
The Potential of 2-Substituted Quinolines as Antileishmanial Drug Candidates

 , , ,
, , ,
Abstract
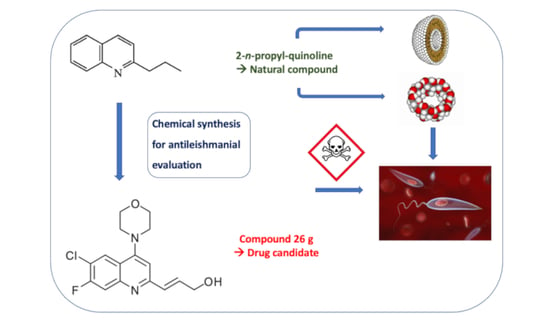
:
1. Introduction
2. The Place of Quinolines as Drug Candidates in the Treatment of Leishmaniases
3. The Potential of 2-Substituted Quinolines as Antileishmanial Agents
3.1. From the Plant to Experimental Models of Leishmaniasis
3.2. From Natural Compounds to Synthetic Derivatives and Their Biological Evaluation
3.3. Formulations of the Natural 2-n-Propyl Quinoline
3.3.1. Preparation, Characterization and Biological Activity of a 2-n-Propylquinoline Salt to Improve Aqueous Solubility
3.3.2. Preparation, Characterization and Biological Activity of a Liposomal Formulation of 2-n-Propylquinoline for the Treatment of Visceral Leishmaniasis by the Intravenous Route
3.3.3. Preparation, Characterization and Biological Activity of a Formulation of 2-n-Propylquinoline with Hydroxypropyl Beta-Cyclodextrin for the Treatment of Different Manifestations of Leishmaniasis
3.4. Entering the DNDi Pipeline to Obtain Second-Generation 2-Substituted Quinolines
3.5. Structure–Activity Relationships
3.5.1. Natural Compounds and Synthetic Compounds of the First Generation
3.5.2. DNDi Series
3.6. Mechanism of Action
3.7. Drug Resistance
3.8. Orientating the Mechanism of Action of 2-Substituted Quinolines: Mechanistic Targeting for a New Series of Compounds
3.8.1. Targeting an Enzyme Involved in Host Cell Recognition
3.8.2. Conferring Chelating Properties on 2-Substituted Quinolines
3.8.3. Obtaining New Metallodrugs
3.8.4. Mechanism of Action of Dual Compounds
4. Antiviral Activities of 2-Substituted Quinolines and the Interest of this Series in Co-Infections
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/data/gho/data/themes/topics/gho-ntd-leishmaniasis (accessed on 28 March 2022).
- Available online: https://dndi.org/diseases/visceral-leishmaniasis/facts/ (accessed on 28 March 2022).
- World Health Organization. Health Topics/Leishmaniasis. Available online: https://www.who.int/health-topics/leishmaniasis#tab=tab_1 (accessed on 28 March 2022).
- Olias-Molero, A.I.; de la Fuente, C.; Cuquerella, M.; Torrado, J.J.; Alunda, J.M. Antileishmanial drug discovery and development: Time to reset the model? Microorganisms 2021, 9, 2500. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 2021, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Van Bocxlaer, K.; Caridha, D.; Black, C.; Vesely, B.; Leed, S.; Sciotti, R.J.; Wijnant, G.J.; Yardley, V.; Braillard, S.; Mowbray, C.E.; et al. Novel benzoxaborole, nitroimidazole and aminopyrazoles with activity against experimental cutaneous leishmaniasis. Int. J. Parasitol.—Drugs Drug Resist. 2019, 11, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, C.E.; Braillard, S.; Glossop, P.A.; Whitlock, G.A.; Jacobs, R.T.; Speake, J.; Pandi, B.; Nare, B.; Maes, L.; Yardley, V.; et al. DNDI-6148: A novel benzoxaborole preclinical candidate for the treatment of visceral leishmaniasis. J. Med. Chem. 2021, 64, 16159–16176. [Google Scholar] [CrossRef]
- Alves, F.; Bilbe, G.; Blesson, S.; Goyal, V.; Monnerat, S.; Mowbray, C.; Muthoni Ouattara, G.; Pécoul, B.; Rijal, S.; Rode, J.; et al. Recent Development of visceral leishmaniasis treatments: Successes, pitfalls, and perspectives. Clin. Microbiol. Rev. 2018, 31, e00048-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyllie, S.; Brand, S.; Thomas, M.; De Rycker, M.; Chung, C.W.; Pena, I.; Bingham, R.P.; Bueren-Calabuig, J.A.; Cantizani, J.; Cebrian, D.; et al. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 9318–9323. [Google Scholar] [CrossRef] [Green Version]
- Silva da Gama, A.N. Quinoline-based compounds as key candidates to tackle drug discovery programs of microbicidal agents. Curr. Pharm. Des. 2021, 27, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.M.; Bray, P.G.; Hawley, S.R.; Ward, S.A.; Park, B.K. 4-aminoquinolines, past, present and future: A chemical perspective. Pharmacol. Ther. 1998, 77, 29–58. [Google Scholar] [CrossRef]
