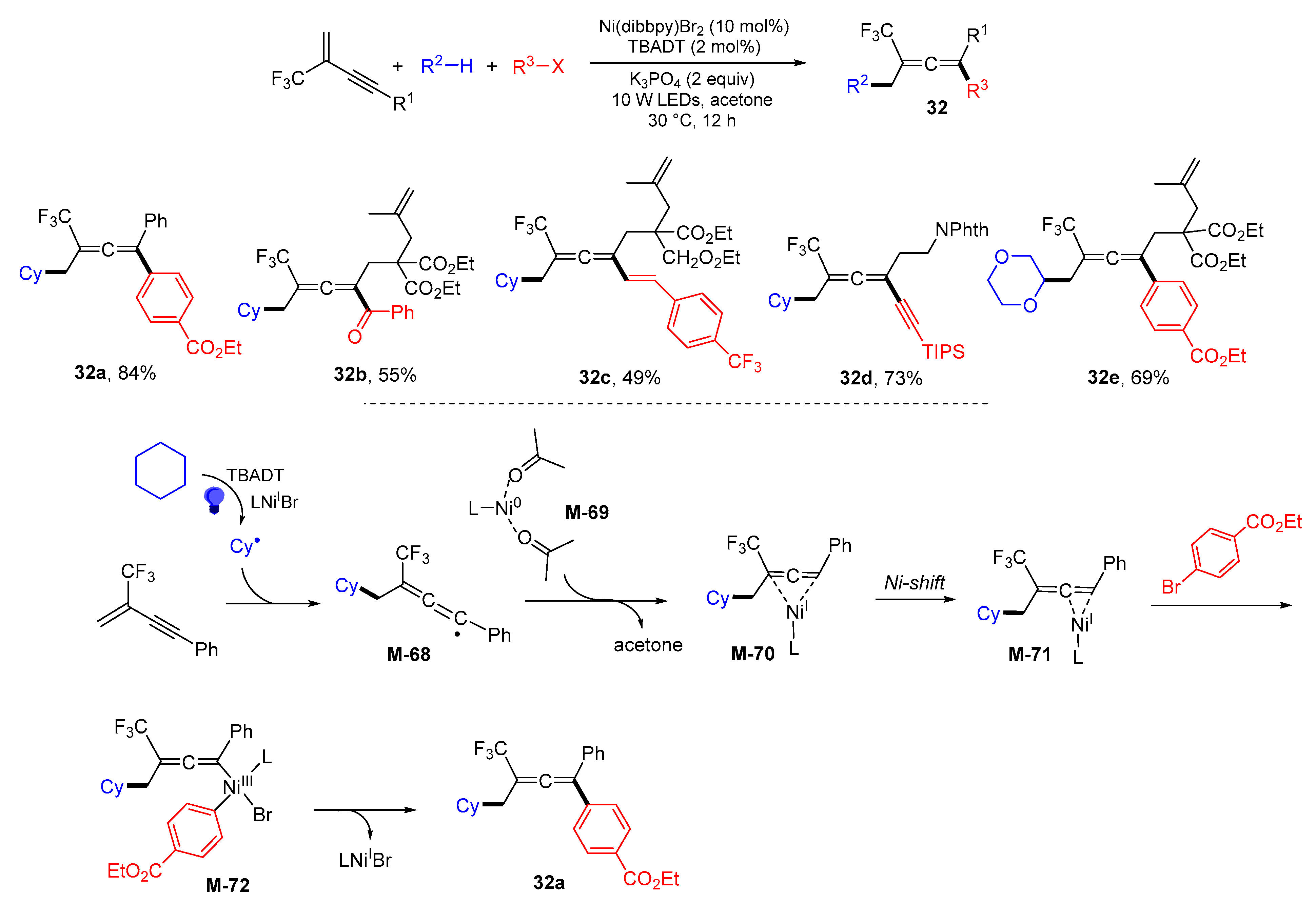{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
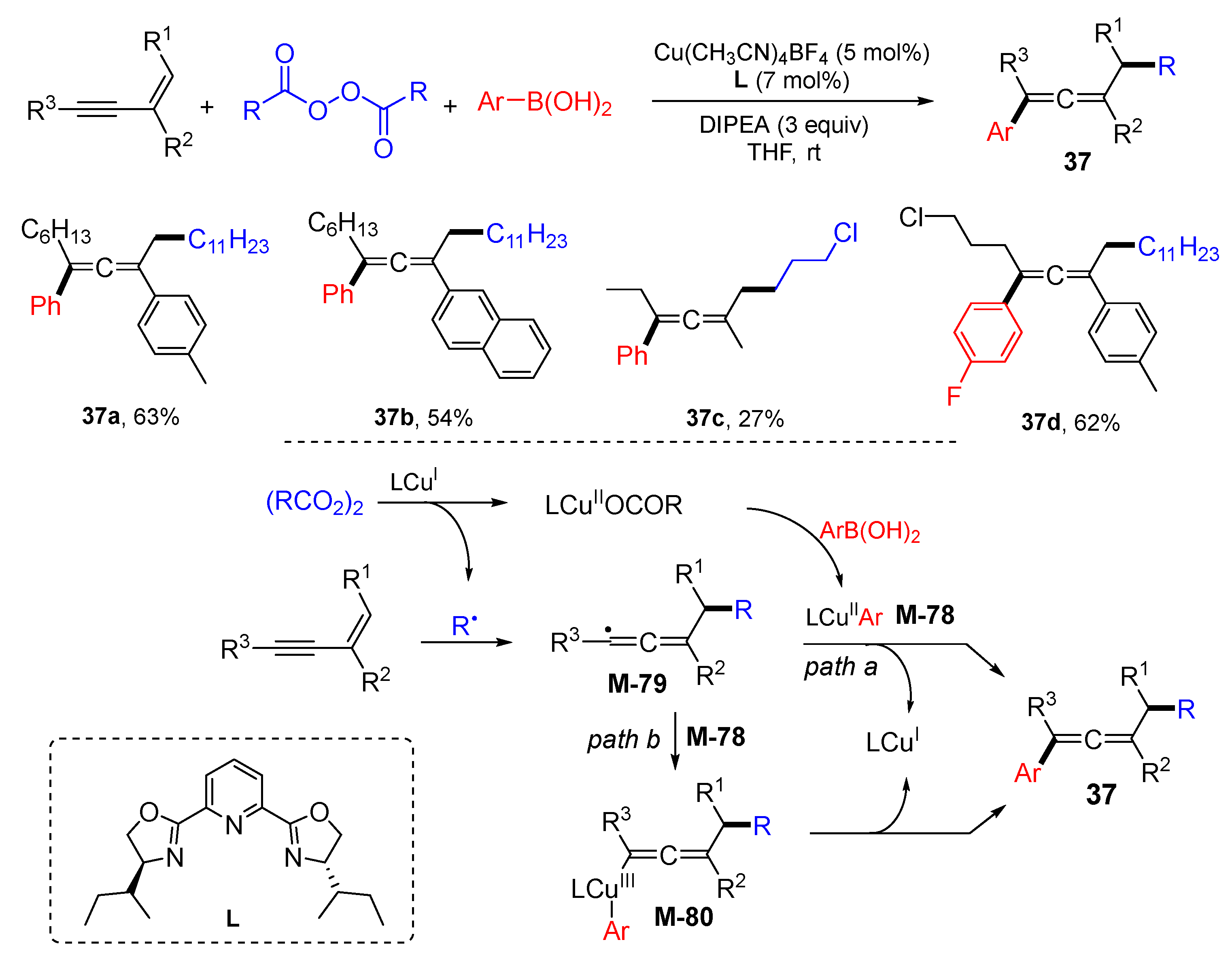{kind=link}
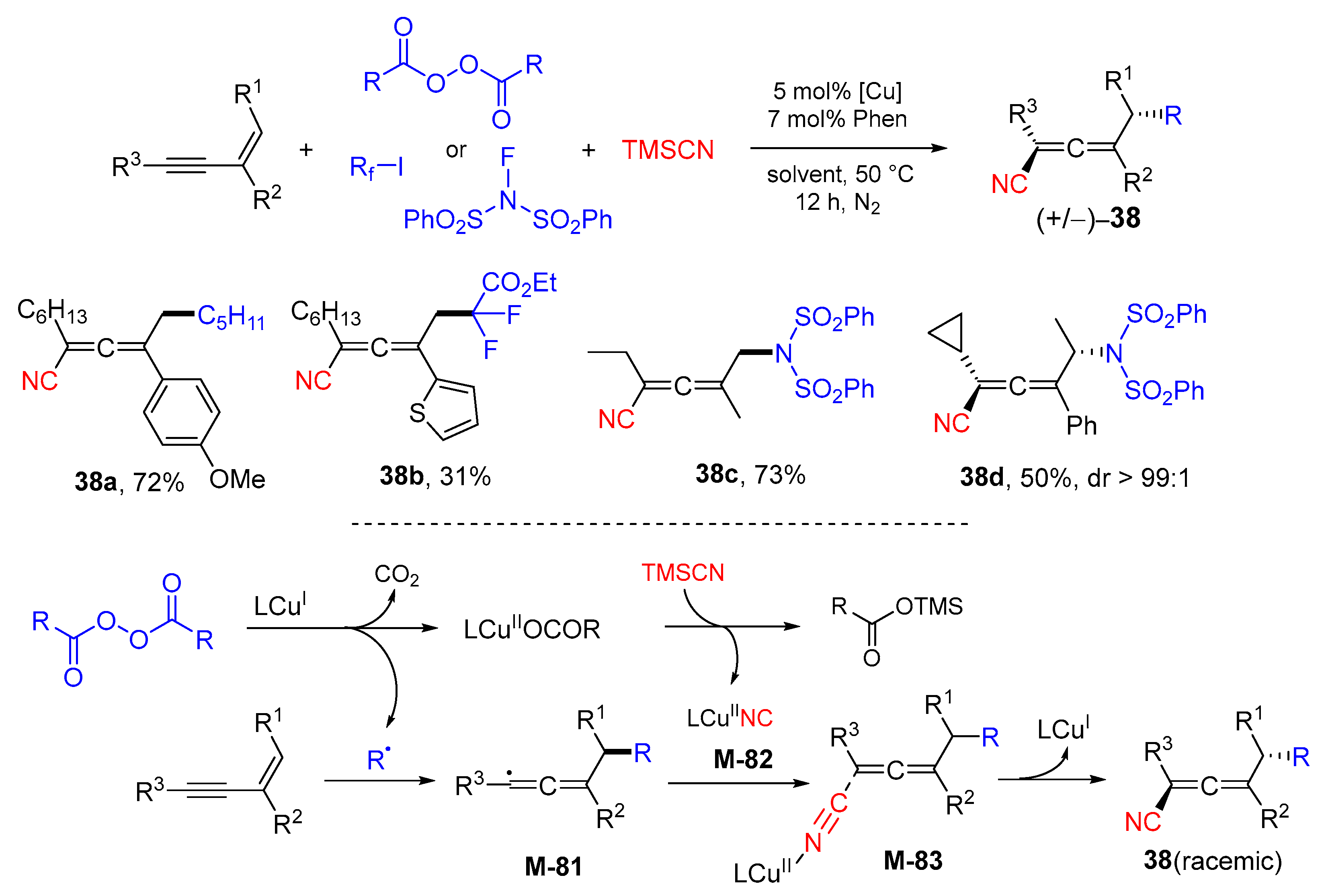{kind=link}
{kind=link}
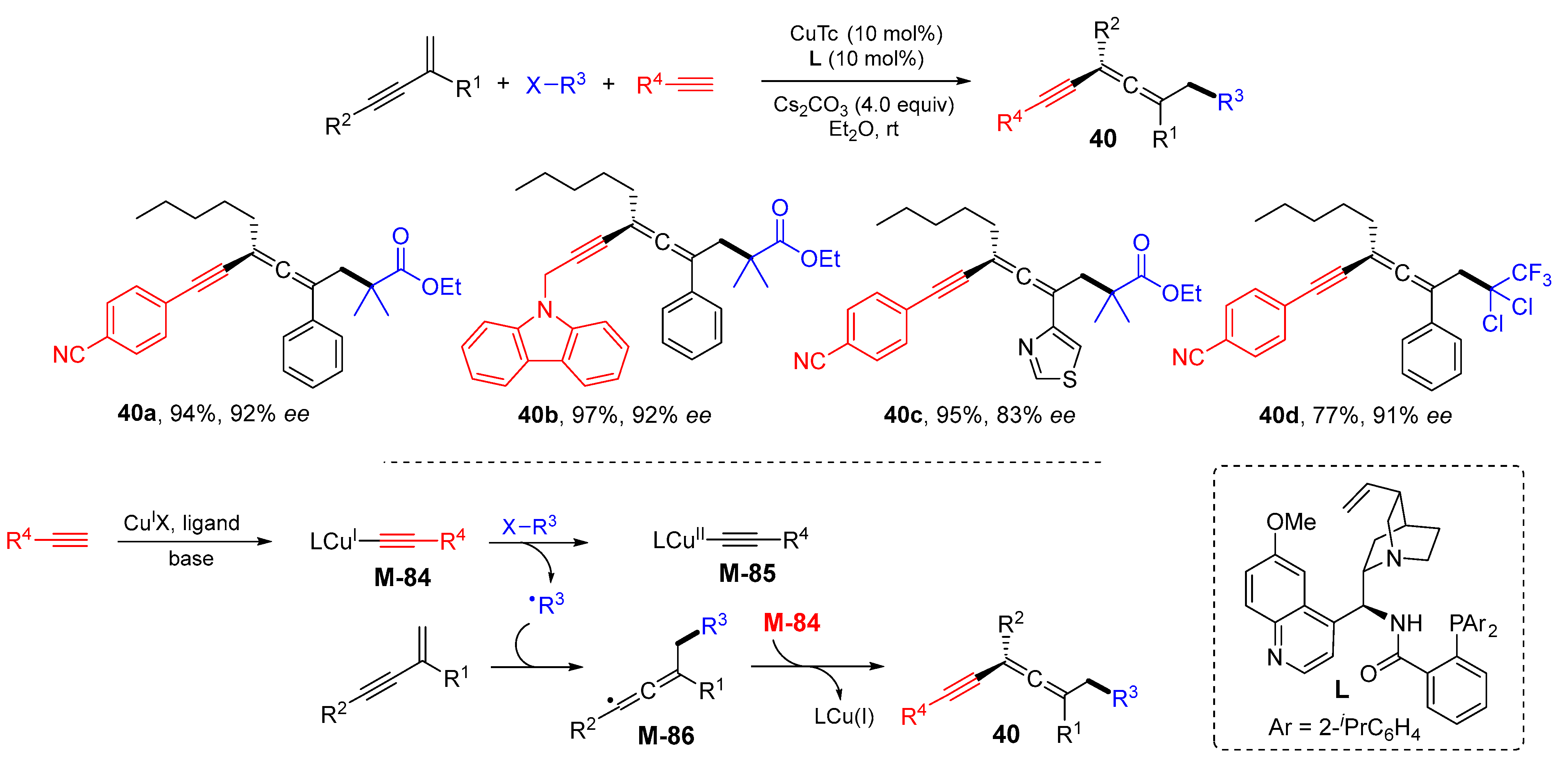{kind=link}
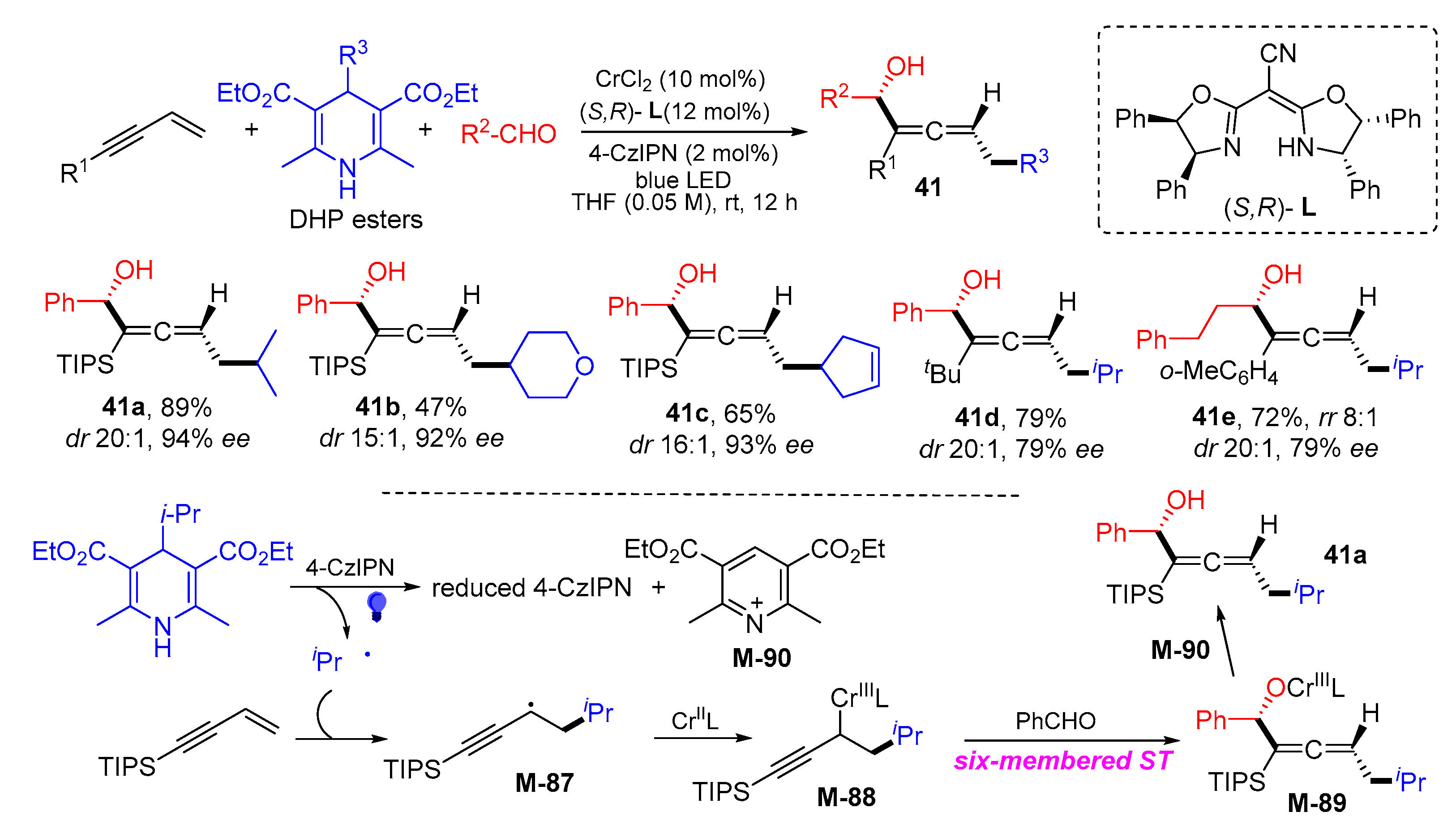{kind=link}
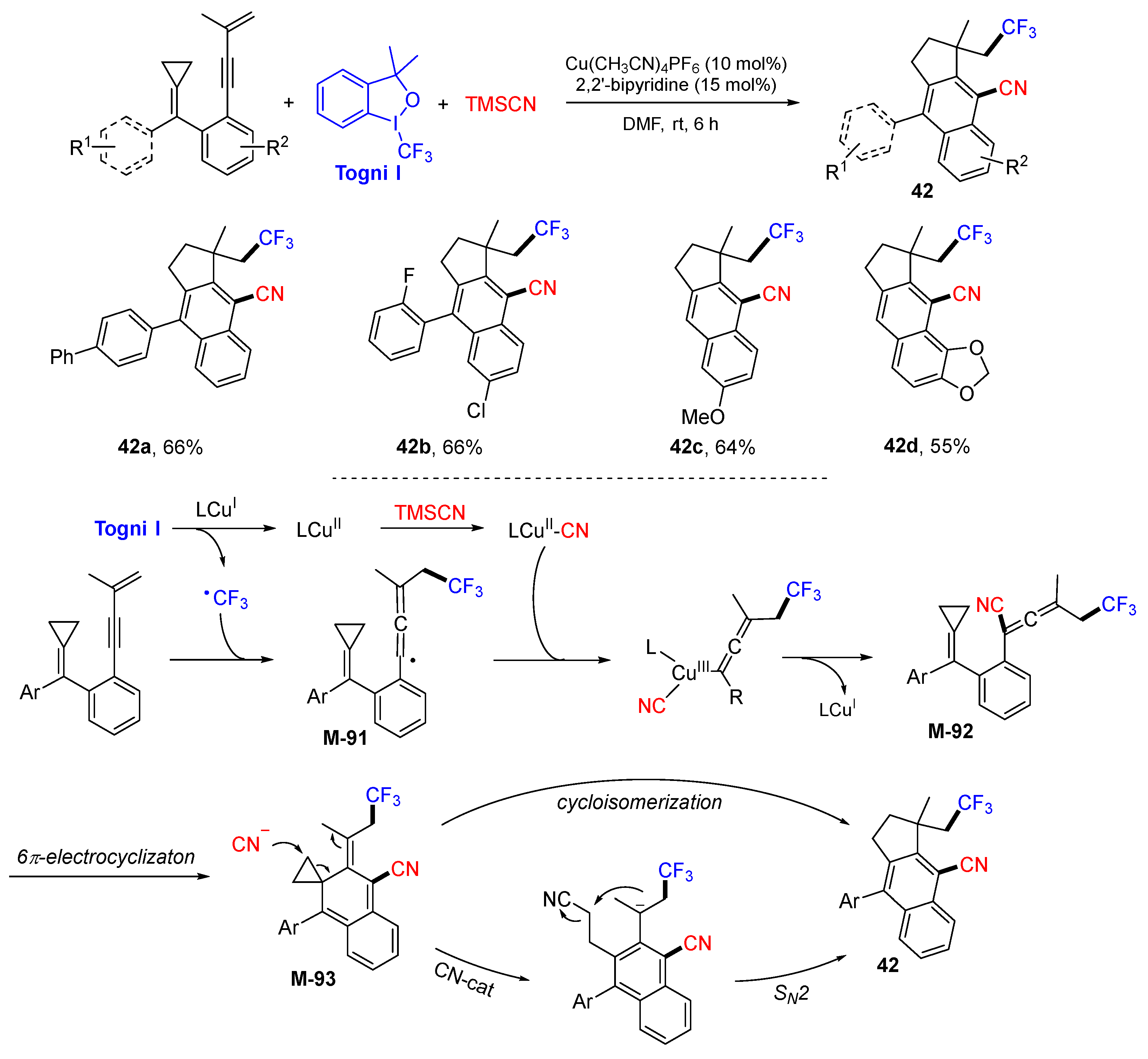{kind=link}
{kind=link}
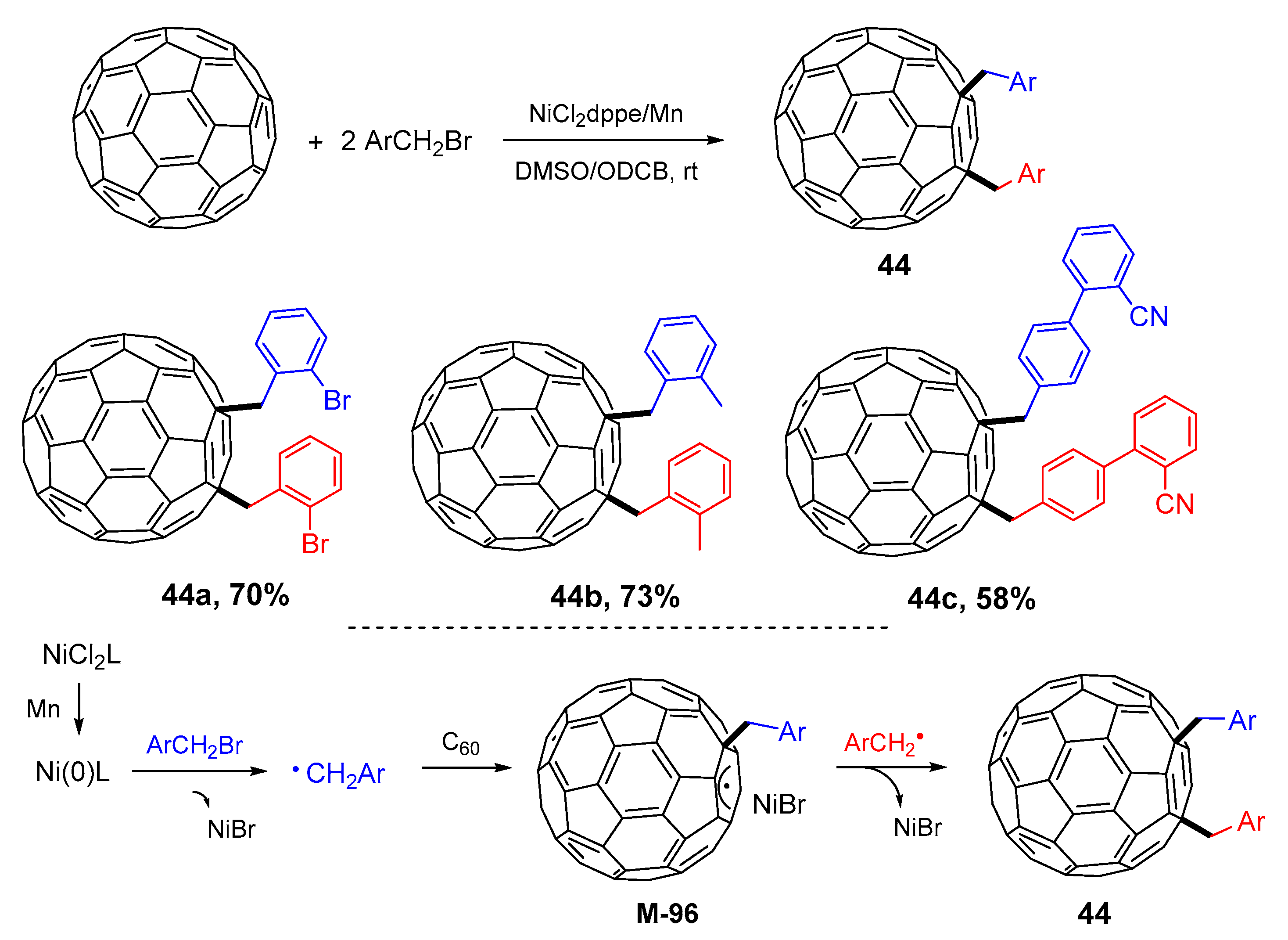{kind=link}
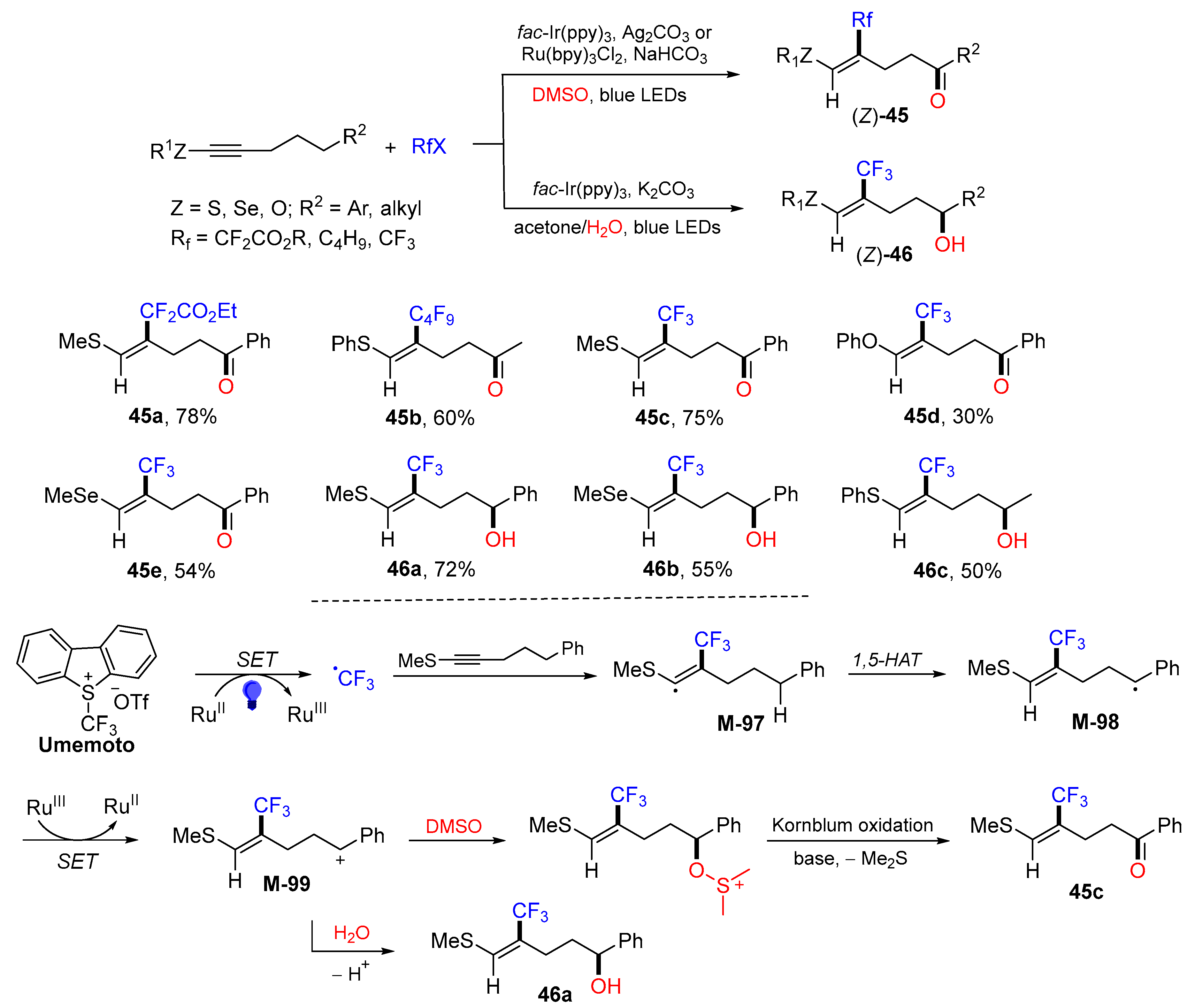{kind=link}
{kind=link}
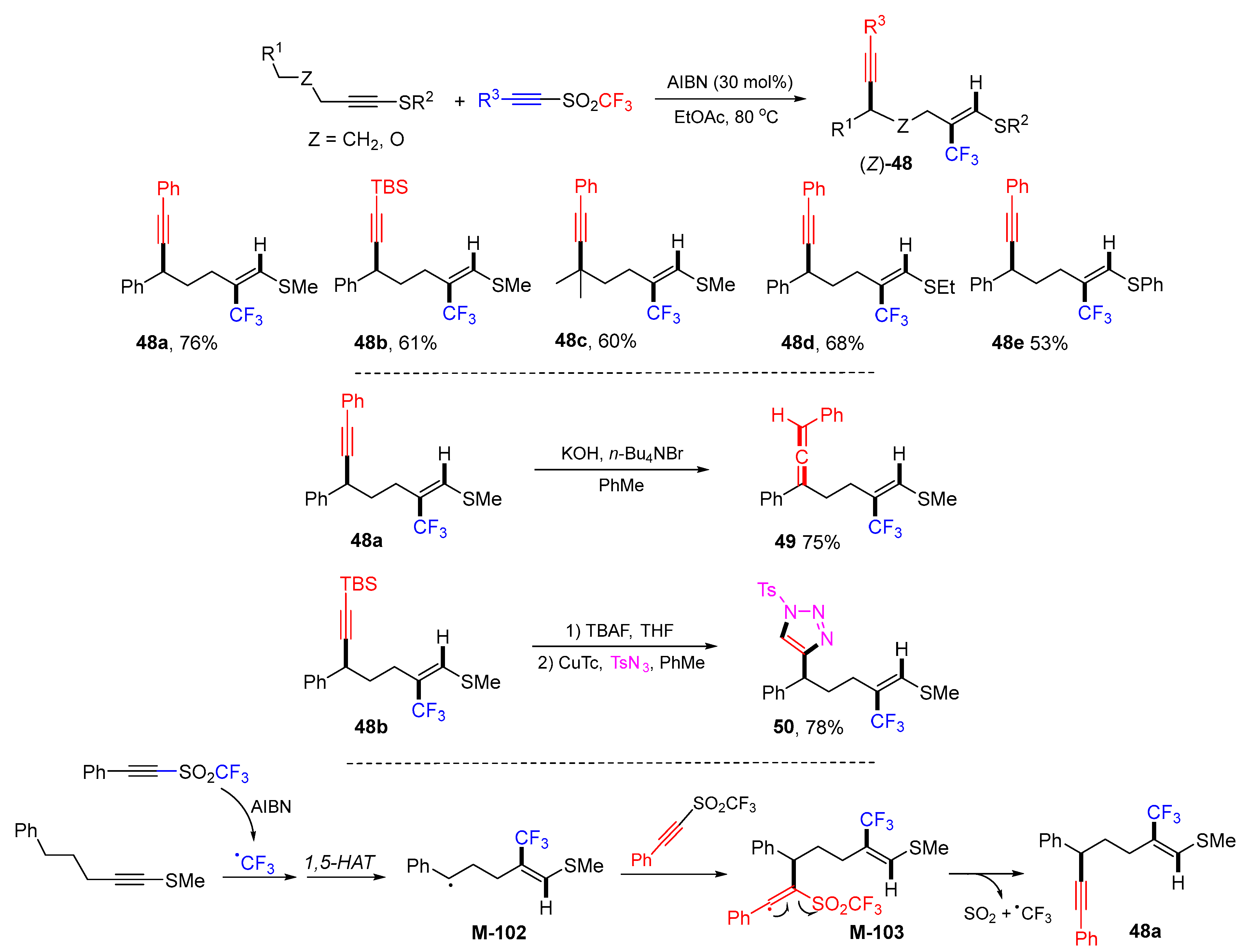{kind=link}
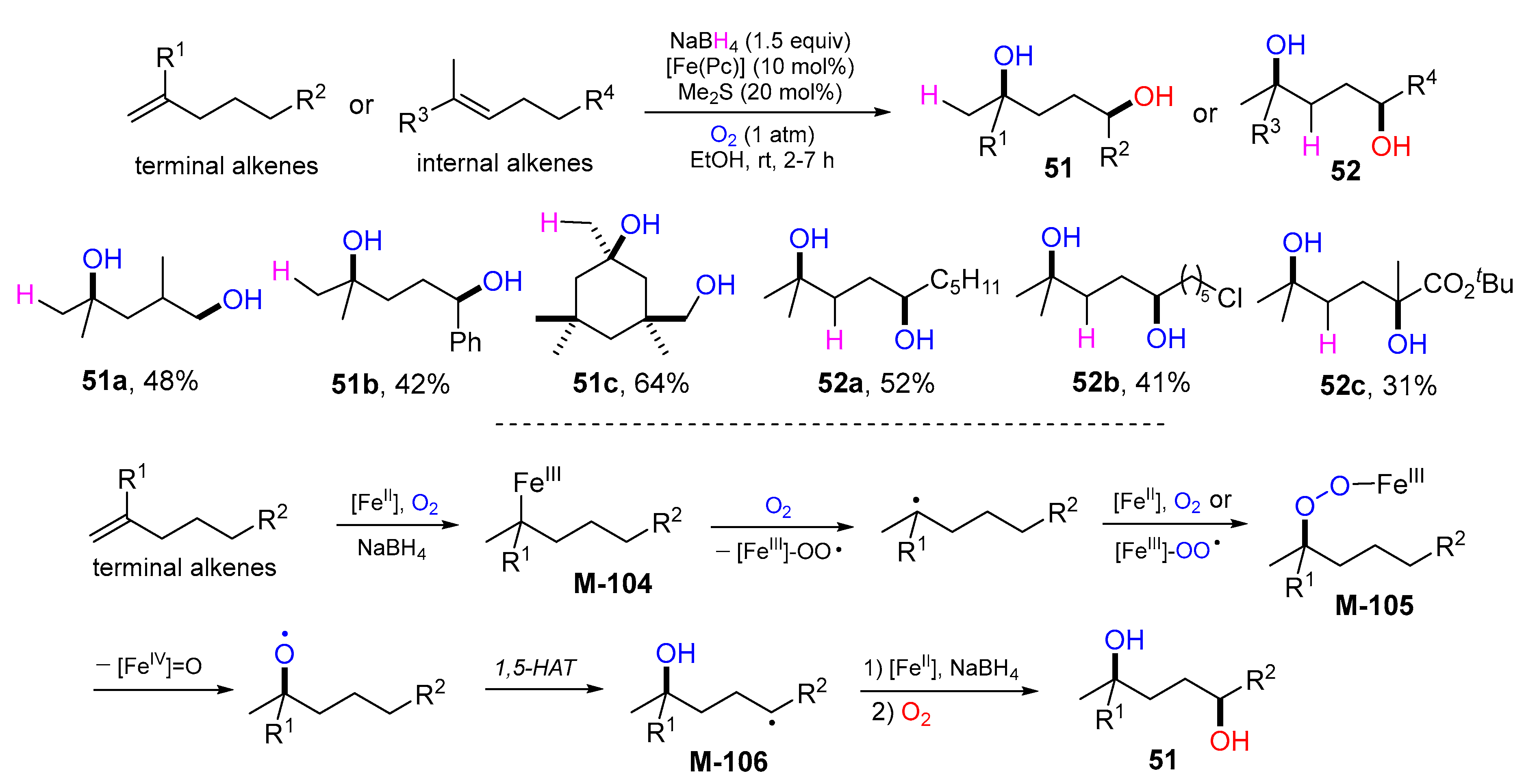{kind=link}
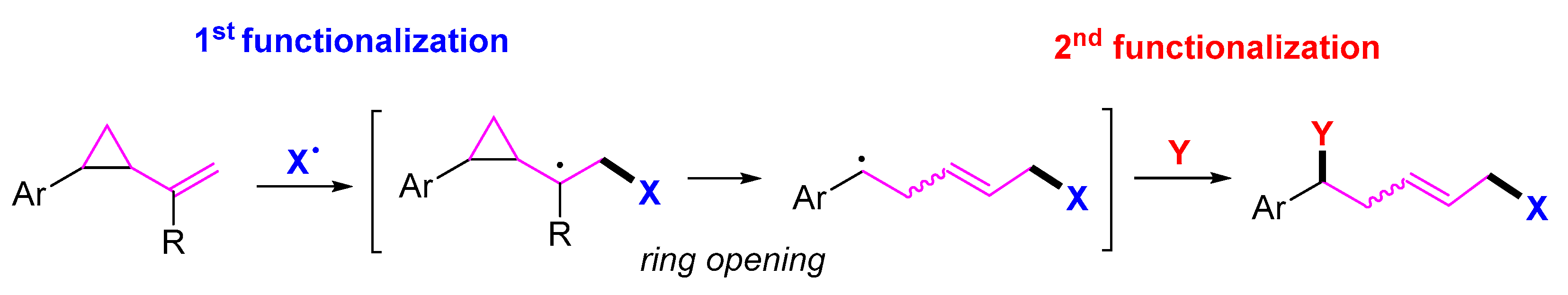{kind=link}
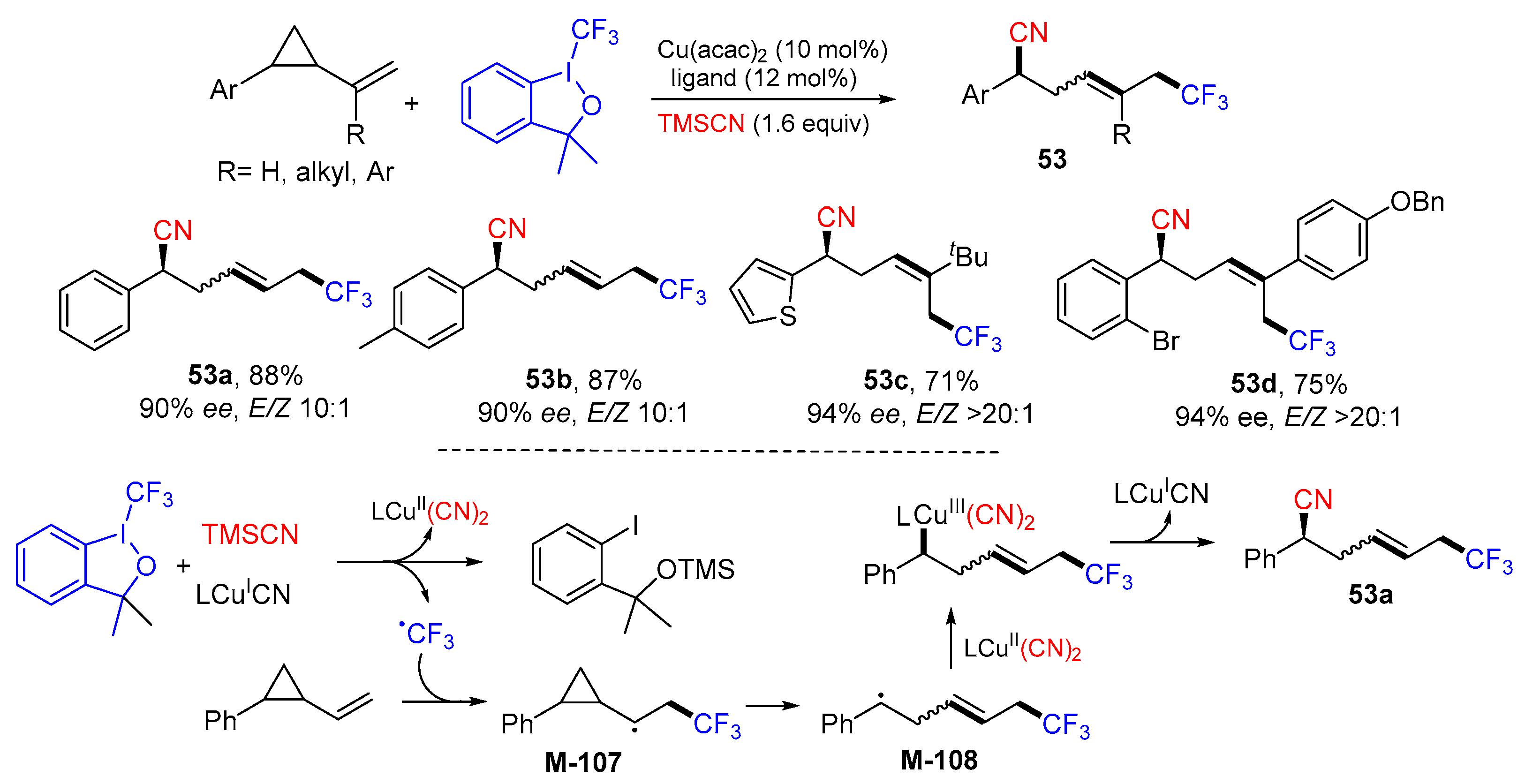{kind=link}
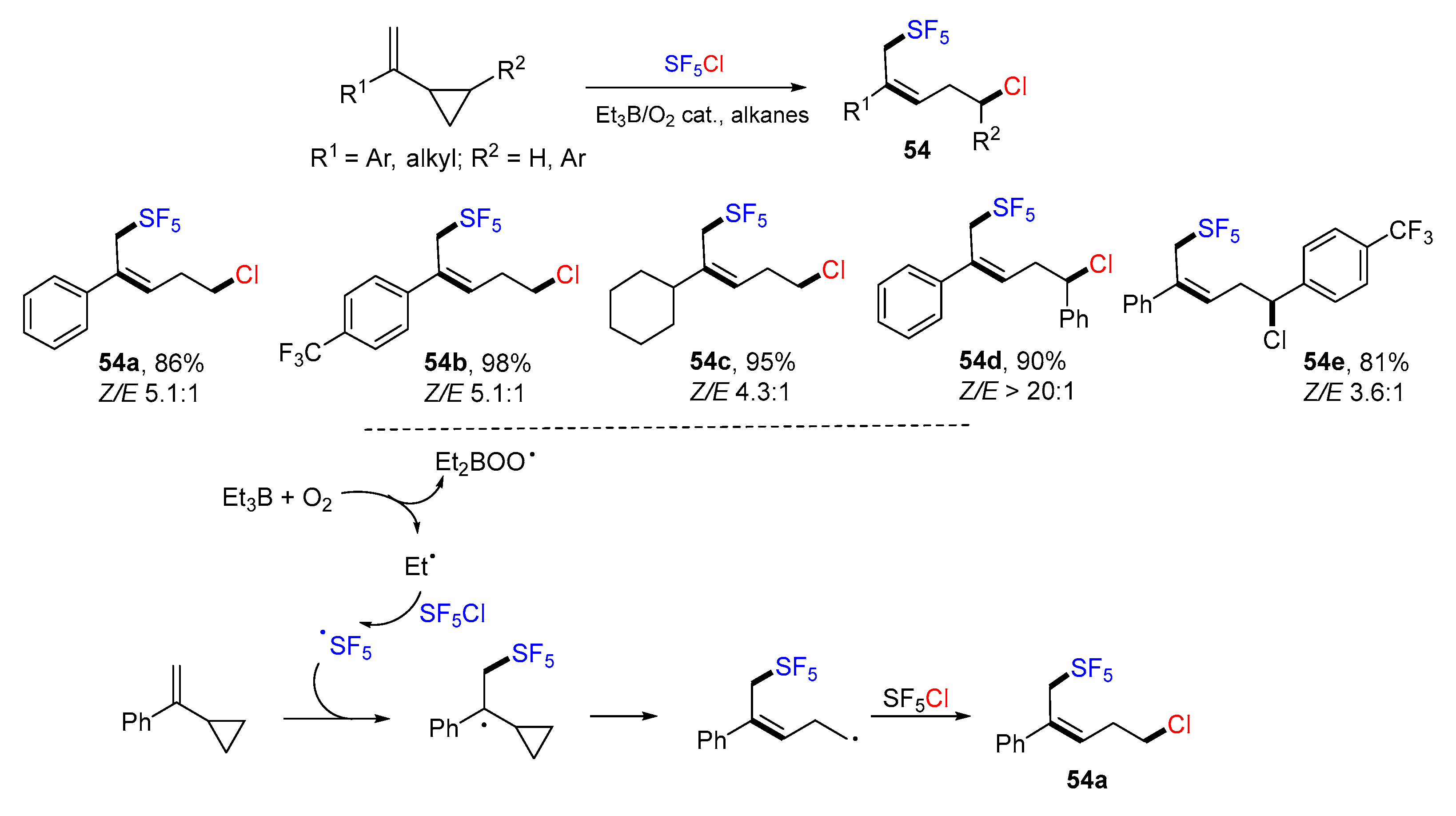{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
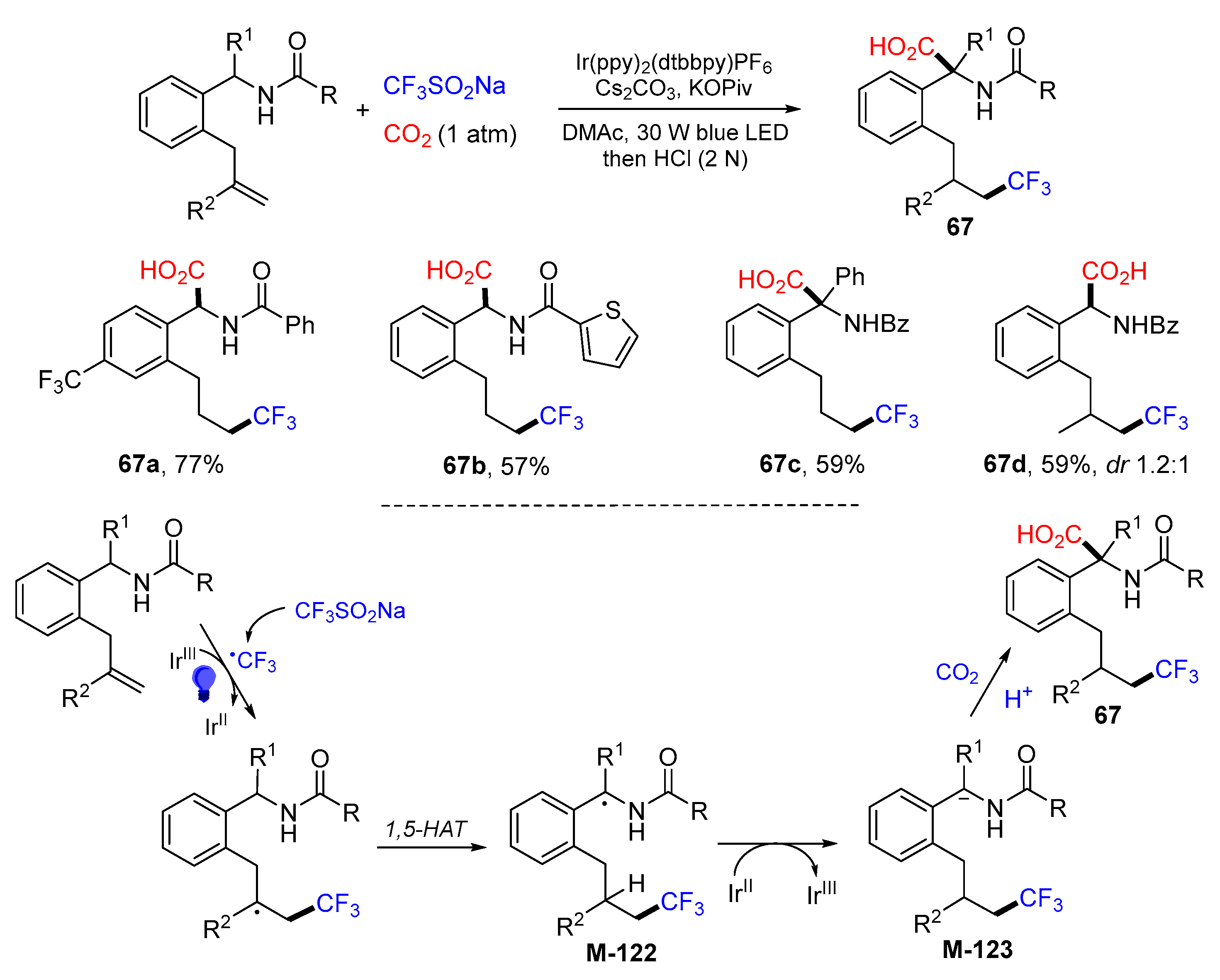{kind=link}
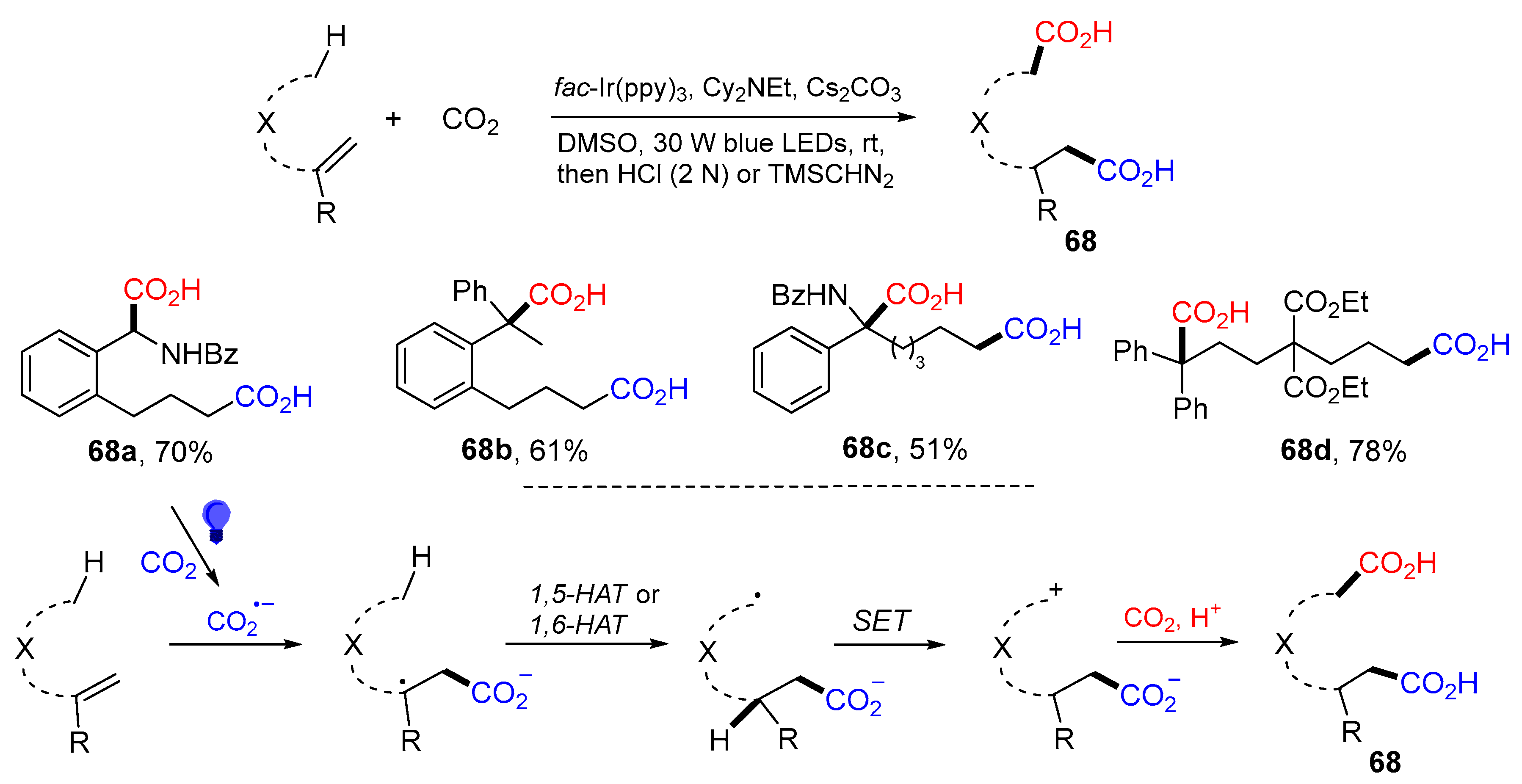{kind=link}
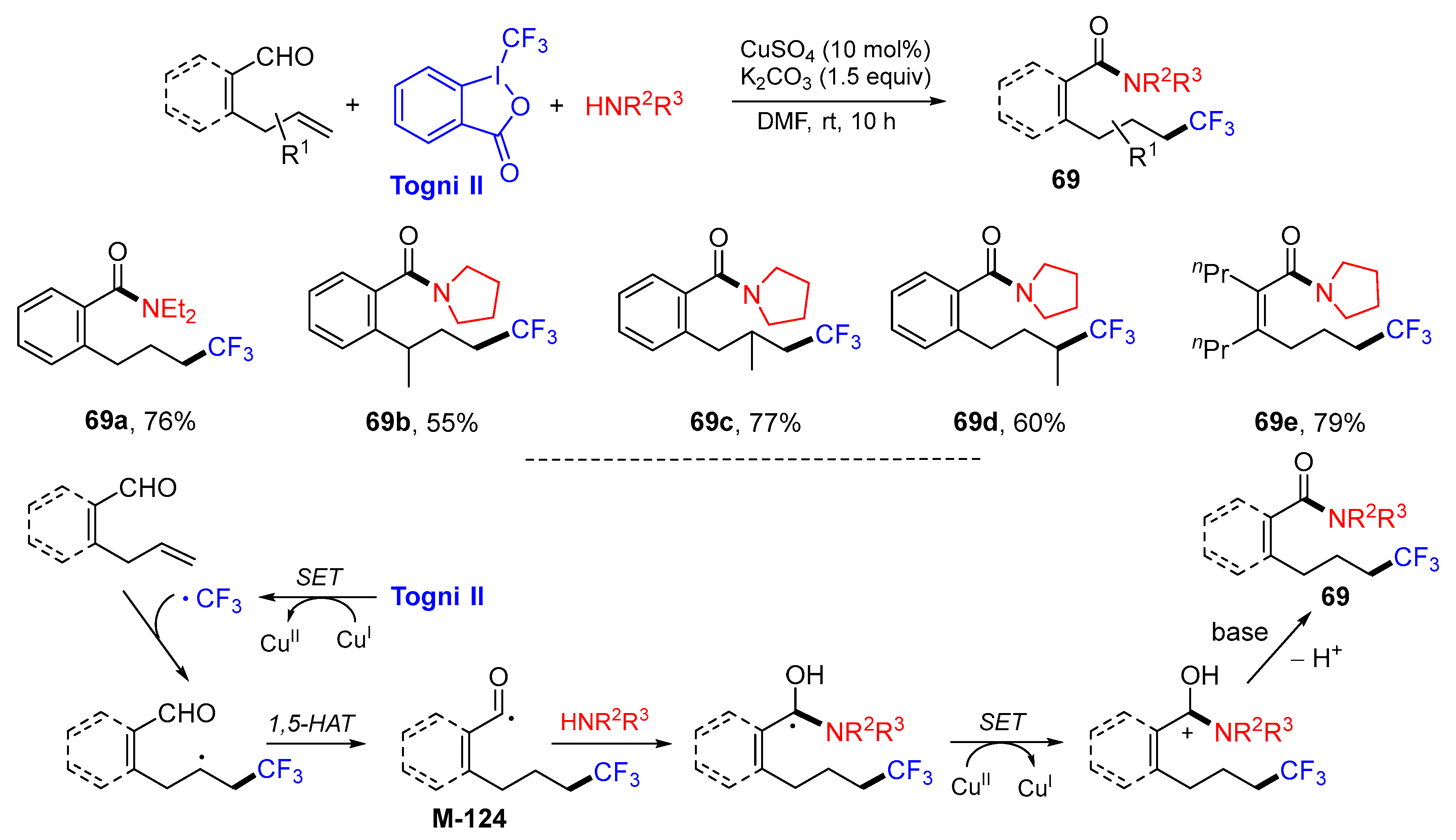{kind=link}
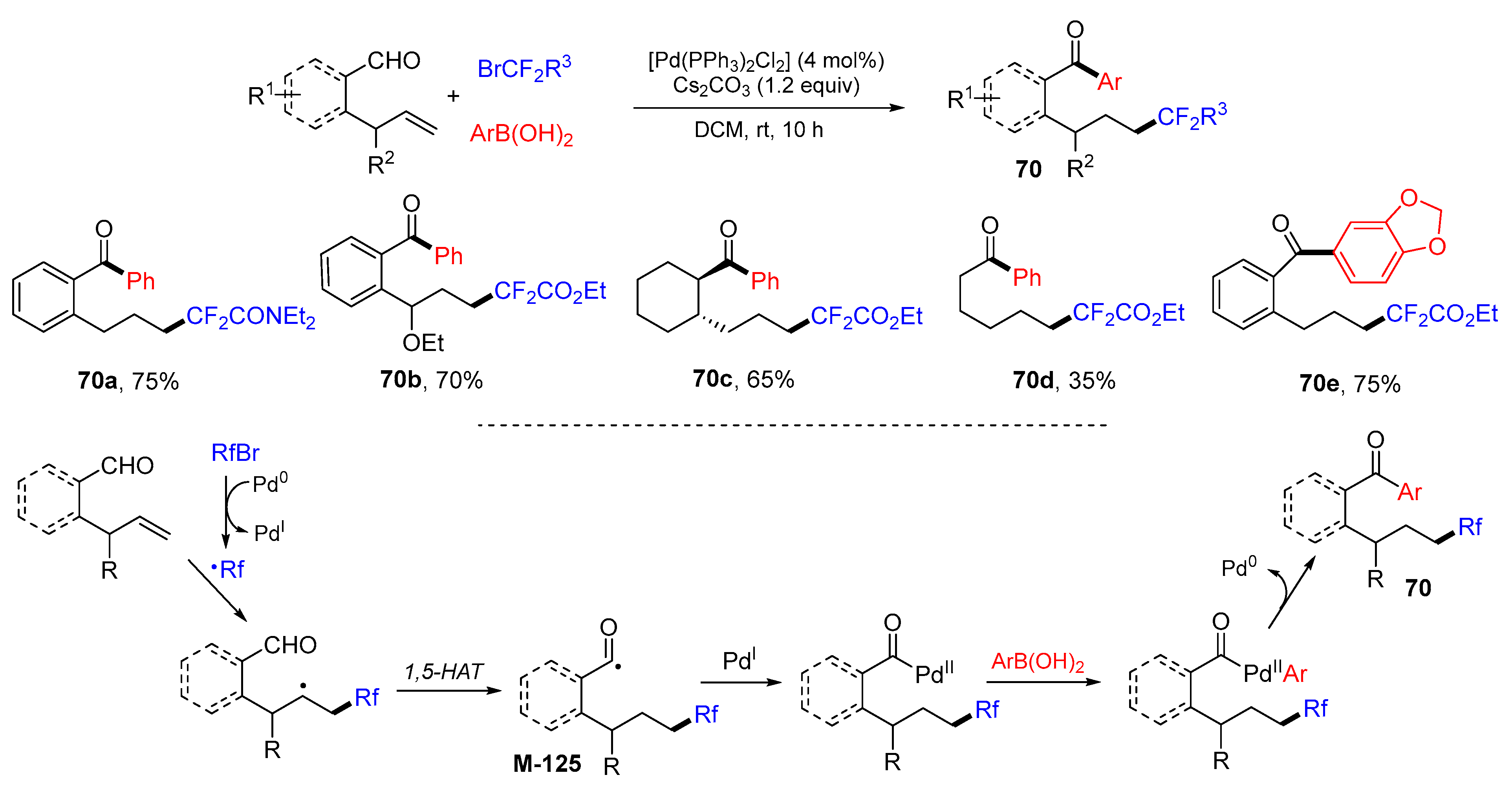{kind=link}
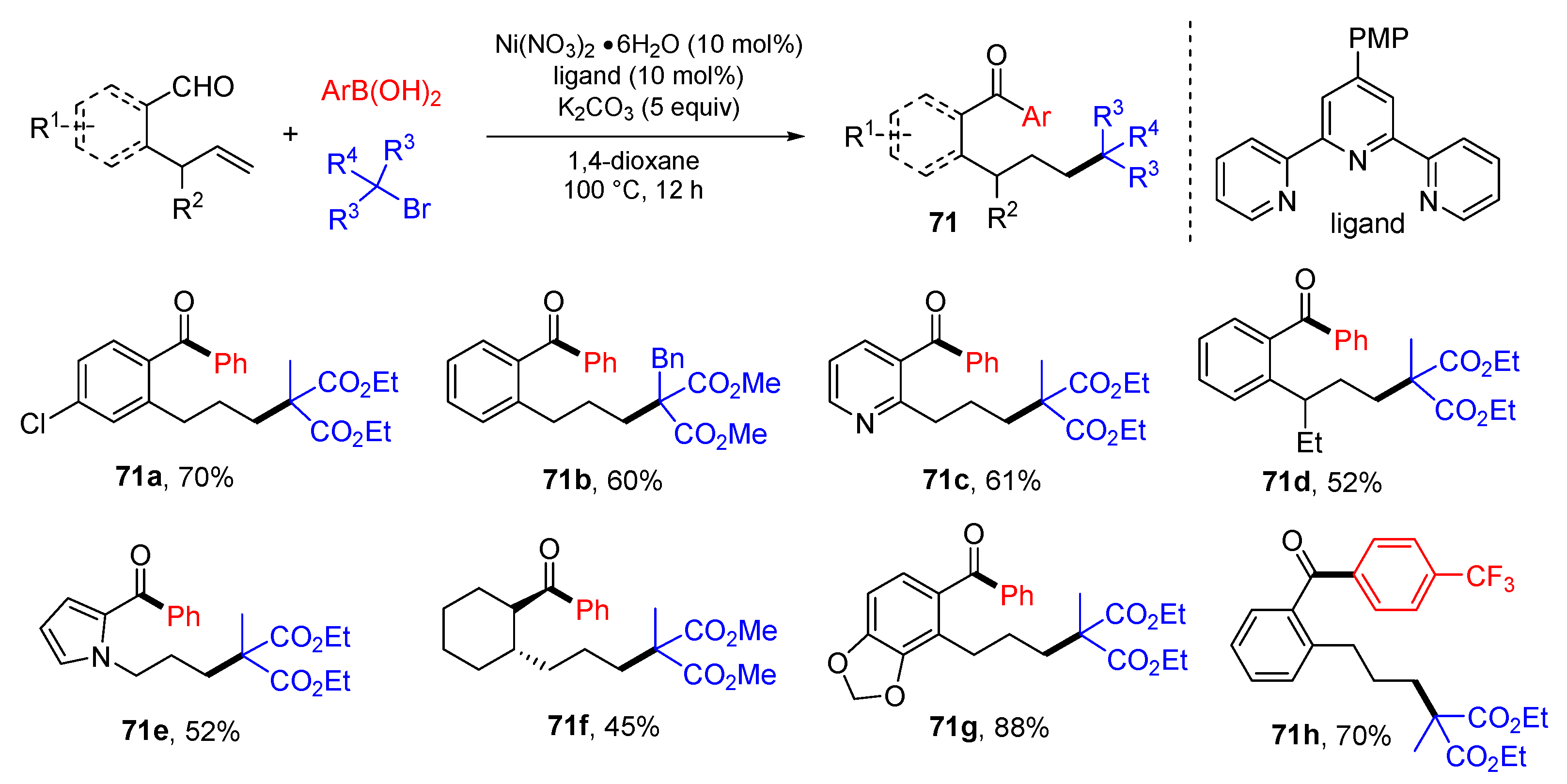{kind=link}
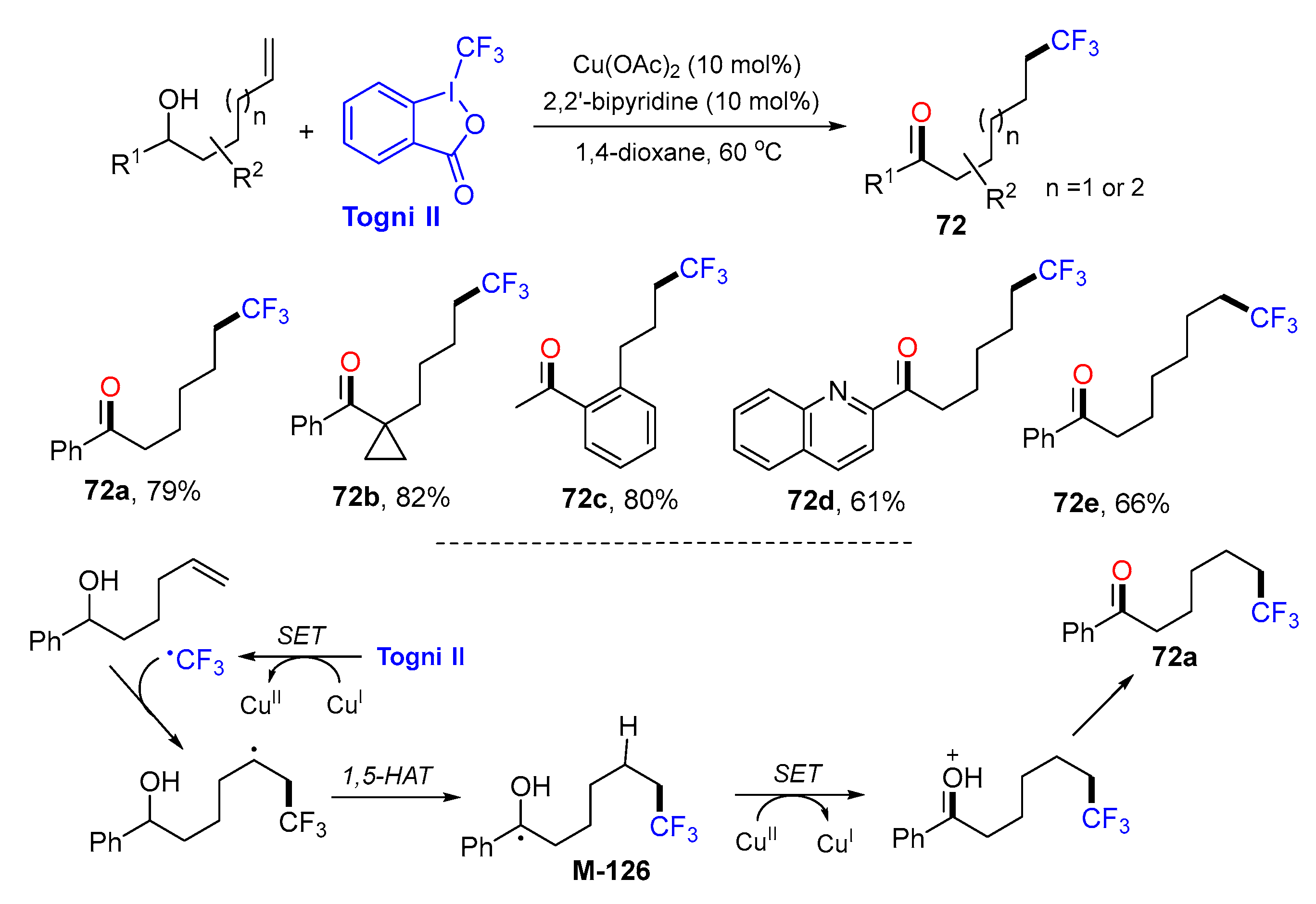{kind=link}
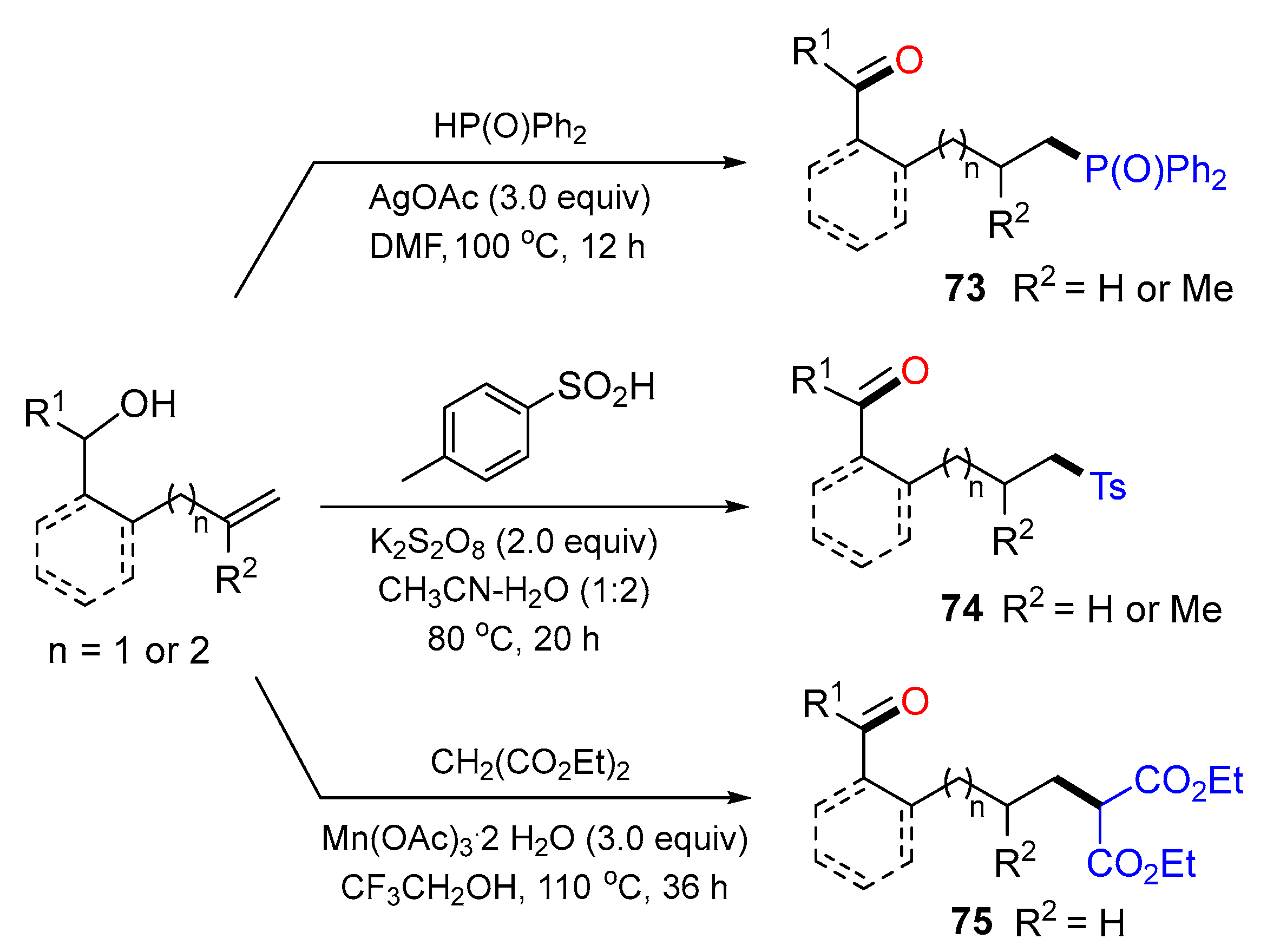{kind=link}
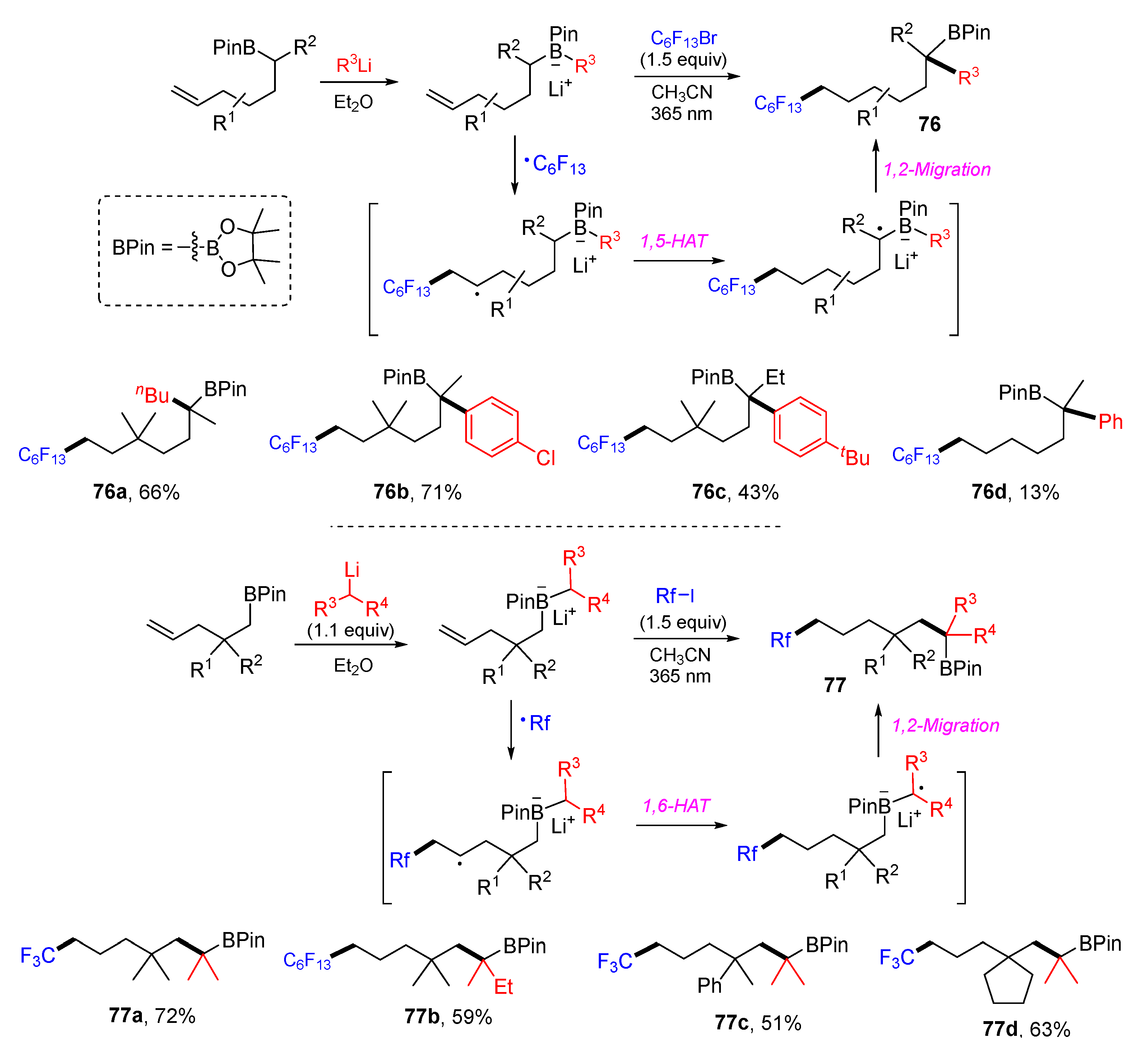{kind=link}
{kind=link}
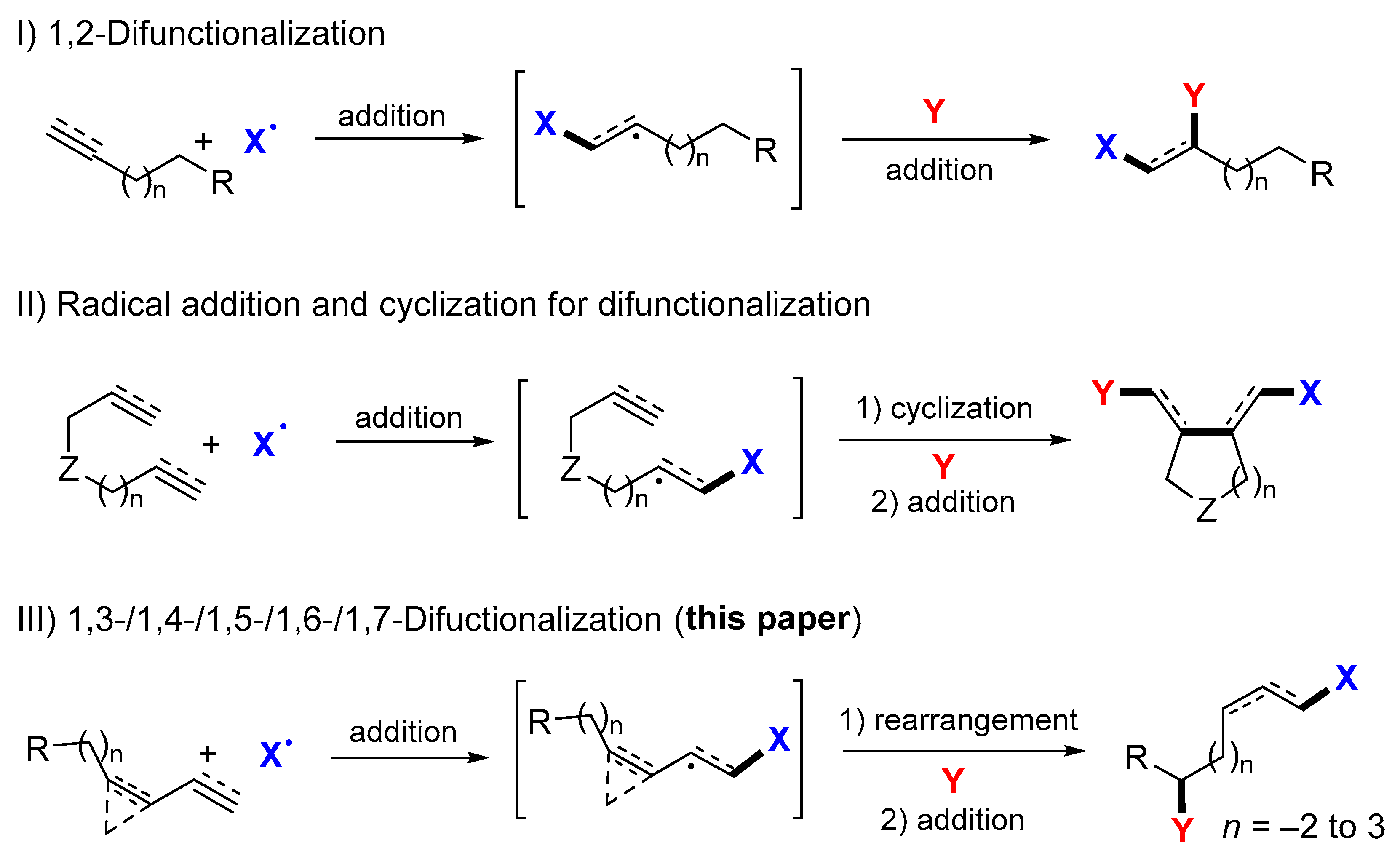
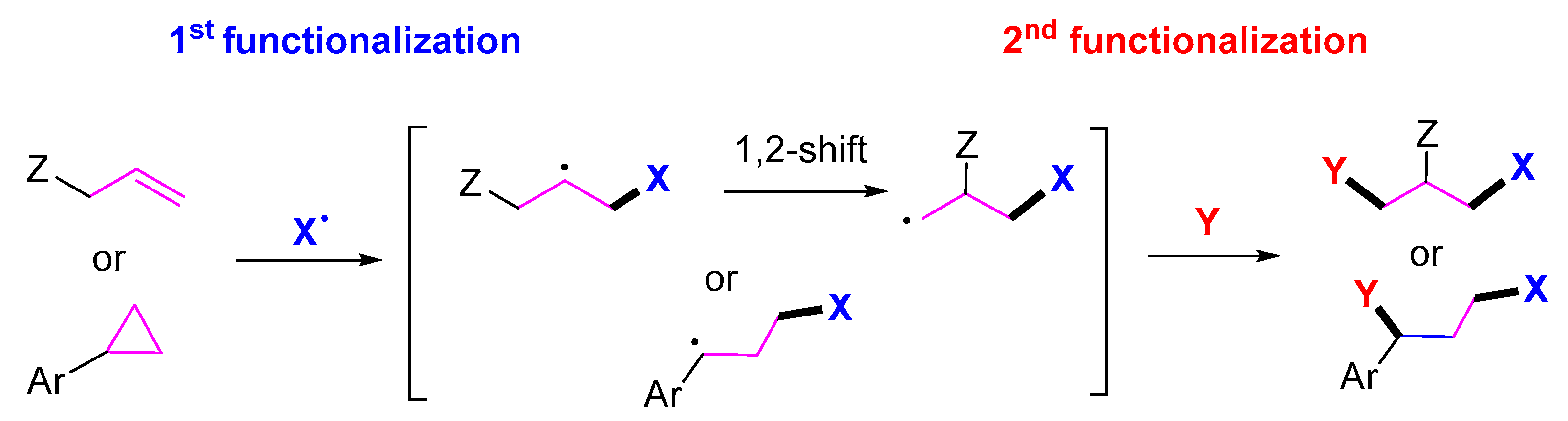
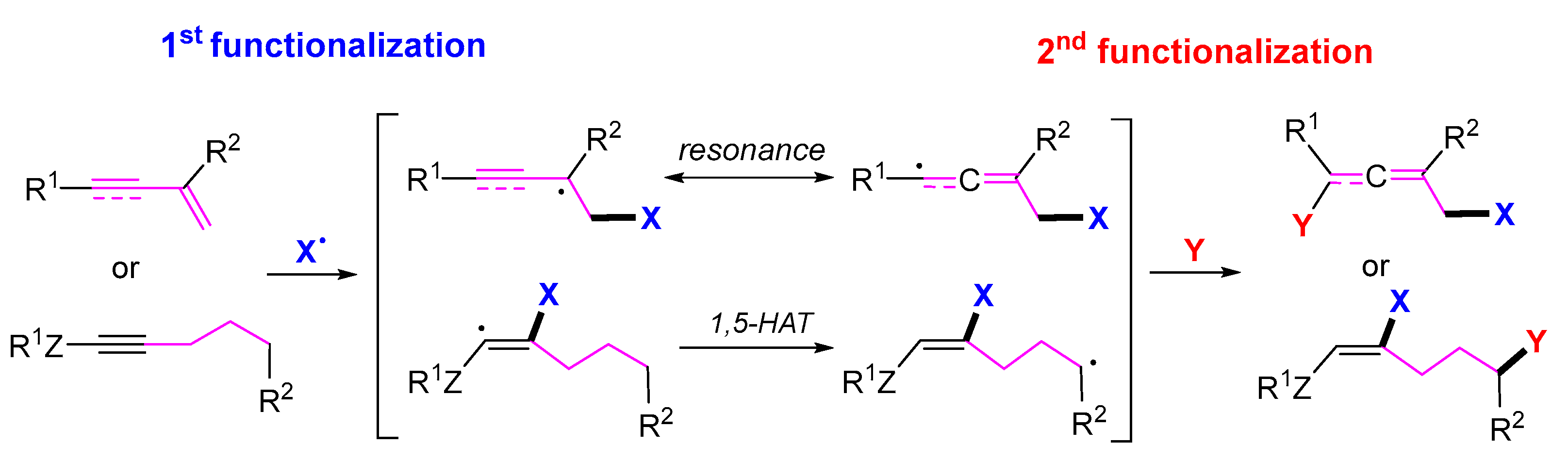
1. Introduction
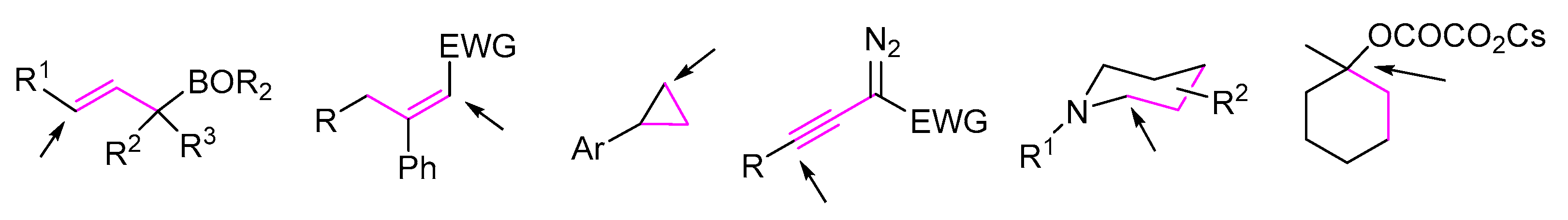
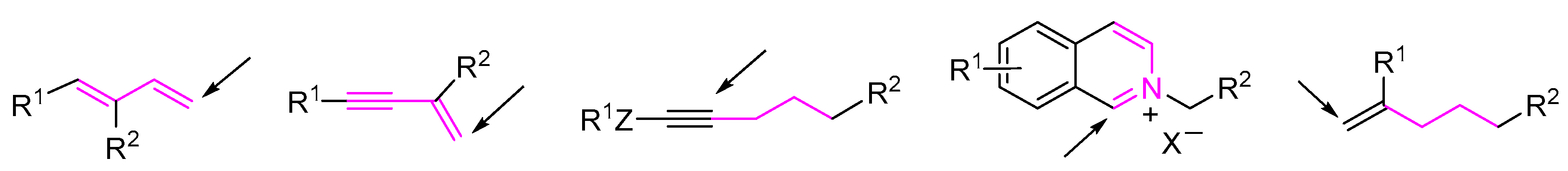
2. 1,3-Difunctionalization Reactions
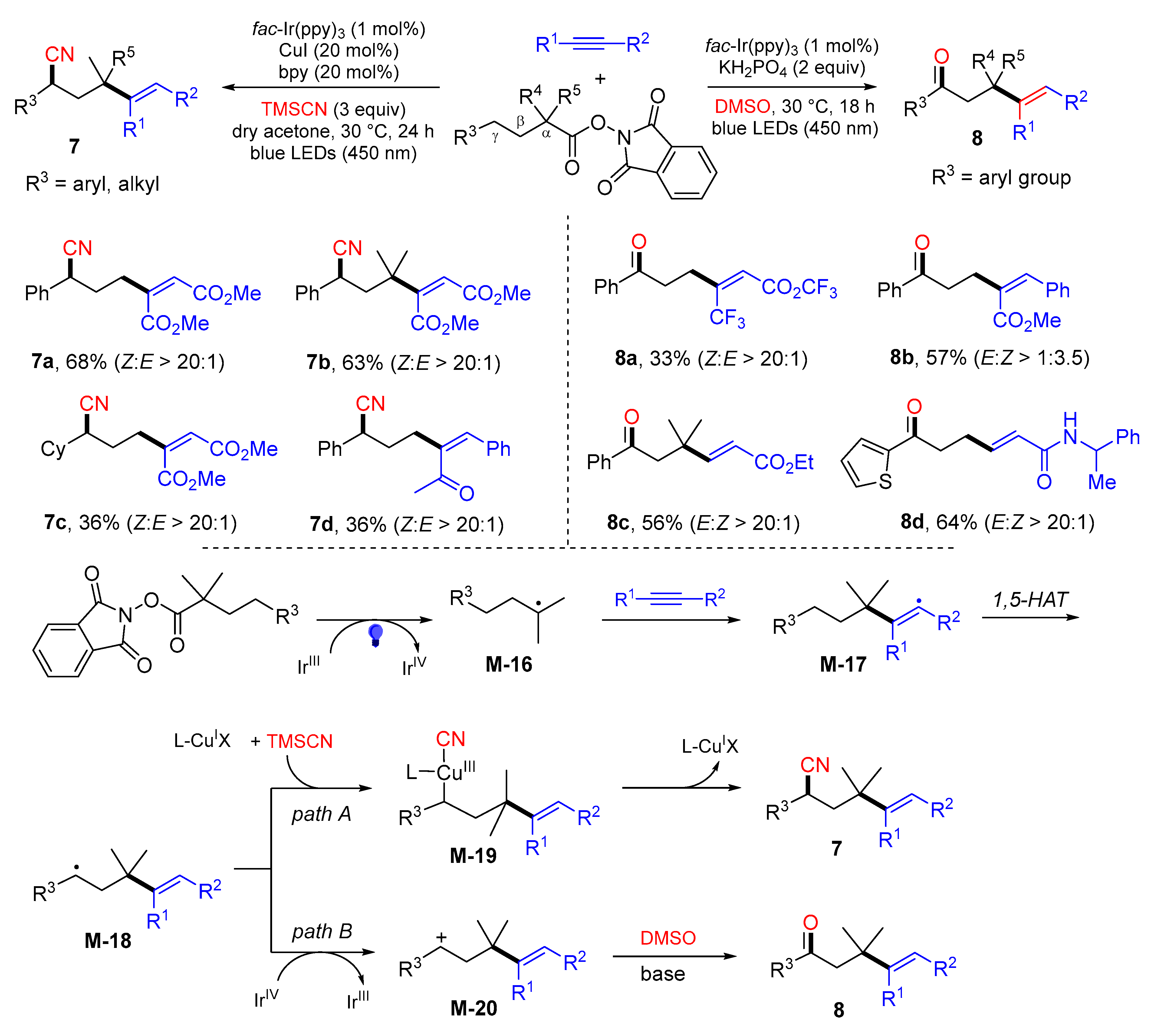
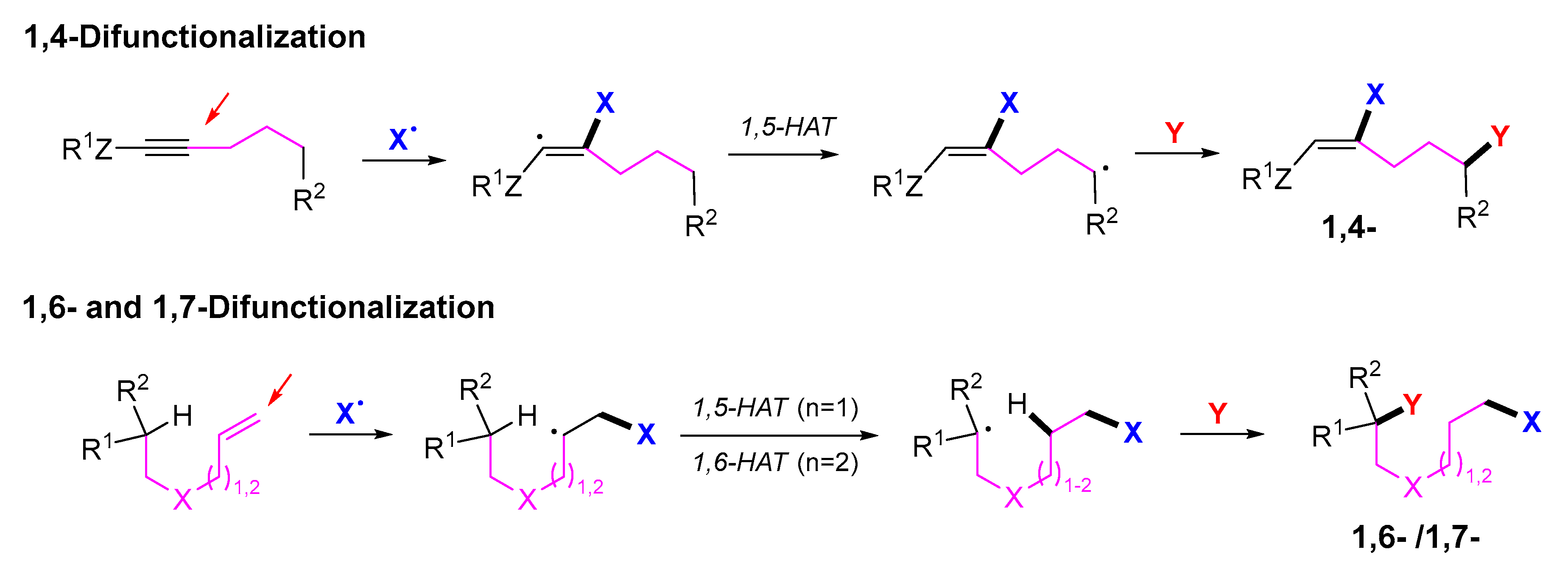
3. 1,4-Difunctionalization Reactions
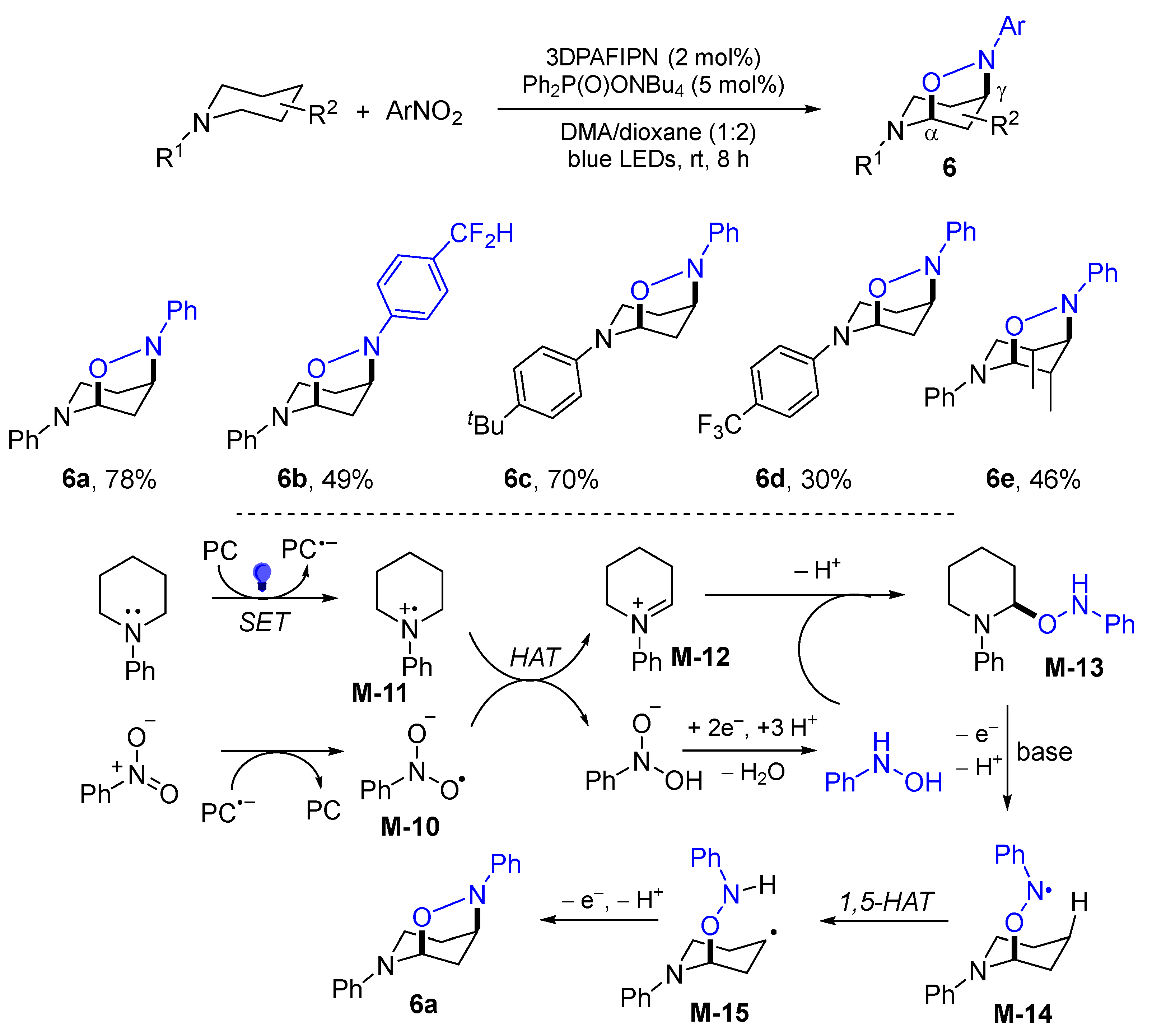
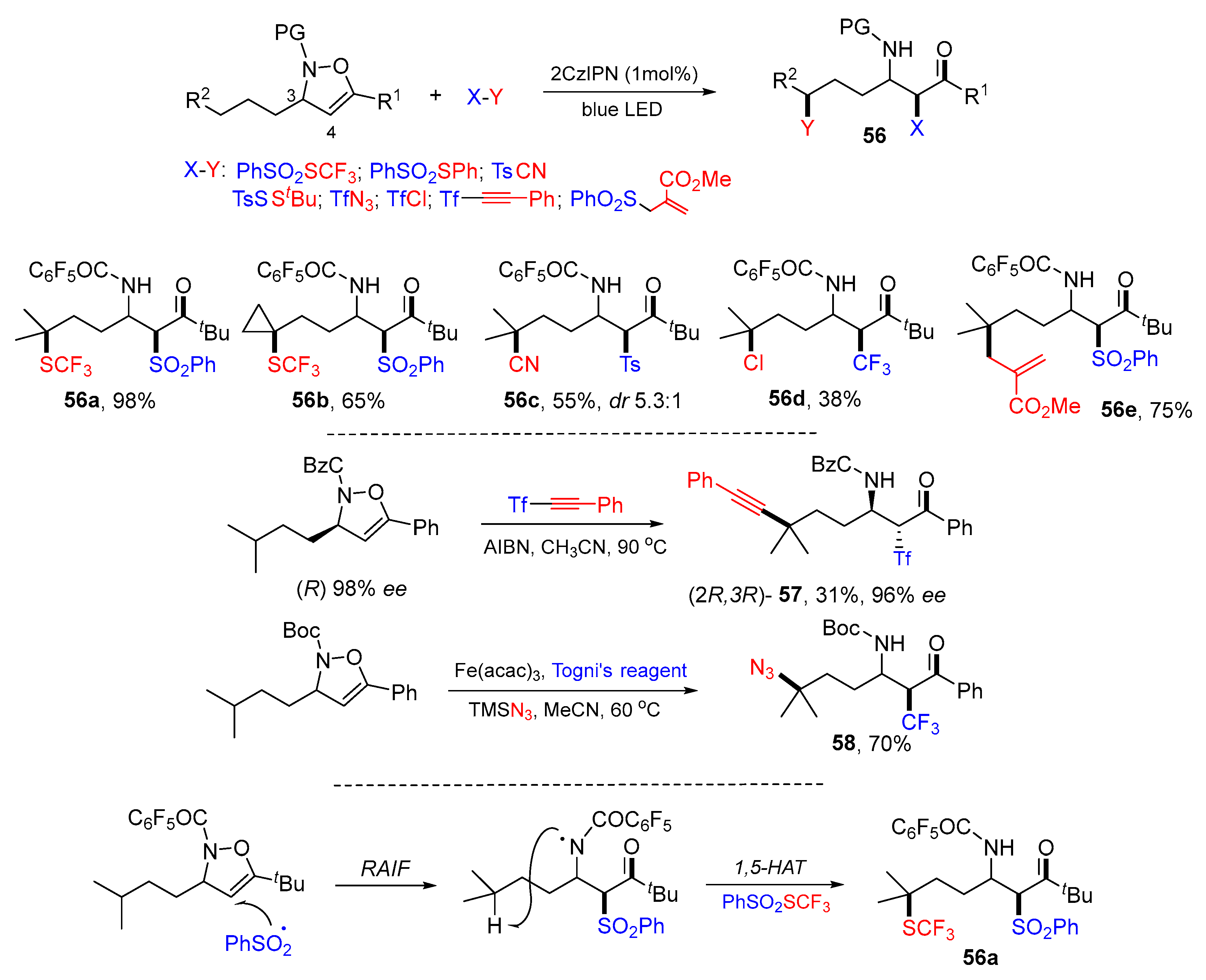
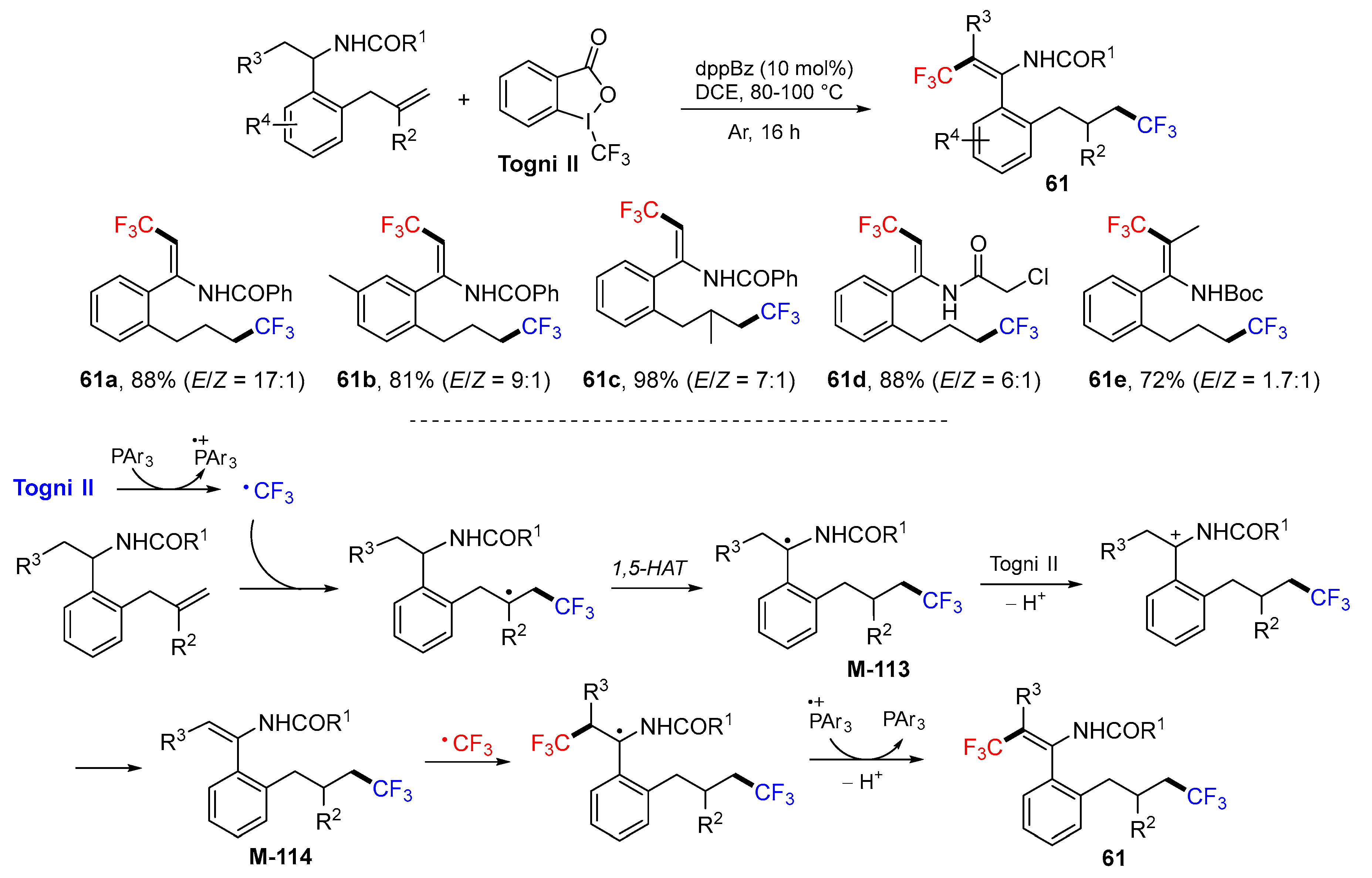
4. 1,5-Difunctionalization Reactions
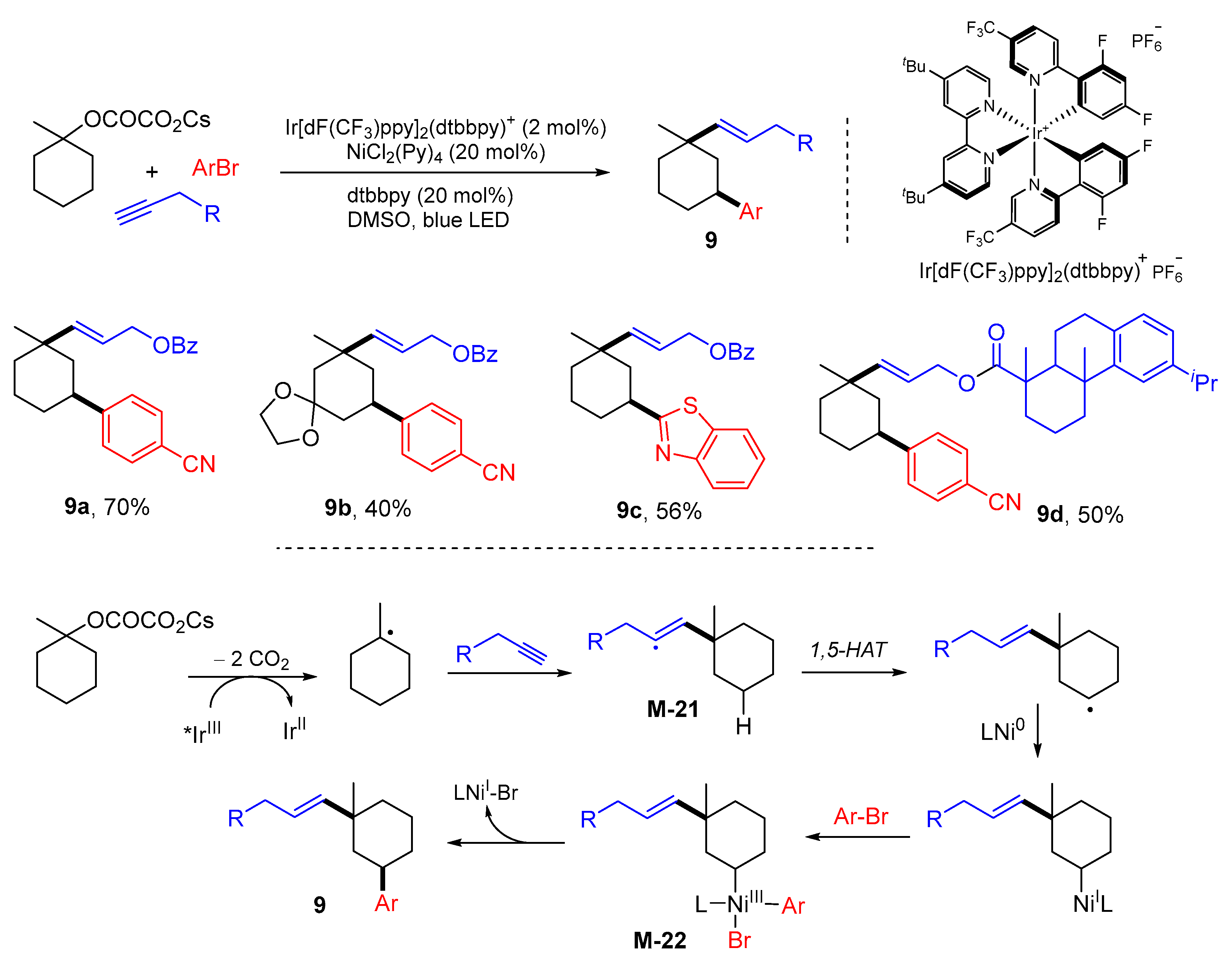
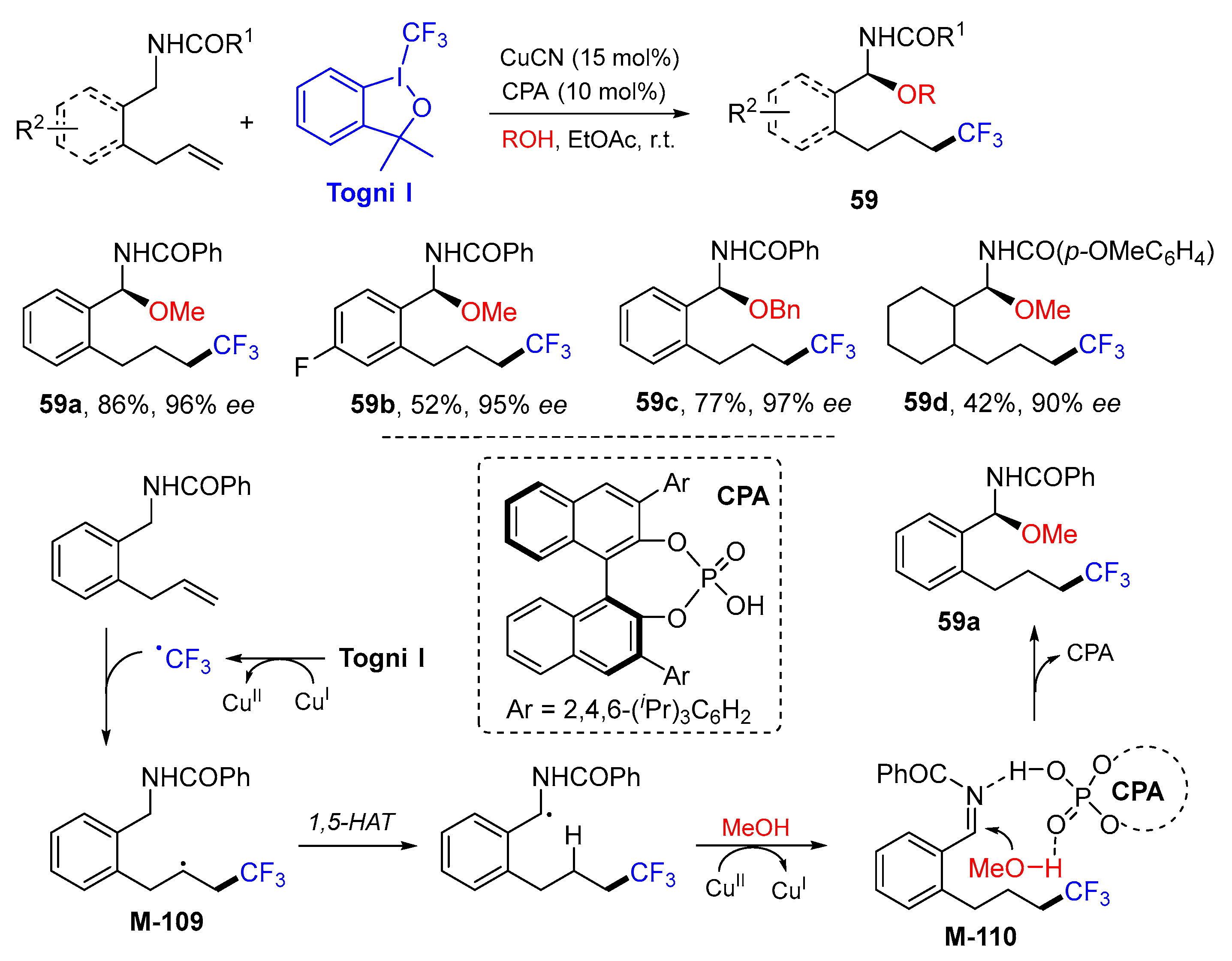
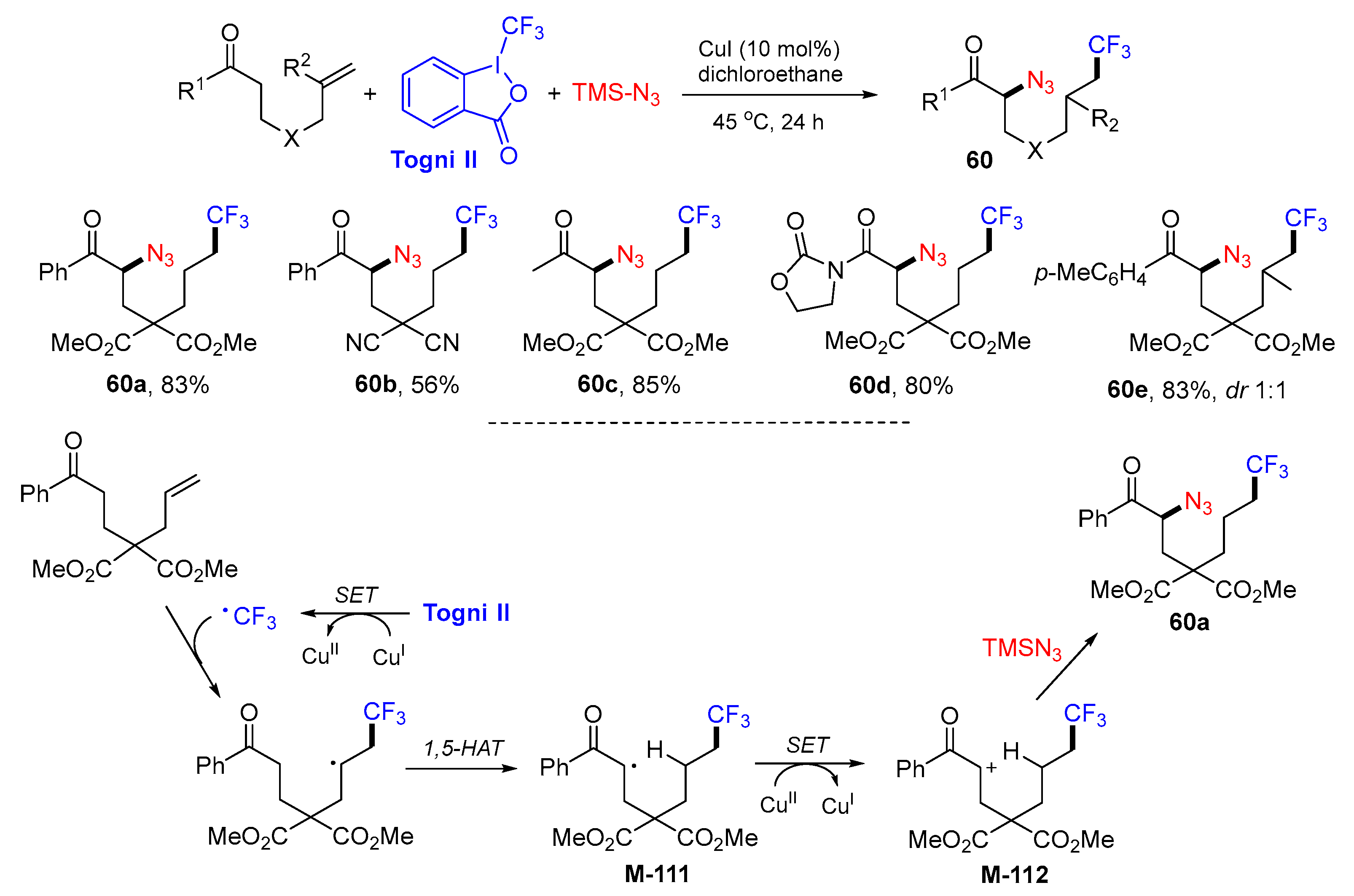
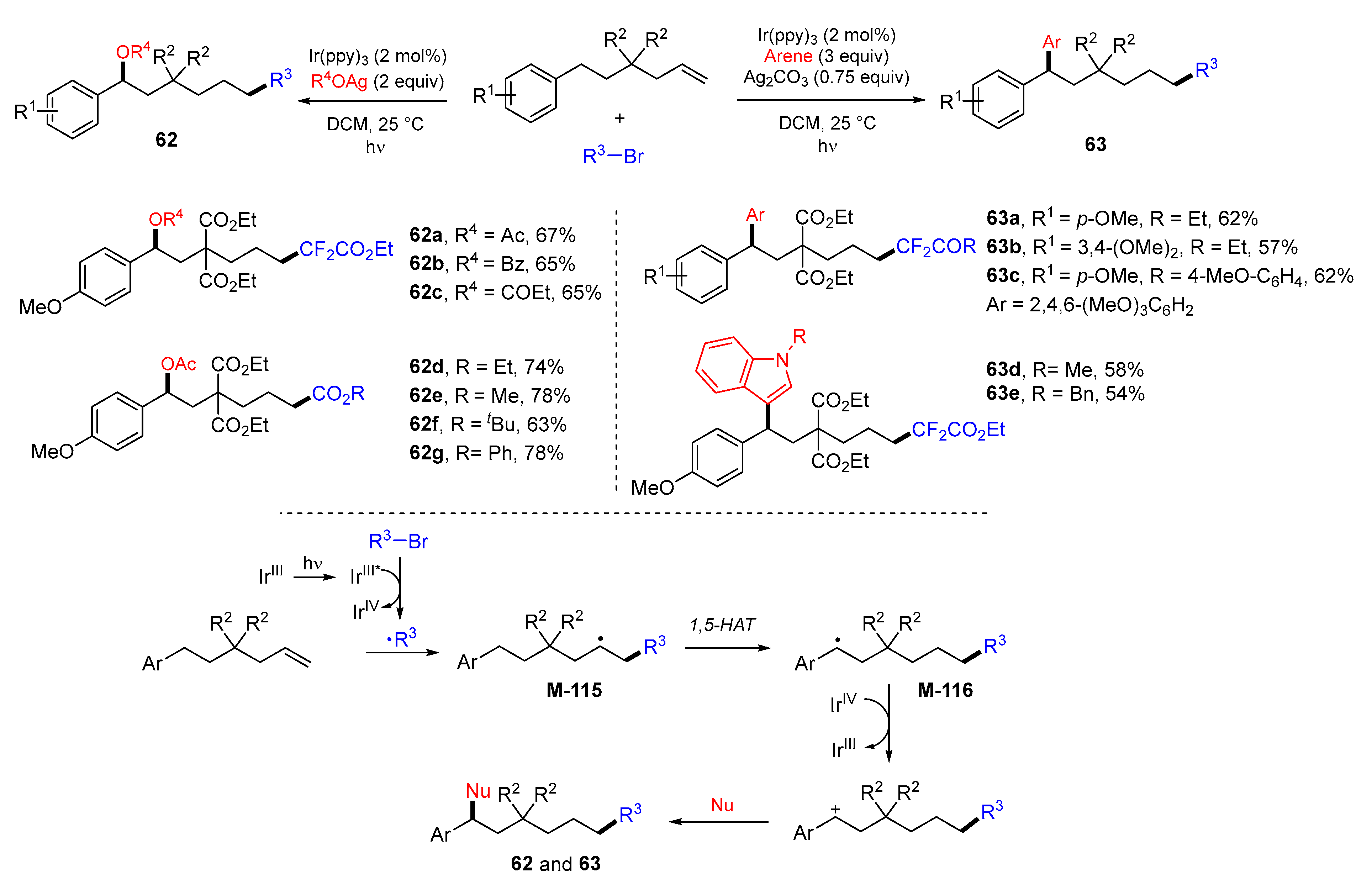
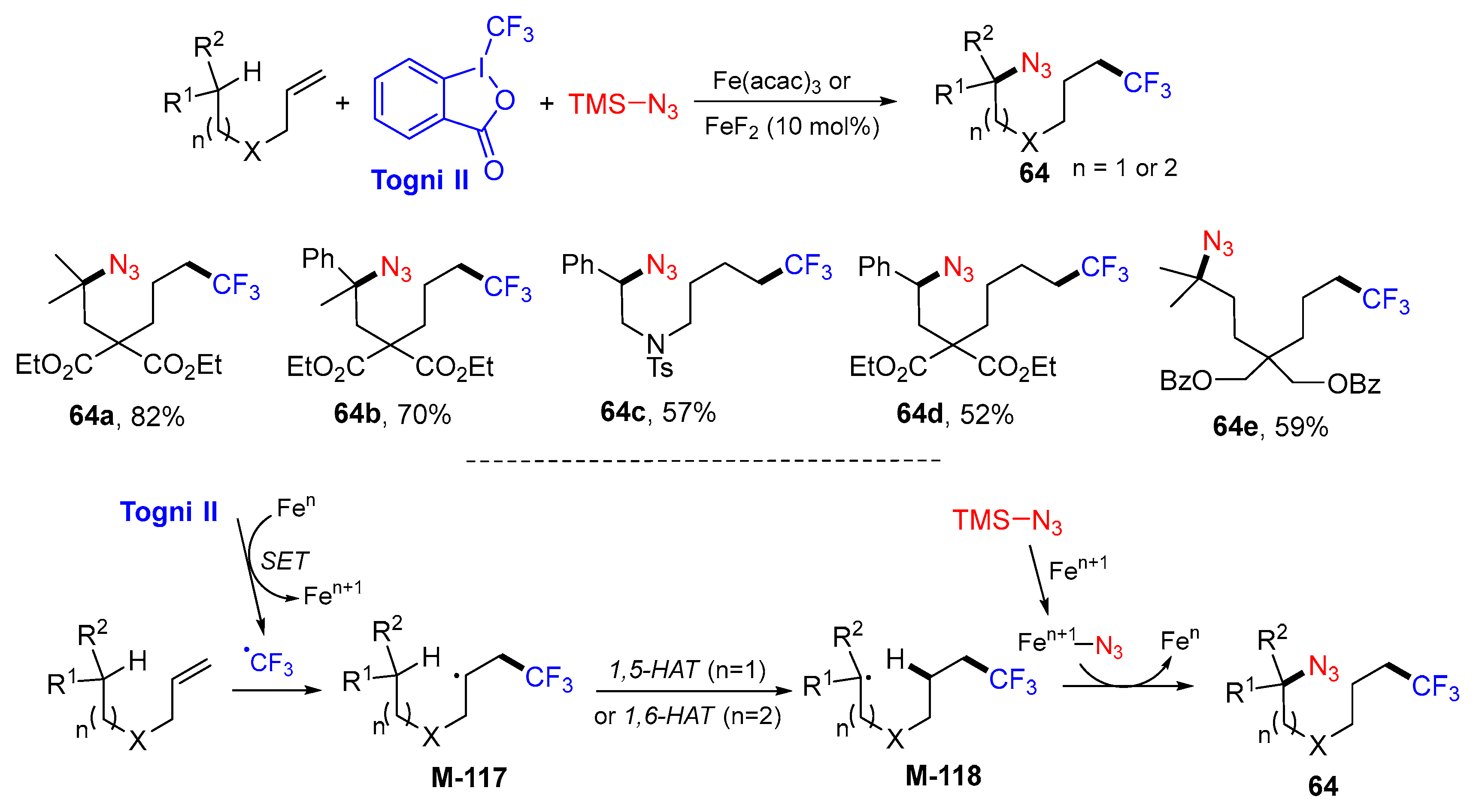
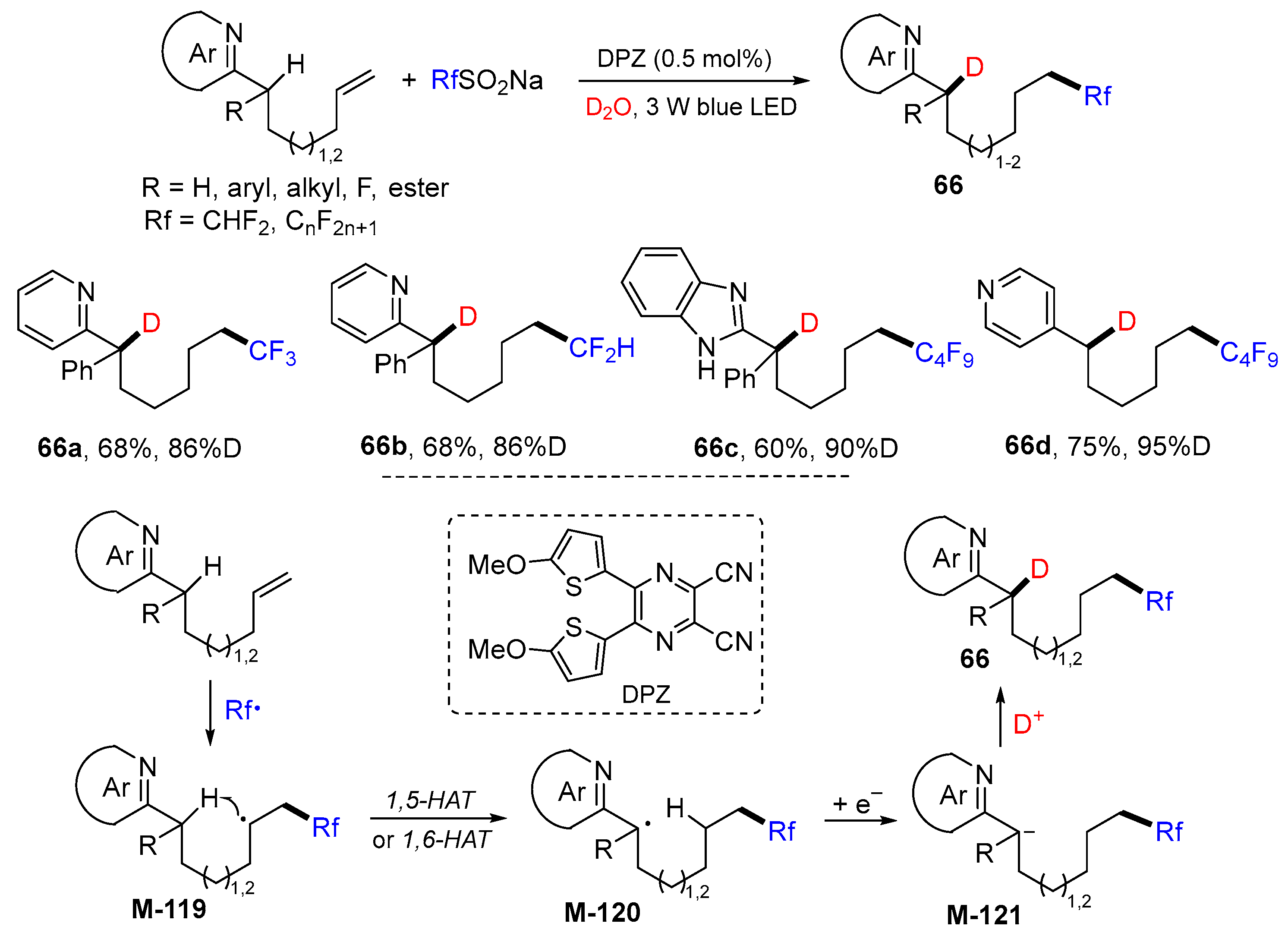
5. 1,6- and 1,7-Difunctionalization Reactions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chatgilialoglu, C.; Studer, A. (Eds.) Encyclopedia of Radicals in Chemistry, Biology and Materials; Wiley: Chichester, UK, 2012. [Google Scholar]
- Zarf, S.Z. Radical Reactions in Organic Synthesis; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Shen, J.; Li, L.; Xu, J.; Shen, C.; Zhang, P. Recent advances in the application of langlois’ reagent in olefin difunctionalization. Org. Biomol. Chem. 2023, 21, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Shen, J.; Xie, L. Recent developments on 1,2-difunctionalization and hydrofunctionalization of unactivated alkenes and alkynes involving C–S bond formation. Org. Chem. Front. 2023, 10, 1322–1345. [Google Scholar] [CrossRef]
- Bouchet, D.; Varlet, T.; Masson, G. Strategies toward the difunctionalizations of enamide derivatives for synthesizing α,β-substituted amines. Accounts Chem. Res. 2022, 55, 3265–3283. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bao, Y.; Tang, M.; Ye, Z.; Yuan, Z.; Zhu, G. Recent advances in difunctionalization of alkenes using pyridinium salts as radical precursors. Chem. Commun. 2022, 58, 3847–3864. [Google Scholar] [CrossRef]