- Tekwani, B.L.; Walker, L.A. 8-aminoquinolines: Future role as antiprotozoal drugs. Curr. Opin. Infect. Dis. 2006, 19, 623–631. [Google Scholar] [CrossRef]
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, H.; Yang, Y.; Chen, Z.S.; Zou, C.; Zhang, J. Chloroquine against malaria, cancers and viral diseases. Drug Discov. Today 2020, 25, 2012–2022. [Google Scholar] [CrossRef]
- Rocha, V.P.C.; Nonato, F.R.; Guimaraes, E.T.; Rodrigues de Freitas, L.A.; Soares, M.B.P. Activity of antimalarial drugs in vitro and in a murine model of cutaneous leishmaniasis. J. Med. Microbiol. 2013, 62, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Malik, F.; Hanif, M.M.; Mustafa, G. Comparing the efficacy of oral chloroquine versus oral tetracycline in the treatment of cutaneous leishmaniasis. J. Coll. Physicians Surg. Pak. 2019, 29, 403–405. [Google Scholar] [CrossRef]
- Mwololo, S.W.; Mutiso, J.M.; Macharia, J.C.; Bourdichon, A.J.; Gicheru, M.M. In vitro activity and in vivo efficacy of a combination therapy of diminazene and chloroquine against murine visceral leishmaniasis. J. Biomed. Res. 2015, 29, 214–223. [Google Scholar]
- Wijnant, G.J.; Van Bocxlaer, K.; Yardley, V.; Murdan, S.; Croft, S.L. Efficacy of paromomycin-chloroquine combination therapy in experimental cutaneous leishmaniasis. Antimicrob. Agents Chemother. 2017, 61, e00358-17. [Google Scholar] [CrossRef] [Green Version]
- Herrera, L.; Llanes, A.; Alvarez, J.; Degracia, K.; Restrepo, C.M.; Rivera, R.; Stephens, D.E.; Dang, H.T.; Larionov, O.V.; Lleonart, R.; et al. Antileishmanial activity of a new chloroquine analog in an animal model of Leishmania panamensis infection. Int. J. Parasitol. Drugs Drug Res. 2020, 14, 56–61. [Google Scholar] [CrossRef]
- Rodrigues, J.M., Jr.; Croft, S.L.; Fessi, H.; Bories, C.; Devissaguet, J.P. The activity and ultrastructural localization of primaquine-loaded poly (d,l-lactide) nanoparticles in Leishmania donovani infected mice. Trop. Med. Parasitol. 1994, 45, 223–228. [Google Scholar]
- Gaspar, R.; Opperdoes, F.R.; Préat, V.; Roland, M. Drug targeting with polyalkylcyanoacrylate nanoparticles: In vitro activity of primaquine-loaded nanoparticles against intracellular Leishmania donovani. Ann Trop Med Parasitol. 1992, 86, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Sangraula, H.; Sharma, K.K.; Rijal, S.; Dwivedi, S.; Koirala, S. Orally effective drugs for kala-azar (visceral leishmaniasis): Focus on miltefosine and sitamaquine. J. Assoc. Physicians India 2003, 51, 686–690. [Google Scholar] [PubMed]
- Taylor, T.; Hawkins, D.R.; Morris, G.R.; Chung, H. Pharmacokinetics of the anti-leishmanian agent WR 6026 in dogs. Eur. J. Drug Metab. Pharmacokinet. 1991, 136–139. Available online: https://pubmed.ncbi.nlm.nih.gov/1820868/ (accessed on 28 March 2022).
- Theoharides, A.D.; Chung, H.; Velazquez, H. Metabolism of a potential 8-aminoquinoline antileishmanial drug in rat liver microsomes. Biochem. Pharmacol. 1985, 34, 181–188. [Google Scholar] [CrossRef]
- Dueñas-Romero, A.M.; Loiseau, P.M.; Saint-Pierre-Chazalet, M. Interaction of sitamaquine with membrane lipids of Leishmania donovani promastigotes. Biochim. Biophys. Acta 2007, 1768, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Coimbra, E.S.; Libong, D.; Cojean, S.; Saint-Pierre-Chazalet, M.; Solgadi, A.; Le Moyec, L.; Duenas-Romero, A.M.; Chaminade, P.; Loiseau, P.M. Mechanism of interaction of sitamaquine with Leishmania donovani. J. Antimicrob. Chemother. 2010, 65, 2548–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, L.; Luque-Ortega, J.R.; López-Martín, C.; Castanys, S.; Rivas, L.; Gamarro, F. The 8-aminoquinoline analogue sitamaquine causes oxidative stress in Leishmania donovani promastigotes by targeting succinate dehydrogenase. Antimicrob. Agents Chemother. 2011, 55, 4204–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Martín, C.; Pérez-Victoria, J.M.; Carvalho, L.; Castanys, S.; Gamarro, F. Sitamaquine sensitivity in Leishmania species is not mediated by drug accumulation in acido-calcisomes. Antimicrob. Agents Chemother. 2008, 52, 4030–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, T.; Brown, M.B.; Lawrence, M.J.; Croft, S.L. In vitro and in vivo studies on a topical formulation of sitamaquine dihydrochloride for cutaneous leishmaniasis. J. Pharm. Pharmacol. 2006, 58, 1043–1054. [Google Scholar] [CrossRef]
- Sherwood, J.A.; Gachihi, G.S.; Muigai, R.K.; Skillman, D.R.; Mugo, M.; Rashid, J.R.; Wasunna, K.M.; Were, J.B.; Kasili, S.K.; Mbugua, J.M.; et al. Phase 2 efficacy trail of an oral 8-aminoquinoline (WR6026) for treatment of visceral leishmaniasis. Clin. Infect. Dis. 1994, 19, 1034–1039. [Google Scholar] [CrossRef]
- Dietze, R.; Carvalho, S.F.; Valli, L.C.; Berman, J.; Brewer, T.; Milhous, W.; Sanchez, J.; Schuster, B.; Grogl, M. Phase 2 trial of WR6026, an orally administered 8-amnioquinoline in the treatment of visceral leishmaniasis caused by Leishmania chagasi. Am. J. Trop. Med. Hyg. 2001, 65, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasunna, M.K.; Rashid, J.R.; Mbui, J.; Kirigi, G.; Kinoti, D.; Lodenyo, H.; Felton, J.M.; Sabin, A.J.; Albert, M.J.; Horton, J. A phase II dose-increasing study of sitamaquine for the treatment of visceral leishmaniasis in Kenya. Am. J. Trop. Med. Hyg. 2005, 73, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Bories, C.; Cojean, S.; Huteau, F.; Loiseau, P.M. Selection and phenotype characterisation of sitamaquine-resistant promastigotes of Leishmania donovani. Biomed. Pharmacother. 2008, 62, 164–167. [Google Scholar] [CrossRef]
- Loiseau, P.M.; Cojean, S.; Schrevel, J. Sitamaquine as a putative antileishmanial drug candidate: From the mechanism of action to the risk of drug resistance. Parasite 2011, 18, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Markus, M.B. Safety and efficacy of tafenoquine for Plasmodium vivax malaria prophylaxis and radical cure: Overview and perspectives. Ther. Clin. Risk Manag. 2021, 17, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Yardley, V.; Gamarro, F.; Croft, S.L. Antileishmanial and antitrypanosomal activities of the 8-aminoquinoline tafenoquine. Antimicrob. Agents Chemother. 2010, 54, 5356–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, L.; Luque-Ortega, J.R.; Manzano, J.I.; Castanys, S.; Rivas, L.; Gamarro, F. Tafenoquine, an antiplasmodial 8-aminoquinoline, targets leishmania respiratory complex III and induces apoptosis. Antimicrob Agents Chemother. 2010, 54, 5344–5351. [Google Scholar] [CrossRef] [Green Version]
- Manzano, J.I.; Carvalho, L.; García-Hernández, R.; Poveda, J.A.; Ferragut, J.A.; Castanys, S.; Gamarro, F. Uptake of the antileishmanial drug tafenoquine follows a sterol-dependent diffusion process in Leishmania. J. Antimicrob. Chemother. 2011, 66, 2562–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano, J.I.; Carvalho, L.; Pérez-Victoria, J.M.; Castanys, S.; Gamarro, F. Increased glycolytic ATP synthesis is associated with tafenoquine resistance in Leishmania major. Antimicrob. Agents Chemother. 2011, 55, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Werbel, L.M. Synthesis and antileishmanial activity of 6-methoxy-4-methyl-n-[6-(substituted-1-piperazinyl)hexyl]-8-quinolinamines and related compounds. J. Med. Chem. 1983, 26, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.; Vagneur, B.; Richomme, P.; Bruneton, J. Aryl et al.kyl-2 quinoléines nouvelles isolées d’une Rutacée bolivienne: Galipea longiflora. Can. J. Chem. 1989, 67, 2116–2118. [Google Scholar] [CrossRef]
- Fournet, A.; Barrios, A.A.; Munoz, V.; Hocquemiller, R.; Cavé, A.; Bruneton, J. 2-substituted quinoline alkaloids as potential antileishmanial drugs. Antimicrob. Agents Chemother. 1993, 37, 859–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournet, A.; Hocquemiller, R.; Roblot, F.; Cavé, A.; Richomme, P.; Bruneton, J. Les chimanines, nouvelles quinoléines substituées en 2, isolées d’une plante bolivienne antiparasitaire: Galipea longiflora. J. Nat. Prod. 1993, 56, 1547–1552. [Google Scholar]