- Sarkar, S.; Cheung, K.P.S.; Gevorgyan, V. C–H functionalization reactions enabled by hydrogen atom transfer to carbon-centered radicals. Chem. Sci. 2020, 11, 12974–12993. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Xiao, Y.-T.; Yang, Y.-Z.; Song, R.-J.; Li, J.-H. Recent advances in silver-mediated radical difunctionalization of alkenes. ChemCatChem 2020, 12, 5312–5329. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Qu, Y.-L.; Li, X.; Wang, C.-C. Recent advances in 1,4-functional group migration-mediated radical fluoroalkylation of alkenes and alkynes. Org. Biomol. Chem. 2020, 18, 8975–8993. [Google Scholar] [CrossRef]
- Fu, L.; Greßies, S.; Chen, P.; Liu, G. Recent advances and perspectives in transition metal-catalyzed 1,4-functionalizations of unactivated 1,3-enynes for the synthesis of allenes. Chin. J. Chem. 2020, 38, 91–100. [Google Scholar] [CrossRef]
- Bao, X.; Li, J.; Jiang, W.; Huo, C. Radical-mediated difunctionalization of styrenes. Synthesis 2019, 51, 4507–4530. [Google Scholar] [CrossRef]
- Sauer, G.S.; Lin, S. An electrocatalytic approach to the radical difunctionalization of alkenes. ACS Catal. 2018, 8, 5175–5187. [Google Scholar] [CrossRef]
- Yao, H.; Hu, W.; Zhang, W. Difunctionalization of alkenes and alkynes via intermolecular radical and nucleophilic additions. Molecules 2021, 26, 105. [Google Scholar] [CrossRef]
- Zhi, S.; Yao, H.; Zhang, W. Difunctionalization of dienes, enynes and related compounds via sequential radical addition and cyclization reactions. Molecules 2023, 28, 1145. [Google Scholar] [CrossRef]
- Shields, B.J.; Doyle, A.G. Direct C(sp3)–H cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc. 2016, 138, 12719–12722. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Wang, J.; Zhang, Y.; Li, Z.; Hu, D.; Zheng, N.; Chen, H. Detection of fleeting amine radical cations and elucidation of chain processes in visible-light-mediated [3 + 2] annulation by online mass spectrometric techniques. J. Am. Chem. Soc. 2017, 139, 12259–12266. [Google Scholar] [CrossRef]
- Park, S.V.; Corcos, A.R.; Jambor, A.N.; Yang, T.; Berry, J.F. Formation of the N≡N triple bond from reductive coupling of a paramagnetic diruthenium nitrido compound. J. Am. Chem. Soc. 2022, 144, 3259–3268. [Google Scholar] [CrossRef]
- Das, A.; Ahmed, J.; Rajendran, N.M.; Adhikari, D.; Mandal, S.K. A bottleable imidazole-based radical as a single electron transfer reagent. J. Org. Chem. 2021, 86, 1246–1252. [Google Scholar] [CrossRef]
- Luo, S.; Weng, C.; Qin, Z.; Li, K.; Zhao, T.; Ding, Y.; Ling, C.; Ma, Y.; An, J. Tandem H/D exchange-set reductive deuteration strategy for the synthesis of α,β-deuterated amines using D2O. J. Org. Chem. 2021, 86, 11862–11870. [Google Scholar] [CrossRef]
- Gong, Q.; Cheng, K.; Wu, Q.; Li, W.; Yu, C.; Jiao, L.; Hao, E. One-pot access to ethylene-bridged BODIPY dimers and trimers through single-electron transfer chemistry. J. Org. Chem. 2021, 86, 15761–15767. [Google Scholar] [CrossRef]
- Laha, J.K.; Gupta, P. Sulfoxylate anion radical-induced aryl radical generation and intramolecular arylation for the synthesis of biarylsultams. J. Org. Chem. 2022, 87, 4204–4214. [Google Scholar] [CrossRef]
- Liu, H.; Laporte, A.G.; Tardieu, D.; Hazelard, D.; Compain, P. Formal glycosylation of quinones with exo-glycals enabled by iron-mediated oxidative radical–polar crossover. J. Org. Chem. 2022, 87, 13178–13194. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, W.; Gao, X.; Ren, X.; Dong, C.; Wang, C.; Liu, L.; Li, J. Synthesis of [1,2,3]triazolo-[1,5-a]quinoxalin-4(5H)-ones through photoredox-catalyzed [3 + 2] cyclization reactions with hypervalent iodine(III) reagents. J. Org. Chem. 2022, 87, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Zhao, W.; Wen, J.; Dong, C.; Hu, C.; Li, J. Photoredox-catalyzed cascade sp2 C–H bond functionalization to construct substituted acridine with diarylamine and hypervalent iodine(III) reagents. Org. Lett. 2023, 25, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; Zhu, C.; Li, L.; Shao, L.; Zhu, S.; Rueping, M.; Chu, L. Ligand-controlled stereodivergent alkenylation of alkynes to access functionalized trans- and cis-1,3-dienes. Nat. Commun. 2023, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, S.; Martin, M.I.; Kim, R.S.; Yap, G.P.A.; Rosenthal, J.; Watson, D.A. Photoredox-nickel dual-catalyzed C-alkylation of secondary nitroalkanes: Access to sterically hindered α-tertiary amines. J. Am. Chem. Soc. 2023, 145, 4707–4715. [Google Scholar] [CrossRef]
- Xian, N.; Yin, J.; Ji, X.; Deng, G.; Huang, H. Visible-light-mediated photoredox carbon radical formation from aqueous sulfoxonium ylides. Org. Lett. 2023, 25, 1161–1165. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; Macmillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.-S.; Zuo, H.-D.; Shen, Y.-T.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Electrochemical selective annulative amino-ketalization and amino-oxygenation of 1,6-enynes. Chem. Commun. 2022, 58, 10420–10423. [Google Scholar] [CrossRef]
- Wan, J.; Huang, J. Electrochemically enabled sulfoximido-oxygenation of alkenes with NH-sulfoximines and alcohols. Org. Lett. 2022, 24, 8914–8919. [Google Scholar] [CrossRef]
- Xiang, H.; He, J.; Qian, W.; Qiu, M.; Xu, H.; Duan, W.; Ouyang, Y.; Wang, Y.; Zhu, C. Electroreductively induced radicals for organic synthesis. Molecules 2023, 28, 857. [Google Scholar] [CrossRef]
- Wang, X.; Li, R.; Deng, Y.; Fu, M.; Zhao, Y.; Guan, Z.; He, Y. Direct hydroxylarylation of benzylic carbons (sp3/sp2/sp) via radical–radical cross-coupling powered by paired electrolysis. J. Org. Chem. 2023, 88, 329–340. [Google Scholar] [CrossRef]
- Tian, Y.; Zheng, L.; Wang, Z.; Li, Z.; Fu, W. Metal-free electrochemical oxidative difluoroethylation/cyclization of olefinic amides to construct difluoroethylated azaheterocycles. J. Org. Chem. 2023, 88, 1875–1883. [Google Scholar] [CrossRef]
- Yao, W.; Lv, K.; Xie, Z.; Qiu, H.; Ma, M. Catalyst-free electrochemical sulfonylation of organoboronic acids. J. Org. Chem. 2023, 88, 2296–2305. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
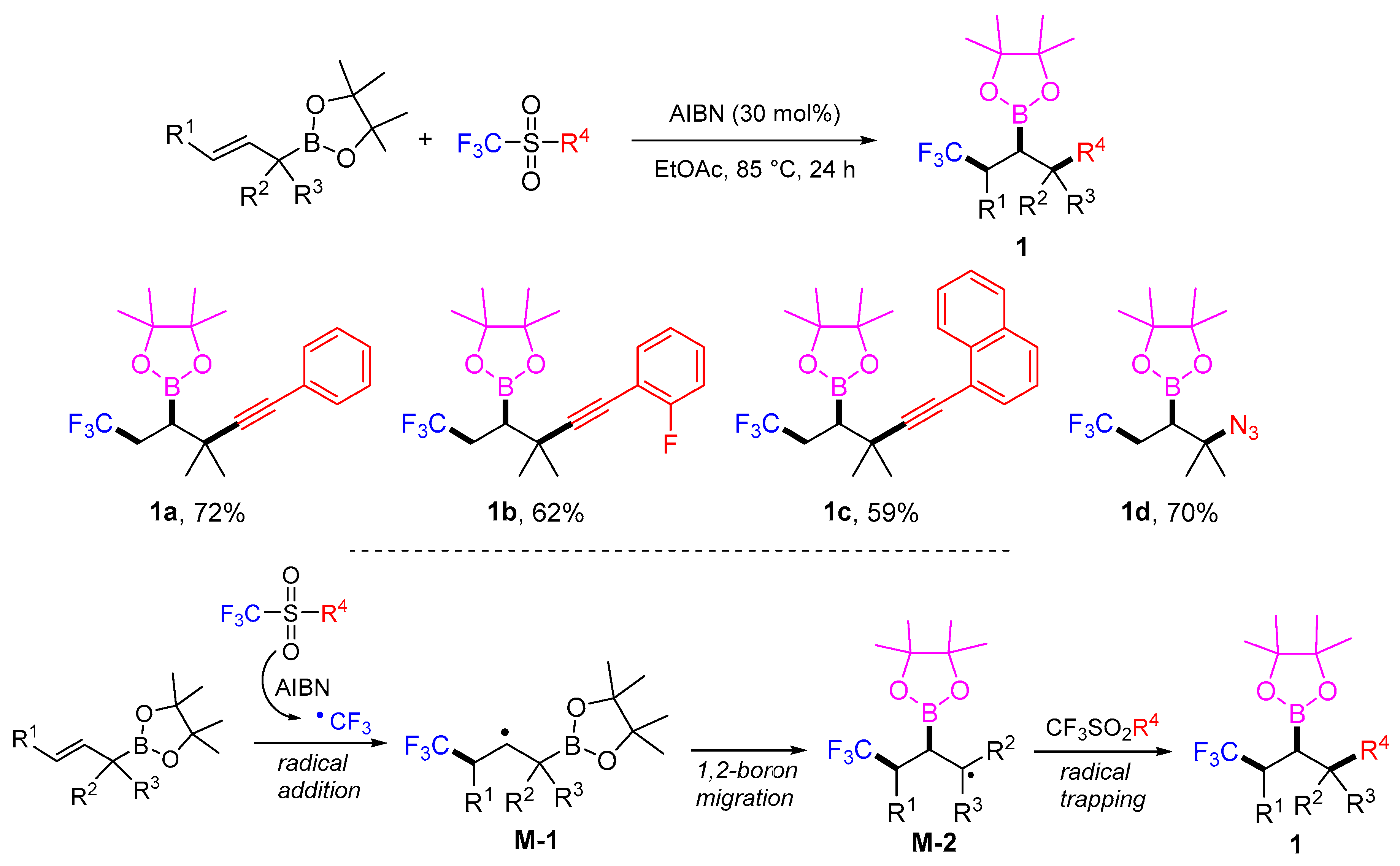
- Jana, K.; Bhunia, A.; Studer, A. Radical 1,3-difunctionalization of allylboronic esters with concomitant 1,2-boron shift. Chem 2020, 6, 512–522. [Google Scholar] [CrossRef]
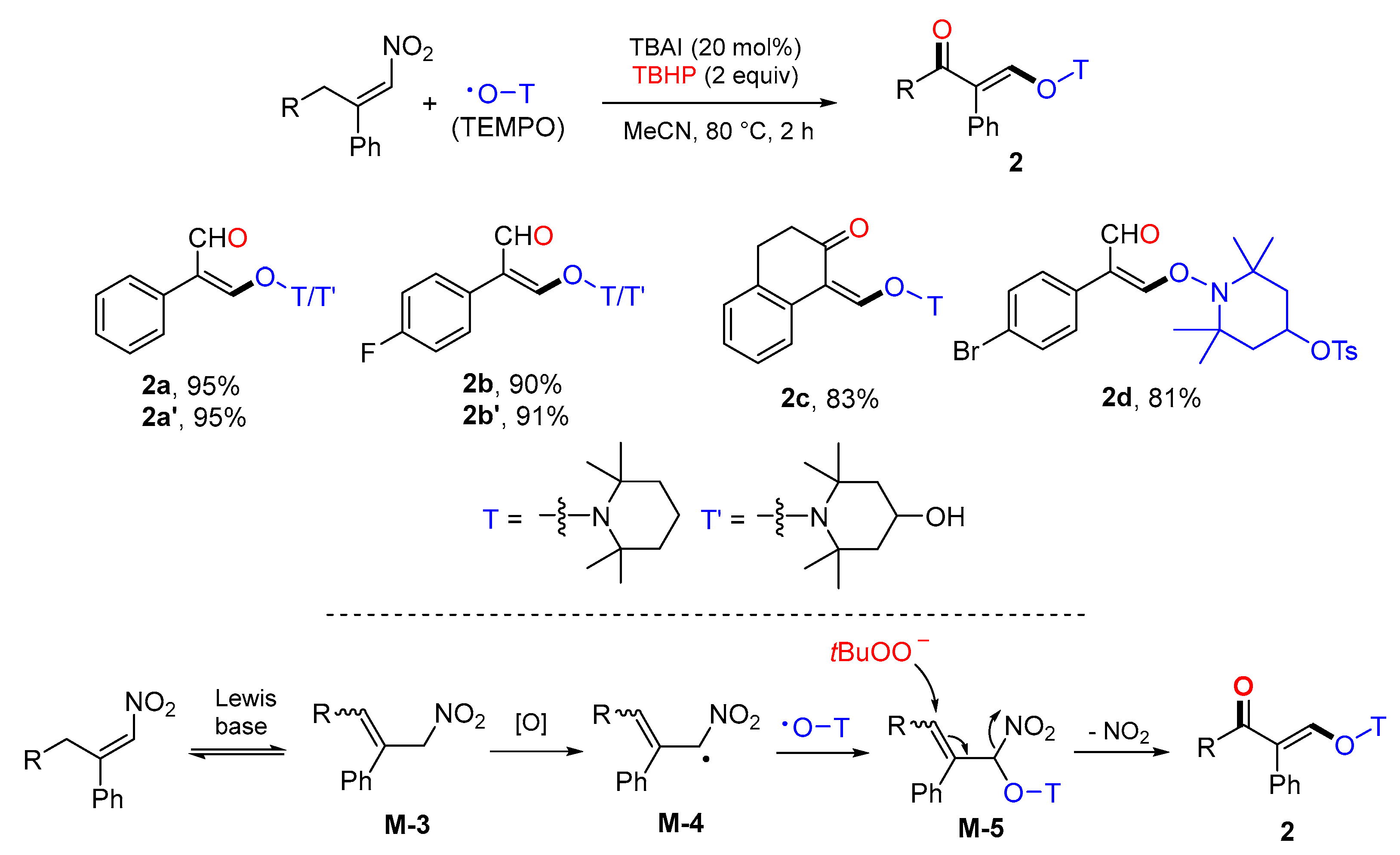
- Wang, Y.; Zheng, L.; Shi, X.; Chen, Y. 1,3-Difunctionalization of β-alkyl nitroalkenes via combination of lewis base catalysis and radical oxidation. Org. Lett. 2021, 23, 886–889. [Google Scholar] [CrossRef]
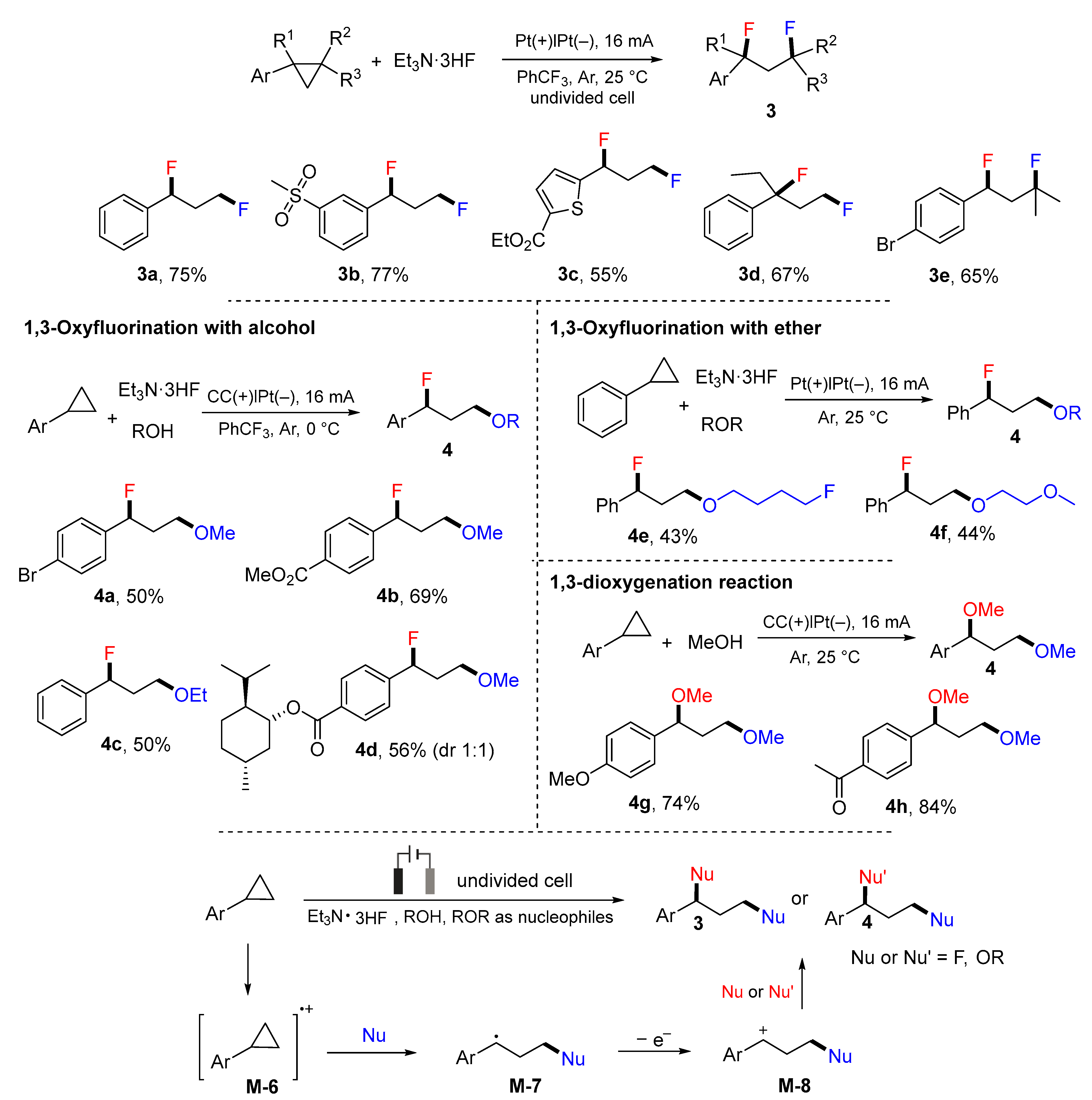
- Peng, P.; Yan, X.; Zhang, K.; Liu, Z.; Zeng, L.; Chen, Y.; Zhang, H.; Lei, A. Electrochemical C−C bond cleavage of cyclopropanes towards the synthesis of 1,3-difunctionalized molecules. Nat. Commun. 2021, 12, 3075. [Google Scholar] [CrossRef]
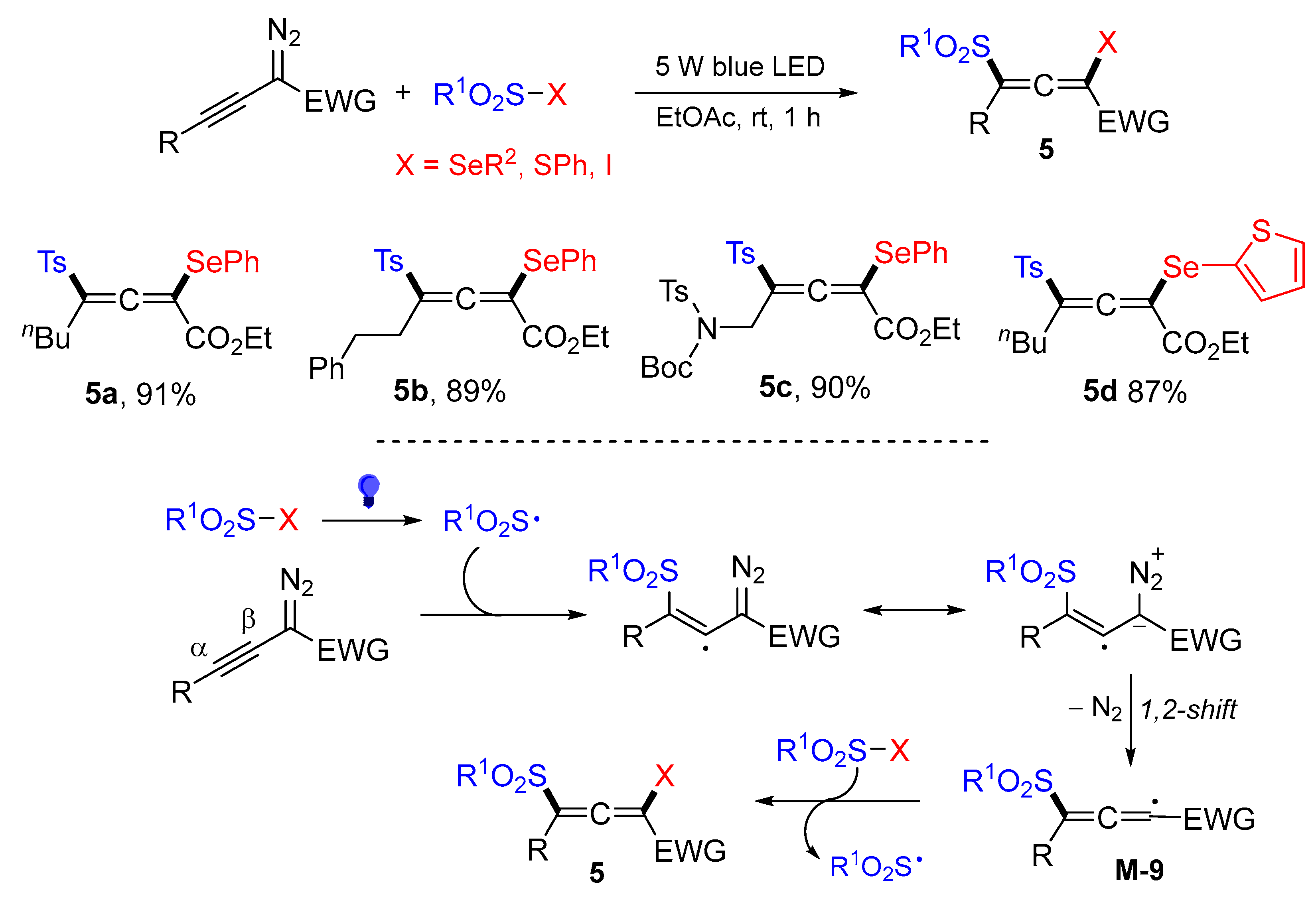
- Li, W.; Zhou, L. Synthesis of tetrasubstituted allenes via visible-light-promoted radical 1,3-difunctionalization of alkynyl diazo compounds. Org. Lett. 2022, 24, 3976–3981. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, M.; Zhou, Q. Visible-light-induced α,γ-C(sp3)–H difunctionalization of piperidines. Org. Lett. 2022, 24, 2894–2898. [Google Scholar] [CrossRef]
- Luo, M.; Zhu, S.; Shi, C.; Du, Y.; Yang, C.; Guo, L.; Xia, W. Photoinduced remote C(sp3)–H cyanation and oxidation enabled by a vinyl radical-mediated 1,5-HAT strategy. Org. Lett. 2022, 24, 6560–6565. [Google Scholar] [CrossRef]
- Li, H.; Guo, L.; Feng, X.; Huo, L.; Zhu, S.; Chu, L. Sequential C–O decarboxylative vinylation/C–H arylation of cyclic oxalates via a nickel-catalyzed multicomponent radical cascade. Chem. Sci. 2020, 11, 4904–4910. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Ouyang, X.; Song, R.; Li, J. Indium controlled regioselective 1,4-alkylarylation of 1,3-dienes with α-carbonyl alkyl bromides and N-heterocycles. Chem. Commun. 2020, 56, 1279–1282. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Bellotti, P.; Pflüger, P.M.; Schwarz, J.L.; Heidrich, B.; Glorius, F. Three-component, interrupted radical heck/allylic substitution cascade involving unactivated alkyl bromides. J. Am. Chem. Soc. 2020, 142, 10173–10183. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, F.; Zhang, G.; Shi, L.; Wang, X. Highly regioselective difluoroalkylarylation of butadiene through a nickel-catalyzed tandem radical process. ACS Catal. 2021, 11, 14848–14853. [Google Scholar] [CrossRef]
- Liu, Z.; Ye, Z.; Chen, Y.; Zheng, Y.; Xie, Z.; Guan, J.; Xiao, J.; Chen, K.; Xiang, H.; Yang, H. Visible-light-induced, palladium-catalyzed 1,4-difunctionalization of 1,3-dienes with bromodifluoroacetamides. Org. Lett. 2022, 24, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Terao, J.; Bando, F.; Kambe, N. Ni-catalyzed regioselective three-component coupling of alkyl halides, arylalkynes, or enynes with R–M (M = MgX′, ZnX′). Chem. Commun. 2009, 2009, 7336–7338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.F.; Bian, K.J.; Li, C.; Sheng, J.; Li, Y.; Wang, X.S. Nickel-catalyzed carbofluoroalkylation of 1,3-enynes to access structurally diverse fluoroalkylated allenes. Angew. Chem. Int. Ed. 2019, 58, 5069–5074. [Google Scholar] [CrossRef]
- Song, Y.; Song, S.; Duan, X.; Wu, X.; Jiang, F.; Zhang, Y.; Fan, J.; Huang, X.; Fu, C.; Ma, S. Copper-catalyzed radical approach to allenyl iodides. Chem. Commun. 2019, 55, 11774–11777. [Google Scholar] [CrossRef]
- Lv, Y.; Han, W.; Pu, W.; Xie, J.; Wang, A.; Zhang, M.; Wang, J.; Lai, J. Copper-catalyzed regioselective 1,4-sulfonylcyanation of 1,3-enynes with sulfonyl chlorides and TMSCN. Org. Chem. Front. 2022, 9, 3775–3780. [Google Scholar] [CrossRef]
- Lv, Y.; Lai, J.; Pu, W.; Wang, J.; Han, W.; Wang, A.; Zhang, M.; Wang, X. Metal-free highly regioselective 1,4-sulfonyliodination of 1,3-enynes. J. Org. Chem. 2023, 88, 2034–2045. [Google Scholar] [CrossRef]
- Hu, D.-D.; Gao, Q.; Dai, J.-C.; Cui, R.; Li, Y.-B.; Li, Y.-M.; Zhou, X.-G.; Bian, K.-J.; Wu, B.-B.; Zhang, K.-F.; et al. Visible-light-induced, autopromoted nickel-catalyzed three-component arylsulfonation of 1,3-enynes and mechanistic insights. Sci. China Chem. 2022, 65, 753–761. [Google Scholar] [CrossRef]
- Wang, F.; Wang, D.; Zhou, Y.; Liang, L.; Lu, R.; Chen, P.; Lin, Z.; Liu, G. Divergent synthesis of CF3-substituted allenyl nitriles by ligand-controlled radical 1,2- and 1,4-addition to 1,3-enynes. Angew. Chem. Int. Ed. 2018, 57, 7140–7145. [Google Scholar] [CrossRef]
- Shen, H.; Xiao, H.; Zhu, L.; Li, C. Copper-catalyzed radical bis(trifluoromethylation) of alkynes and 1,3-enynes. Synlett 2020, 31, 41–44. [Google Scholar] [CrossRef]