- Fournet, A.; Ferreira, M.E.; Rojas de Arias, A.; Torres de Ortiz, S.; Fuentes, S.; Nakayama, H.; Schinini, A.; Hocquemiller, R. In vivo efficacy of oral and intralesional administration of 2-substitutted quinolines in experimental treatment on New World cutaneous leishmaniasis caused by Leishmania amazonensis. Antimicrob. Agents Chemother. 1996, 40, 2447–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournet, A.; Gantier, J.C.; Gautheret, A.; Leysalles, L.; Munos, M.H.; Mayrargue, J.; Moscovitz, H.; Cavé, A.; Hocquemiller, R. The activity of 2-substituted quinoline alkaloids in BALB/c mice infected with Leishmania donovani. J. Antimicrob. Chemother. 1994, 33, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Vieira, N.C.; Herrenknecht, C.; Vacus, J.; Fournet, A.; Bories, C.; Figadère, B.; Espindola, L.S.; Loiseau, P.M. Selection of the most promising 2-substituted quinoline as antileishmanial candidate for clinical trials. Biomed. Pharmacother. 2008, 62, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Gantier, J.C.; Fournet, A.; Munos, M.H.; Hocquemiller, R. The effect of some 2-substituted quinolines isolated from Galipea longiflora on Plasmodium vinckei petteri infected mice. Planta Med. 1996, 62, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Munos, M.H.; Mayrargue, J.; Fournet, A.; Gantier, J.C.; Hocquemiller, R.; Moskowitz, H. Synthesis of an antileishmanial alkaloid isolated from Galipea longiflora and of related compounds. Chem. Pharm. Bull. 1994, 42, 1914–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhfakh, M.A.; Franck, X.; Fournet, A.; Hocquemiller, R.; Figadère, B. Expeditious preparation of 2-substituted quinolines. Tetrahedron Lett. 2001, 42, 3847–3850. [Google Scholar] [CrossRef]
- Quintin, J.; Franck, X.; Hocquemiller, R.; Figadère, B. Iron-catalysed arylation of heteroaryl halides by Grignard reagents. Tetrahedron Lett. 2002, 43, 3547–3549. [Google Scholar] [CrossRef]
- Dade, J.; Provot, O.; Moskowitz, H.; Mayrargue, J.; Prina, E. Synthesis of 2-substituted trifluoromethylquinolines for the evaluation of leishmanicidal activity. Chem. Pharm. Bull. 2001, 49, 480–483. [Google Scholar] [CrossRef] [Green Version]
- Fakhfakh, M.A.; Franck, X.; Hocquemiller, R.; Figadère, B. Iron catalyzed hydrodebromination of 2-aryl-1,1-dibromo-1-alkenes. J. Organomet. Chem. 2001, 624, 131–135. [Google Scholar] [CrossRef]
- Seck, M.; Franck, X.; Hocquemiller, R.; Figadère, B.; Peyrat, J.F.; Provot, O.; Brion, J.D.; Alami, M. Synthesis of substituted quinolines by iron-catalyzed coupling reactions between chloroenynes and Grignard reagents. Tetrahedron Lett. 2004, 45, 1881–1884. [Google Scholar] [CrossRef]
- Dos Santos, M.; Franck, X.; Hocquemiller, R.; Figadère, B.; Peyrat, J.F.; Provot, O.; Brion, J.D.; Alami, M. Iron-catalyzed coupling reaction between 1,1-dichloro-1-alkenes and Grignard reagents. Synlett 2004, 15, 2697–2700. [Google Scholar] [CrossRef]
- Fakhfakh, M.A.; Fournet, A.; Prina, E.; Mouscadet, J.F.; Franck, X.; Hocquemiller, R.; Figadère, B. Synthesis and biological evaluation of substituted quinolines: Potential treatment of protozoal and retro-viral co-infections. Bioorg. Med. Chem. 2003, 11, 5013–5023. [Google Scholar] [CrossRef]
- Nakayama, H.; Loiseau, P.M.; Bories, C.; Torres de Ortiz, S.; Schinini, A.; Serna, E.; Rojas de Arias, A.; Fakhfakh, M.A.; Franck, X.; Figadère, B.; et al. Efficacy of orally administered 2-substituted quinolines in experimental murine cutaneous and visceral leismaniases. Antimicrob. Agents Chemother. 2005, 49, 4950–4956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, H.; Desrivot, J.; Bories, C.; Franck, X.; Figadère, B.; Hocquemiller, R.; Fournet, A.; Loiseau, P.M. In vitro and in vivo antileishmanial efficacy of a new nitrilquinoline against Leishmania donovani. Biomed. Pharmacother. 2007, 61, 186–188. [Google Scholar] [CrossRef]