- Huang, J.; Jia, Y.; Li, X.; Duan, J.; Jiang, Z.; Yang, Z. Halotrifluoromethylation of 1,3-enynes: Access to tetrasubstituted allenes. Org. Lett. 2021, 23, 2314–2319. [Google Scholar] [CrossRef]
- Li, S.; Yang, W.; Shi, J.; Dan, T.; Han, Y.; Cao, Z.; Yang, M. Synthesis of trifluoromethyl-substituted allenols via catalytic trifluoromethylbenzoxylation of 1,3-enynes. ACS Catal. 2023, 13, 2142–2148. [Google Scholar] [CrossRef]
- Song, Y.; Fu, C.; Ma, S. Copper-catalyzed syntheses of multiple functionalizatized allenes via three-component reaction of enynes. ACS Catal. 2021, 11, 10007–10013. [Google Scholar] [CrossRef]
- He, F.; Bao, P.; Yu, F.; Zeng, L.; Deng, W.; Wu, J. Copper-catalyzed regioselective 1,4-selenosulfonylation of 1,3-enynes to access cyanoalkylsulfonylated allenes. Org. Lett. 2021, 23, 7472–7476. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, J.; Cui, S.; Xie, X.; Wang, X.; Wu, L. Visible-light-induced 1,4-hydroxysulfonylation of vinyl enynes with sulfonyl chlorides: The bridge of chloride linking water and enynes. Org. Lett. 2021, 23, 3530–3535. [Google Scholar] [CrossRef]
- Xie, X.Y.; Xu, Y.F.; Li, Y.; Wang, X.D.; Zhu, J.; Wu, L. Radical modulated regioselective difunctionalization of vinyl enynes: Tunable access to naphthalen-1(2H)-ones and allenic alcohols. Chem. Commun. 2022, 58, 3031–3034. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Lu, Y. Decarboxylative 1,4-carbocyanation of 1,3-enynes to access tetra-substituted allenes via copper/photoredox dual catalysis. Chem. Sci. 2021, 12, 11316–11321. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, K.; Huang, Q.; Lu, Y. Regiodivergent sulfonylarylation of 1,3-enynes via nickel/photoredox dual catalysis. Chem. Sci. 2021, 12, 13564–13571. [Google Scholar] [CrossRef]
- Xu, T.; Wu, S.; Zhang, Q.; Wu, Y.; Hu, M.; Li, J. Dual photoredox/nickel-catalyzed 1,4-sulfonylarylation of 1,3-enynes with sulfinate salts and aryl halides: Entry into tetrasubstituted allenes. Org. Lett. 2021, 23, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hu, D.D.; Jin, R.X.; Wu, B.B.; Wang, C.Y.; Ke, Z.F.; Wang, X.S. Selective 1,4-arylsulfonation of 1,3-enynes via photoredox/nickel dual catalysis. Org. Chem. Front. 2022, 9, 788–794. [Google Scholar] [CrossRef]
- Liu, W.; Liu, C.; Wang, M.; Kong, W. Modular synthesis of multifunctionalized CF3-allenes through selective activation of saturated hydrocarbons. ACS Catal. 2022, 12, 10207–10221. [Google Scholar] [CrossRef]
- Chen, L.; Lin, C.; Zhang, S.; Zhang, X.; Zhang, J.; Xing, L.; Guo, Y.; Feng, J.; Gao, J.; Du, D. 1,4-alkylcarbonylation of 1,3-enynes to access tetra-substituted allenyl ketones via an NHC-catalyzed radical relay. ACS Catal. 2021, 11, 13363–13373. [Google Scholar] [CrossRef]
- Chen, L.; Wang, J.; Lin, C.; Zhu, Y.; Du, D. CF2Br2 as a source for difluoroolefination of 1,3-enynes via N-heterocyclic carbene catalysis. Org. Lett. 2022, 24, 7047–7051. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, J.; Huang, Y. N-heterocyclic carbene-catalyzed 1,4-alkylacylation of 1,3-enynes. Org. Lett. 2021, 23, 9251–9255. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.; Kou, X.; Zeng, R.; Qi, T.; Zhang, X.; Peng, C.; Han, B.; Li, J. Radical acylalkylation of 1,3-enynes to access allenic ketones via N-heterocyclic carbene organocatalysis. J. Org. Chem. 2022, 87, 5229–5241. [Google Scholar] [CrossRef]
- Wang, L.; Ma, R.; Sun, J.; Zheng, G.; Zhang, Q. NHC and visible light-mediated photoredox Co-catalyzed 1,4-sulfonylacylation of 1,3-enynes for tetrasubstituted allenyl ketones. Chem. Sci. 2022, 13, 3169–3175. [Google Scholar] [CrossRef]