- Desrivot, J.; Herrenknecht, C.; Ponchel, G.; Garbi, N.; Prina, E.; Fournet, A.; Bories, C.; Figadère, B.; Hocquemiller, R.; Loiseau, P.M. Antileishmanial 2-substituted quinolines: In vitro behaviour towards biological components. Biomed. Pharmacother. 2007, 61, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Campos-Vieira, N.; Vacus, J.; Fournet, A.; Baudouin, R.; Bories, C.; Séon-Méniel, B.; Figadère, B.; Loiseau, P.M. Antileishmanial activity of a formulation of 2-n-propylquinoline by oral route in mice model. Parasite 2011, 18, 333–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, W.; Daligaux, P.; Lazar, N.; Ha-Duong, T.; Cavé, C.; van Tilbeurgh, H.; Loiseau, P.M.; Pomel, S. Biochemical analysis of leishmanial and human GDP-mannose-pyrophosphorylases and selection of inhibitors as new leads. Sci. Rep. 2017, 7, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinath, V.S.; Pinjari, J.; Dere, R.T.; Verma, A.; Vishwakarma, P.; Shivahare, R.; Moger, M.; Goud, P.S.K.; Ramanathan, V.; Bose, P.; et al. Design, synthesis and biological evaluation of2-substituted quinolines as potential antileishmanial agents. Eur. J. Med. Chem. 2013, 69, 527–536. [Google Scholar] [CrossRef]
- Loiseau, P.M.; Gupta, S.; Verma, A.; Srivastava, S.; Puri, S.K.; Sliman, F.; Normand-Bayle, M.; Desmaele, D. In vitro activities of new 2-substituted quinolines against Leishmania donovani. Antimicrob. Agents Chemother. 2011, 55, 1777–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, E.L.; Santafé, G.G.; Torres, O.L.; Munoz, D.L.; Robledo, S.M. Styrylquinolines-type synthetic compounds with leishmanicidal and cytotoxic activities. Biomedica 2014, 34, 605–611. [Google Scholar]
- Sahu, N.P.; Pal, C.; Mandal, N.B.; Banerjee, S.; Raha, M.; Kundu, A.P.; Basu, A.; Ghosh, M.; Roy, K.; Bandyopadhyay, S. Synthesis of a novel quinoline derivative, 2-(2-methylquinolin-4-ylamino)-n-phenylacetamide-a potential antileishmanial agent. Bioorg. Med. Chem. 2002, 10, 1687–1693. [Google Scholar] [CrossRef]
- Upadhyay, A.; Kushwaha, P.; Gupta, S.; Prasad Dodda, R.; Ramalingam, K.; Kant, R.; Goyal, N.; Sashidhara, K.V. Synthesis and evaluation of novel triazolyl quinoline derivatives as potential antileishmanial agents. Eur. J. Med. Chem. 2018, 154, 172–181. [Google Scholar] [CrossRef]
- Carroll, F.I.; Berrang, B.D.; Linn, C.P. Synthesis of 2,4-disubstituted 6-methoxy-8-aminoquinoline analogues as potential antiparasitics. J. Med. Chem. 1980, 23, 581–584. [Google Scholar] [CrossRef]
- Shetty, R.V.; Blanton, C.D. Synthesis of 2-substituted primaquine analogues as potential antimalarials. J. Med. Chem. 1978, 21, 995–998. [Google Scholar] [CrossRef]
- Kieffer, C.; Cohen, A.; Verhaeghe, P.; Paloque, L.; Hutter, S.; Castera-Ducros, C.; Laget, M.; Rault, S.; Valentin, A.; Rathelot, P.; et al. Antileishmanial pharmacomodulation in 8-nitroquinolin-2(1H)-one series. Biorg. Med. Chem. 2015, 23, 2377–2386. [Google Scholar] [CrossRef]
- Pedron, J.; Boudot, C.; Bourgeade-Delmas, S.; Sournia-Saquet, A.; Paloque, L.; Rastegari, M.; Abdoulaye, M.; El-Kashef, H.; Bonduelle, C.; Pratviel, G.; et al. Antitrypanosomal pharmacomodulation at position 3 of the 8-nitroquinolin-2(1H)-one scaffold using pallado-catalyzed cross coupling reactions. ChemMedChem 2018, 13, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Pedron, J.; Boudot, C.; Hutter, S.; Bourgeade-Delmas, S.; Stigliani, J.L.; Sournia-Saquet, A.; Moreau, A.; Boutet-Robinet, E.; Paloque, L.; Mothes, E.; et al. Novel 8-nitroquinolin-2(1H)-ones as NTR-bioactivated antikinetoplastid molecules: Synthesis, electrochemical and SAR study. Eur. J. Med. Chem. 2018, 155, 135–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paloque, L.; Verhaeghe, P.; Casanova, M.; Castera-Ducros, C.; Dumètre, A.; Mbatchi, L.; Hutter, S.; Kraiem-M’rabe, M.; Laget, M.; Remusat, V.; et al. Discovery of a new antileishmanial hit in 8-nitroquinoline series. Eur. J. Med. Chem. 2012, 54, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, K.; Barratt, G.; Pomel, S.; Cojean, S.; Pilcher, B.; Kesavan, V.; Vacus, J.; Figadère, B.; Loiseau, P.M. New formulations of 2-n-propylquinoline for the treatment of visceral leishmaniasis. In Proceedings of the 2nd Conference of the European COST Action CM1307 “Targeted Chemotherapy towards Diseases Caused by Endoparasites”, Belgrade, Serbia, 26–28 October 2015. [Google Scholar]
- Balaraman, K.; Campos Vieira, N.; Moussa, F.; Vacus, J.; Cojean, S.; Pomel, S.; Bories, C.; Figadère, B.; Kesavan, V.; Loiseau, P.M. In vitro and in vivo antileishmanial properties of a 2-n-propylquinoline hydroxypropyl beta-cyclodextrin formulation and pharmacokinetics via intravenous route. Biomed. Pharmacother. 2015, 76, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, V.S.; Rao, M.; Shivahare, R.; Vishwakarma, P.; Ghose, S.; Pradhan, A.; Hindupur, R.; Das Sarma, K.; Gupta, S.; Puri, S.K.; et al. Design, synthesis, ADME characterization and antileishmanial evaluation of novel substituted quinoline analogs. Bioorg. Med. Chem. Lett. 2014, 24, 2046–2052. [Google Scholar] [CrossRef] [PubMed]
- Desrivot, J.; Moussa, F.; Champy, P.; Fournet, A.; Figadère, B.; Herrenknecht, C. Development of a SPE/HPLC/DAD method for the determination of antileishmanial 2-substituted quinolines and metabolites in rat plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 854, 230–238. [Google Scholar] [CrossRef]
- Desrivot, J.; Edlund, P.O.; Svensson, R.; Baranczewski, P.; Fournet, A.; Figadère, B.; Herrenknecht, C. Metabolism of 2-substituted quinolines with antileishmanial activity studied in vitro with liver microsomes, hepatocytes and recombinantly expressed enzymes analyzed by LC/MS. Toxicology 2007, 235, 27–38. [Google Scholar] [CrossRef]
- Bompart, D.; Nunez-Duran, J.; Rodriguez, D.; Kouznetsov, V.V.; Melendez-Gomez, C.M.; Sojo, F.; Arvelo, F.; Visbal, G.; Alvarez, A.; Serrano-Martin, X.; et al. Anti-leishmanial evaluation of C2-aryl quinolines: Mechanistic insight on bioenergetics and sterol biosynthetic pathway of Leishmania braziliensis. Bioorg. Med. Chem. 2013, 21, 4426–4431. [Google Scholar] [CrossRef]
- Ray, S.; Sadhukhan, P.K.; Mandal, N.B.; Mahato, S.B.; Majunder, H.K. Dual inhibition of DNA topoisomerases of Leishmania donovani by novel indolyl quinolines. Biochem. Biophys. Res. Comm. 1997, 230, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Reguera, R.M.; Dia-Gonzalez, R.; Perez-Pertejo, Y.; Balana-Fouce, R. Characterizing the bi-subunit type IB DNA topoisomerase of leishmania parasites; a novel scenario for drug intervention in trypanosomatids. Curr. Drug Targets 2008, 9, 966–978. [Google Scholar] [CrossRef]
- Tejeria, A.; Perez-Pertejo, Y.; Reguera, R.M.; Carbajo-Andres, R.; Balana-Fouce, R.; Alonso, C.; Martin-Encinas, E.; Selas, A.; Rubiales, G.; Palacios, F. Antileishmanial activity of new hybrid tetrahydroquinoline and quinoline derivatives with phosphorus substituents. Eur. J. Med. Chem. 2019, 162, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Calla-Magarinos, J.; Quispe, T.; Gimenez, A.; Freysdottir, J.; Troye-Blomberg, M.; Fernandez, C. Quinolinic alkaloids from Galipea longiflora Krause suppress production of proinflammatory cytokines in vitro and control inflammation in vivo upon Leishmania infection in mice. Scand. J. Immunol. 2013, 77, 30–38. [Google Scholar] [CrossRef]
- Belliard, A.M.; Leroy, C.; Banide, H.; Farinotti, R.; Lacour, B. Decrease of intestinal P-glycoprotein activity by 2-n-propylquinoline, a new oral treatment for visceral leishmaniasis. Exp. Parasitol. 2003, 103, 51–56. [Google Scholar] [CrossRef]
- Garami, A.; Ilg, T. Disruption of mannose activation in Leishmania mexicana: GDP-mannose pyrophosphorylase is required for virulence, but not for viability. EMBO J. 2001, 20, 3657–3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.; Curtis, J.; Spurck, T.P.; Ilg, T.; Garami, A.; Baldwin, T.; Courret, N.; McFadden, G.I.; Davis, A.; Handman, E. Characterisation of a Leishmania mexicana knockout lacking guanosine diphosphate-mannose pyrophosphorylase. EMBO J. 2005, 35, 861–873. [Google Scholar] [CrossRef]
- Pomel, S.; Rodrigo, J.; Hendra, F.; Cavé, C.; Loiseau, P.M. In silico analysis of a therapeutic target in Leishmania infantum: The guanosine-diphospho-D-mannose pyrophosphorylase. Parasite 2012, 19, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Lazar, N.; van Tilbeurgh, H.; Loiseau, P.M.; Pomel, S. Minor impact of A258D mutation on biochemical and enzymatic properties of Leishmania infantum GDP-mannose pyrophosphorylase. Microorganisms 2022, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Pomel, S.; Mao, W.; Ha-Duong, T.; Cavé, C.; Loiseau, P.M. GDP-mannose pyrophosphorylase: A biologically validated target for drug development against leishmaniasis. Front. Cell. Infect. Microbiol. 2019, 9, 186. [Google Scholar] [CrossRef]