- Ye, C.; Li, Y.; Zhu, X.; Hu, S.; Yuan, D.; Bao, H. Copper-catalyzed 1,4-alkylarylation of 1,3-enynes with masked alkyl electrophiles. Chem. Sci. 2019, 10, 3632–3636. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Deng, W.; Chiou, M.; Ye, C.; Jian, W.; Zeng, Y.; Jiao, Y.; Ge, L.; Li, Y.; Zhang, X.; et al. Copper-catalyzed radical 1,4-difunctionalization of 1,3-enynes with alkyl diacyl peroxides and N-fluorobenzenesulfonimide. J. Am. Chem. Soc. 2019, 141, 548–559. [Google Scholar] [CrossRef]
- Zeng, Y.; Chiou, M.; Zhu, X.; Cao, J.; Lv, D.; Jian, W.; Li, Y.; Zhang, X.; Bao, H. Copper-catalyzed enantioselective radical 1,4-difunctionalization of 1,3-enynes. J. Am. Chem. Soc. 2020, 142, 18014–18021. [Google Scholar] [CrossRef]
- Dong, X.; Zhan, T.; Jiang, S.; Liu, X.; Ye, L.; Li, Z.; Gu, Q.; Liu, X. Copper-catalyzed asymmetric coupling of allenyl radicals with terminal alkynes to access tetrasubstituted allenes. Angew. Chem. Int. Ed. 2021, 60, 2160–2164. [Google Scholar] [CrossRef]
- Zhang, F.; Guo, X.; Zeng, X.; Wang, Z. Asymmetric 1,4-functionalization of 1,3-enynes via dual photoredox and chromium catalysis. Nat. Commun. 2022, 13, 5036. [Google Scholar] [CrossRef]
- Inoue, M.; Nakada, M. Studies into asymmetric catalysis of the nozaki-hiyama allenylation. Angew. Chem. Int. Ed. 2006, 45, 252–255. [Google Scholar] [CrossRef]
- Li, P.H.; Wei, Y.; Shi, M. Cu(I)-catalyzed addition-cycloisomerization difunctionalization reaction of 1,3-enyne-alkylidenecyclopropanes (ACPs). Org. Biomol. Chem. 2020, 18, 7127–7138. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, Y.; Sun, J.; Han, Y.; Jia, X.; Yan, C. Construction of C(sp2)–X (X = Br, Cl) bonds through a copper-catalyzed atom-transfer radical process: Application for the 1,4-difunctionalization of isoquinolinium salts. Org. Lett. 2018, 20, 987–990. [Google Scholar] [CrossRef]
- Si, W.; Zhang, X.; Asao, N.; Yamamoto, Y.; Jin, T. Ni-catalyzed direct 1,4-difunctionalization of [60]fullerene with benzyl bromides. Chem. Commun. 2015, 51, 6392–6394. [Google Scholar] [CrossRef]
- Shang, T.; Zhang, J.; Zhang, Y.; Zhang, F.; Li, X.; Zhu, G. Photocatalytic remote oxyfluoroalkylation of heteroalkynes: Regio-, stereo-, and site-selective access to complex fluoroalkylated (Z)-alkenes. Org. Lett. 2020, 22, 3667–3672. [Google Scholar] [CrossRef]
- Shu, C.; Feng, J.; Zheng, H.; Cheng, C.; Yuan, Z.; Zhang, Z.; Xue, X.; Zhu, G. Internal alkyne-directed fluorination of unactivated C(sp3)–H bonds. Org. Lett. 2020, 22, 9398–9403. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhang, F.; Yu, Y.; Tan, Z.; Zhu, G. AIBN-induced remote trifluoromethyl-alkynylation of thioalkynes. Org. Lett. 2020, 22, 4088–4092. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hirose, D.; Taniguchi, T. Direct synthesis of 1,4-diols from alkenes by iron-catalyzed aerobic hydration and C–H hydroxylation. Angew. Chem. Int. Ed. 2014, 53, 2730–2734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Meng, X.; Sheng, J.; Lan, Q.; Wang, X. Enantioselective copper-catalyzed 1,5-cyanotrifluoromethylation of vinylcyclopropanes. Org. Lett. 2019, 21, 8256–8260. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Ma, J.; Cahard, D. Radical 1,5-chloropentafluorosulfanylation of unactivated vinylcyclopropanes and transformation into α-SF5 ketones. J. Org. Chem. 2021, 86, 13808–13816. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Sun, Q.; Ye, R.; Sun, J.; Han, Y.; Yan, C. Copper-catalyzed selective difunctionalization of N-heteroarenes through a halogen atom transfer radical process. N. J. Chem. 2019, 43, 13832–13836. [Google Scholar] [CrossRef]
- Li, X.; Shui, Y.; Shen, P.; Wang, Y.; Zhang, C.; Feng, C. A novel type of radical-addition-induced β-fragmentation and ensuing remote functionalization. Chem 2022, 8, 2245–2259. [Google Scholar] [CrossRef]
- Li, W.; Xu, W.; Xie, J.; Yu, S.; Zhu, C. Distal radical migration strategy: An emerging synthetic means. Chem. Soc. Rev. 2018, 47, 654–667. [Google Scholar] [CrossRef]
- Xi, J.-M.; Liao, W.-W. Radical addition to the C=C bond meets (1,n)-HAT: Recent advances in the remote C(sp3)–H or C(sp2)–H functionalization of alkenes. Org. Chem. Front. 2022, 9, 4490–4506. [Google Scholar] [CrossRef]
- Yu, P.; Lin, J.; Li, L.; Zheng, S.; Xiong, Y.; Zhao, L.; Tan, B.; Liu, X. Enantioselective C–H bond functionalization triggered by radical trifluoromethylation of unactivated alkene. Angew. Chem. Int. Ed. 2014, 53, 11890–11894. [Google Scholar] [CrossRef]
- Li, T.; Yu, P.; Du, Y.; Lin, J.; Zhi, Y.; Liu, X. Enantioselective α-C–H functionalization of amides with indoles triggered by radical trifluoromethylation of alkenes: Highly selective formation of C–CF3 and C–C bonds. J. Fluor. Chem. 2017, 203, 210–214. [Google Scholar] [CrossRef]
- Li, T.; Yu, P.; Lin, J.; Zhi, Y.; Liu, X. Copper-catalyzed redox-triggered remote C–H functionalization: Highly selective formation of C–CF3 and C=O bonds. Chin. J. Chem. 2016, 34, 490–494. [Google Scholar] [CrossRef]
- Huang, L.; Zheng, S.; Tan, B.; Liu, X. Metal-free direct 1,6- and 1,2-difunctionalization triggered by radical trifluoromethylation of alkenes. Org. Lett. 2015, 17, 1589–1592. [Google Scholar] [CrossRef]
- Huang, L.; Lin, J.; Tan, B.; Liu, X. Alkene trifluoromethylation-initiated remote α-azidation of carbonyl compounds toward trifluoromethyl γ-lactam and spirobenzofuranone-lactam. ACS Catal. 2015, 5, 2826–2831. [Google Scholar] [CrossRef]
- Yu, P.; Zheng, S.; Yang, N.; Tan, B.; Liu, X. Phosphine-catalyzed remote β-C–H functionalization of amines triggered by trifluoromethylation of alkenes: One-pot synthesis of bistrifluoromethylated enamides and oxazoles. Angew. Chem. Int. Ed. 2015, 54, 4041–4045. [Google Scholar] [CrossRef]
- Shu, W.; Merino, E.; Nevado, C. Visible light mediated, redox neutral remote 1,6-difunctionalizations of alkenes. ACS Catal. 2018, 8, 6401–6406. [Google Scholar] [CrossRef]
- Bian, K.; Li, Y.; Zhang, K.; He, Y.; Wu, T.; Wang, C.; Wang, X. Iron-catalyzed remote functionalization of inert C(sp3)–H bonds of alkenes via 1,n-hydrogen-atom-transfer by C-centered radical relay. Chem. Sci. 2020, 11, 10437–10443. [Google Scholar] [CrossRef]
- Li, L.; Luo, H.; Zhao, Z.; Li, Y.; Zhou, Q.; Xu, J.; Li, J.; Ma, Y. Photoredox-catalyzed remote difunctionalizations of alkenes to synthesize fluoroalkyl ketones with dimethyl sulfoxide as the oxidant. Org. Lett. 2019, 21, 9228–9231. [Google Scholar] [CrossRef]
- Chen, X.; Wei, W.; Li, C.; Zhou, H.; Qiao, B.; Jiang, Z. Photoredox-catalyzed synthesis of remote fluoroalkylated azaarene derivatives and the α-deuterated analogues via 1,n-hydrogen-atom-transfer-involving radical reactions. Org. Lett. 2021, 23, 8744–8749. [Google Scholar] [CrossRef]
- Song, L.; Fu, D.M.; Chen, L.; Jiang, Y.X.; Ye, J.H.; Zhu, L.; Lan, Y.; Fu, Q.; Yu, D.G. Visible-light photoredox-catalyzed remote difunctionalizing carboxylation of unactivated alkenes with CO2. Angew. Chem. Int. Ed. 2020, 59, 21121–21128. [Google Scholar] [CrossRef]