- Daligaux, P.; Pomel, S.; Leblanc, K.; Loiseau, P.M.; Cavé, C. Simple and efficient synthesis of 5′-aryl-5′-deoxyguanosine analogs by azide-alkyne click reaction and their antileishmanial activities. Mol. Divers. 2016, 20, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Daligaux, P.; Bernadat, G.; Tran, L.; Cavé, C.; Loiseau, P.M.; Pomel, S.; Ha-Duong, T. Comparative study of structural models of Leishmania donovani and human GDP-mannose pyrophosphorylases. Eur. J. Med. Chem. 2016, 107, 109–118. [Google Scholar] [CrossRef]
- Coa, J.C.; Cardona-Galeano, W.; Restrepo, A. Fe3+ chelating quinoline–hydrazone hybrids with proven cytotoxicity, leishmanicidal, and trypanocidal activities. Phys. Chem. Chem. Phys. 2018, 20, 20382–20390. [Google Scholar] [CrossRef] [PubMed]
- Dahal, U.P.; Joswig-Jones, C.; Jones, J.P. Comparative study of the affinity and metabolism of type I and type II binding quinoline carboxamide analogues by cytochrome P450 3A4. J. Med. Chem. 2012, 55, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Chanquia, S.N.; Larregui, F.; Puente, V.; Labriola, C.; Lombardo, E.; Linares, G.G. Synthesis and biological evaluation of new quinoline derivatives as antileishmanial and antitrypanosomal agents. Bioorg. Chem. 2019, 83, 526–534. [Google Scholar] [CrossRef]
- Paloque, L.; Hemmert, C.; Valentin, A.; Gornitzka, H. Synthesis, characterization, and antileishmanial activities of gold(I) complexes involving quinoline functionalized N-heterocyclic carbenes. Eur. J. Med. Chem. 2015, 94, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Chandrakar, P.; Gupta, S.; Parmar, N.; Singh, S.K.; Rashid, M.; Kushwaha, P.; Wahajuddin, M.; Sashidhara, K.V.; Kar, S. Synthesis, biological evaluation, structure-activity relationship, and mechanism of action studies of quinoline-metronidazole derivatives against experimental visceral leishmaniasis. J. Med. Chem. 2019, 62, 5655–5671. [Google Scholar] [CrossRef]
- Kaur, R.; Kumar, K. Synthetic and medicinal perspective of quinolines as antiviral agents. Eur. J. Med. Chem. 2021, 215, 113220. [Google Scholar] [CrossRef] [PubMed]
- Persoons, L.; Vanderlinden, E.; Vangeel, L.; Wang, X.; Do, N.D.T.; Foo, S.Y.C.; Leyssen, P.; Neyts, J.; Jochmans, D.; Schols, D.; et al. Broad spectrum anti-coronavirus activity of a series of anti-malaria quinoline analogues. Antiviral Res. 2021, 193, 105127. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.; Pu, S.Y.; Zuercher, W.J.; Einav, S.; Asquith, C.R.M. Potent antiviral activity of novel multi-substituted 4-anilinoquin(az)olines. Biorg. Med. Chem. Lett. 2020, 30, 127284. [Google Scholar] [CrossRef]
- Saul, S.; Huang, P.T.; Einav, S.; Asquith, C.R.M. Identification and evaluation of 4-anilino-quin(az)olines as potent inhibitors of both dengue virus (DENV) and Venezuelan equine encephalitis virus (VEEV). Bioorg. Med. Chem. Lett. 2021, 52, 128407. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.; Mahieux, R.; Fakhfakh, M.A.; Franck, X.; Hocquemiller, R.; Figadère, B. Substituted quinolines induce inhibition of proliferation of HTLV-1 infected cells. Bioorg. Med. Chem. Lett. 2003, 13, 891–894. [Google Scholar] [CrossRef]
- Franck, X.; Fournet, A.; Prina, E.; Mahieux, R.; Hocquemiller, R.; Figadère, B. Biological evaluation of substituted quinolines. Bioorg. Med. Chem. Lett. 2004, 14, 3635–3638. [Google Scholar] [CrossRef]
- Grassi, F.; Guimaraes Correa, A.B.; Mascarenhas, R.; Galvao, B.; Séon-Méniel, B.; Schmidt, F.; Franck, X.; Hocquemiller, R.; Figadère, B.; Fournet, A. Quinoline compounds decrease in vit1ro spontaneous proliferation of peripheral blood mononuclear cells (PBMC) from human T-cell lymphotropic virus (HTLV) type-1-infected patients. Biomed. Pharmacother. 2008, 62, 430–435. [Google Scholar] [CrossRef]
- Veljkovic, V.; Loiseau, P.M.; Figadère, B.; Glisic, S.; Vejkovic, N.; Perovic, V.R.; Cavanaugh, D.P.; Branch, D.R. Virtual screen for repurposing approved and experimental drugs for candidate inhibitors of EBOLA virus infection. F1000 Res. 2015, 4, 34. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Formula | In Vitro Activity Expressed as IC50 (µM) | Selectivity | In Vivo Significant Activity Monitored | References | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| L. donovani | L. infantum | L. amazonensis | Index = CC50/IC50 | on the Leishmania sp./BALB/c Mice Model | ||||||
| Oral | Sub-Cutaneous | Intralesional | Intraperitoneal | |||||||
| 2-n-propylquinoline |  | >100 (pro./i.a.) | >100 (i.a.) | / | 10 mg/kg/day × 10 (L. d.) | 85 mg/kg/day × 14 (L.a.) | 35 mg/kg/day × 15 (L.a.) | 100 mg/kg/day × 5 (L. d.) | Fournet et al., 1993 [42]; Fakhfakh et al., 2003 [55]; Desrivot et al., 2007 [58]; Campos-Vieira et al., 2008 [46] | |
| 2-n-propylquinoline camphorsulfonic acid |  | >100 (pro.) | / | / | / | 10 mg/kg/day × 10 (L. d.) | / | / | / | Campos-Vieira et al. 2011 [59] |
| 2-(2-hydroxyprop-2-enyl)quinoline |  | 7.8 (pro.) | 2 | 4 | > 25 | 25 mg/kg/day × 15 (L.a.); × 10 (L. i.); 12.5 mg/kg/day × 5 (L.d.) | / | / | / | Campos-Vieira et al., 2008 [46]; Fakhfakh et al., 2003 [55]; Nakayama et al., 2005 [56] |
| (E)-3-quinolin-2-yl-acrylonitrile |  | 38.6 (pro.); 2.4 (i.a.) | / | / | / | 12.5 mg/kg/day × 10 (L.d.) | / | / | / | Nakayama et al., 2007 [57] |
| tetraisopropyl (1-(1-(2-(quinolin-2-ylmethoxy)ethyl)-1H-1,2,3-triazol-4-yl)but-3-yne-1,1-53 diyl)bisphosphonate = Compound 99 |  | 0.63 (i.a.) | / | / | 2.4 | In progress | / | / | / | Mao et al., 2017 [60] |
| 3-(6- chloro-7-fluoro-4-morpholino) quinoline prop-2-en-1-ol = Compound 26 g |  | 0.22 (i.a.) | / | / | 187.5 | 50 mg/kg/twice daily × 5 (L.d.) | / | / | / | Gopinath et al., 2013 [61] |
| Miltefosine |  | 3.6 (pro.); 7.5 (i.a.) | / | / | 55 | 7.5 mg/kg/day × 10 (L. i. and L.d.) | / | / | / | Campos-Vieira et al., 2008 [46]; Nakayama et al., 2005 [56]; Nakayama et al., 2007 [57] |
| Compound/Formulation | In Vitro Activity on L. donovani | Cytotoxicity CC50 (µM ± SD) RAW 264.7 Cells | SI = CC50/IC50 | Treatment Regimen (Intravenous Route) × 5 Consecutive Days | Number of Mice | In Vivo Activity Reduction of Parasite Burden (%) | |
|---|---|---|---|---|---|---|---|
| IC50 (µM ± SD) Axenic Amastigotes | Intramacrophage Amastigotes | ||||||
| 2-n-PQ-Lip | 3.10 ± 0.25 Eq 2-n-PQ | 5.84 ± 0.31 Eq 2-n-PQ | 74.09 ± 6.47 Eq 2-n-PQ | 12.7 | 3 mg/kg Eq 2-n-PQ | 8 | 83.8 a |
| 1.5 mg/kg Eq 2-n-PQ | 8 | 32.5 a | |||||
| 0.75 mg/kg Eq 2-n-PQ | 8 | 5.2 | |||||
| 2-n-PQ-AmB-Lip | 2.02 ± 0.23 Eq 2-n-PQ | 4.50 ± 0.23 Eq 2-n-PQ | 58.31 ± 7.32 Eq 2-n-PQ | 4.3 | (1.5 mg Eq 2-n-PQ + 0.012 mg Eq AmB)/kg | 8 | 89.0 a |
| 0.003 Eq AmB | 0.006 Eq AmB | 0.08 Eq AmB | (0.75 mg Eq 2-n-PQ + 0.006 mg Eq AmB)/kg | 8 | 86.5 a | ||
| (0.37 mg Eq 2-n-PQ + 0.003 mg Eq AmB)/kg | 8 | 10.3 | |||||
| AmBisome® | 2.54 ± 0.70 Eq AmB | 1.51 ± 0.22 Eq AmB | 38.50 ± 2.37 Eq 2-n-PQ | 25.5 | 1 mg Eq AmB/kg | 8 | 88.7 a |
| 0.25 mg Eq AmB/kg | 8 | 27.1 | |||||
| 0.006 mg Eq AmB/kg | 8 | 2.3 | |||||
| Blank liposomes | Inactive | Inactive | / | / | Same suspension | 10 | 5.7 |
| 2-n-propylquinoline (2PQ) | >100 | >100 | / | / | / | / | / |
| Control (vehicle) | Inactive | Inactive | Inactive | / | 0.2 mL | 12 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loiseau, P.M.; Balaraman, K.; Barratt, G.; Pomel, S.; Durand, R.; Frézard, F.; Figadère, B. The Potential of 2-Substituted Quinolines as Antileishmanial Drug Candidates. Molecules 2022, 27, 2313. https://doi.org/10.3390/molecules27072313
Loiseau PM, Balaraman K, Barratt G, Pomel S, Durand R, Frézard F, Figadère B. The Potential of 2-Substituted Quinolines as Antileishmanial Drug Candidates. Molecules. 2022; 27(7):2313. https://doi.org/10.3390/molecules27072313
Chicago/Turabian StyleLoiseau, Philippe M., Kaluvu Balaraman, Gillian Barratt, Sébastien Pomel, Rémy Durand, Frédéric Frézard, and Bruno Figadère. 2022. "The Potential of 2-Substituted Quinolines as Antileishmanial Drug Candidates" Molecules 27, no. 7: 2313. https://doi.org/10.3390/molecules27072313