- Song, L.; Wang, W.; Yue, J.; Jiang, Y.; Wei, M.; Zhang, H.; Yan, S.; Liao, L.; Yu, D. Visible-light photocatalytic di- and hydro-carboxylation of unactivated alkenes with CO2. Nat. Catal. 2022, 5, 832–838. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, S.; Lu, D.; Zhu, G. Copper-catalyzed trifluoromethylation of alkenes with redox-neutral remote amidation of aldehydes. Org. Lett. 2016, 18, 2852–2855. [Google Scholar] [CrossRef]
- Nie, X.; Cheng, C.; Zhu, G. Palladium-catalyzed remote aryldifluoroalkylation of alkenyl aldehydes. Angew. Chem. Int. Ed. 2017, 56, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Zhou, Y.; Zhao, Y.; Ma, Q.; Kong, L.; Zhu, G. Nickel-catalyzed remote arylation of alkenyl aldehydes initiated by radical alkylation with tertiary α-carbonyl alkyl bromides. Org. Lett. 2018, 20, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Lonca, G.H.; Ong, D.Y.; Tran, T.M.H.; Tejo, C.; Chiba, S.; Gagosz, F. Anti-markovnikov hydrofunctionalization of alkenes: Use of a benzyl group as a traceless redox-active hydrogen donor. Angew. Chem. Int. Ed. 2017, 56, 11440–11444. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ye, L.; Ni, S.; Li, Z.; Chen, S.; Du, Y.; Li, X.; Dang, L.; Liu, X. Phosphine-catalyzed remote α-C–H bond activation of alcohols or amines triggered by the radical trifluoromethylation of alkenes: Reaction development and mechanistic insights. Org. Chem. Front. 2017, 4, 2139–2146. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, W.; Cheng, C.; Luo, F. Copper-catalyzed remote oxidation of alcohols initiated by radical difluoroalkylation of alkenes: Facile access to difluoroalkylated carbonyl compounds. Org. Biomol. Chem. 2018, 16, 3876–3880. [Google Scholar] [CrossRef]
- Wang, N.; Ye, L.; Li, Z.; Li, L.; Li, Z.; Zhang, H.; Guo, Z.; Gu, Q.; Liu, X. Hydrofunctionalization of alkenols triggered by the addition of diverse radicals to unactivated alkenes and subsequent remote hydrogen atom translocation. Org. Chem. Front. 2018, 5, 2810–2814. [Google Scholar] [CrossRef]
- Leonori, D.; Aggarwal, V.K. Lithiation–borylation methodology and its application in synthesis. Accounts Chem. Res. 2014, 47, 3174–3183. [Google Scholar] [CrossRef]
- Kischkewitz, M.; Friese, F.W.; Studer, A. Radical-induced 1,2-migrations of boron ate complexes. Adv. Synth. Catal. 2020, 362, 2077–2087. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Jana, K.; Studer, A. Intramolecular hydrogen atom transfer induced 1,2-migration of boronate complexes. Org. Lett. 2021, 23, 5876–5879. [Google Scholar] [CrossRef]
- Luo, X.-L.; Li, S.-S.; Jiang, Y.-S.; Liu, F.; Li, S.-H.; Xia, P.-J. Photocatalytic 1,2-iminosulfonylation and remote 1,6-iminosulfonylation of olefins. Org. Lett. 2023, 25, 1742–1747. [Google Scholar] [CrossRef]









































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Zhang, Q.; Zhang, W. Remote Radical 1,3-, 1,4-, 1,5-, 1,6- and 1,7-Difunctionalization Reactions. Molecules 2023, 28, 3027. https://doi.org/10.3390/molecules28073027
Ma X, Zhang Q, Zhang W. Remote Radical 1,3-, 1,4-, 1,5-, 1,6- and 1,7-Difunctionalization Reactions. Molecules. 2023; 28(7):3027. https://doi.org/10.3390/molecules28073027
Chicago/Turabian StyleMa, Xiaoming, Qiang Zhang, and Wei Zhang. 2023. "Remote Radical 1,3-, 1,4-, 1,5-, 1,6- and 1,7-Difunctionalization Reactions" Molecules 28, no. 7: 3027. https://doi.org/10.3390/molecules28073027