
Structure-Based Bioisosterism Design, Synthesis, Biological Evaluation and In Silico Studies of Benzamide Analogs as Potential Anthelmintics

 , , and
, , and
Abstract
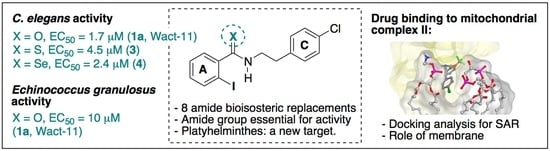
:
1. Introduction
2. Results and Discussion
2.1. Chemistry: Rational and Synthesis
2.1.1. In Silico Design
2.1.2. Synthesis
2.2. Biological Evaluation
2.2.1. C. elegans L1 Assay
2.2.2. Echinococcus granulosus Protoscolex Assay
2.3. In Silico Studies
2.3.1. Docking Analysis: Benzamides Interaction with Nematode Complex II
2.3.2. Simulated Mitochondrial Membrane
3. Materials and Methods
3.1. General Experimental Parameters
3.2. Compound Synthesis
3.3. Anthelmintic Activity Assays
3.3.1. C. elegans Motility Assay
3.3.2. E. granulosus Protoscolex Assay
3.4. In Silico Methods
3.4.1. Homology Modeling
3.4.2. Docking
3.4.3. Membrane Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Caffrey, C.R. Parasitic Helminths: Targets, Screens, Drugs and Vaccines; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2012; pp. 121–216. [Google Scholar] [CrossRef]
- Sangster, N.C.; Cowling, A.; Woodgate, R.G. Ten Events That Defined Anthelmintic Resistance Research. Trends Parasitol. 2018, 34, 553–563. [Google Scholar] [CrossRef]
- Salinas, G.; Risi, G. Caenorhabditis elegans: Nature and nurture gift to nematode parasitologists. Parasitology 2018, 145, 979–987. [Google Scholar] [CrossRef]
- Blaxter, M.; Koutsovoulos, G. The evolution of parasitism in Nematoda. Parasitology 2015, 142, S26–S39. [Google Scholar] [CrossRef]
- Charlier, J.; Höglund, J.; Morgan, E.R.; Geldhof, P.; Vercruysse, J.; Claerebout, E. Biology and Epidemiology of Gastrointestinal Nematodes in Cattle. Vet. Clin. N. Am. Food Anim. Pract. 2020, 36, 1–15. [Google Scholar] [CrossRef]
- Preston, S.; Jiao, Y.; Jabbar, A.; McGee, S.L.; Laleu, B.; Willis, P.; Wells, T.N.; Gasser, R.B. Screening of the ‘Pathogen Box’; identifies an approved pesticide with major anthelmintic activity against the barber’s pole worm. Int. J. Parasitol. Drugs Drug Resist. 2016, 6, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Risi, G.; Aguilera, E.; Ladós, E.; Suárez, G.; Carrera, I.; Álvarez, G.; Salinas, G. Caenorhabditis elegans Infrared-Based Motility Assay Identified New Hits for Nematicide Drug Development. Vet. Sci. 2019, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Burns, A.R.; Luciani, G.; Musso, G.; Bagg, R.; Yeo, M.; Zhang, Y.; Rajendran, L.; Glavin, J.; Hunter, R.; Redman, E.; et al. Caenorhabditis elegans is a useful model for anthelmintic discovery. Nat. Commun. 2015, 6, 7485. [Google Scholar] [CrossRef]
- Mathew, M.D.; Mathew, N.D.; Miller, A.; Simpson, M.; Au, V.; Garland, S.; Gestin, M.; Edgley, M.L.; Flibotte, S.; Balgi, A.; et al. Using C. elegans Forward and Reverse Genetics to Identify New Compounds with Anthelmintic Activity. PLoS Negl. Trop. Dis. 2016, 10, e0005058. [Google Scholar] [CrossRef]
- Shimizu, H.; Osanai, A.; Sakamoto, K.; Inaoka, D.K.; Shiba, T.; Harada, S.; Kita, K. Crystal structure of mitochondrial quinol-fumarate reductase from the parasitic nematode Ascaris suum. J. Biochem. 2012, 151, 589–592. [Google Scholar] [CrossRef]
- Hungenberg, H.; Fürsch, H.; Rieck, H.; Hellwege, E. Use of Fluopyram for Controlling Nematodes in Crops and for Increasing Yield. Patent WO2012072660A1, 7 June 2012. [Google Scholar]
- Chen, J.; Li, Q.X.; Song, B. Chemical Nematicides: Recent Research Progress and Outlook. J. Agric. Food Chem. 2020, 68, 12175–12188. [Google Scholar] [CrossRef]
- Brown, N.; Mannhold, E.R.; Kubinyi, H.; Folkers, G.; Ed, H.; Ed, C. Bioisosteres in Medicinal Chemistry; Wiley-VCH Verlag & Co., KGaA: Weinheim, Germany, 2013. [Google Scholar] [CrossRef]
- Kumari, S.; Carmona, A.; Tiwari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef]
- Wirth, M.; Zoete, V.; Michielin, O.; Sauer, W.H.B. SwissBioisostere: A database of molecular replacements for ligand design. Nucleic Acids Res. 2012, 41, D1137–D1143. [Google Scholar] [CrossRef]
- Matsumoto, J.; Sakamoto, K.; Shinjyo, N.; Kido, Y.; Yamamoto, N.; Yagi, K.; Miyoshi, H.; Nonaka, N.; Katakura, K.; Kita, K.; et al. Anaerobic NADH-Fumarate Reductase System Is Predominant in the Respiratory Chain of Echinococcus multilocularis, Providing a Novel Target for the Chemotherapy of Alveolar Echinococcosis. Antimicrob. Agents Chemother. 2008, 52, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Cuozzo, A.; Daina, A.; Perez, M.A.S.; Michielin, O.; Zoete, V. SwissBioisostere 2021: Updated structural, bioactivity and physicochemical data delivered by a reshaped web interface. Nucleic Acids Res. 2021, 50, D1382–D1390. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, Z.; Liang, P.; Wang, S.; Li, F.; Jin, S.; Zhang, J. Identification of Novel Nematode SDH Inhibitors: Virtual Screening Based on Ligand-Pocket Interactions. Chem. Biol. Drug Des. 2022, 1–15. [Google Scholar] [CrossRef]
- Inaoka, D.K.; Shiba, T.; Sato, D.N.; Balogun, E.O.; Sasaki, T.; Nagahama, M.; Oda, M.; Matsuoka, S.; Ohmori, J.; Honma, T.; et al. Structural Insights into the Molecular Design of Flutolanil Derivatives Targeted for Fumarate Respiration of Parasite Mitochondria. Int. J. Mol. Sci. 2015, 16, 15287–15308. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.J.; Faust, R.; Garratt, P.J.; Marivingt-Mounir, C.; Davidson, K.; Teh, M.-T.; Sugden, D. Binding affinity and biological activity of oxygen and sulfur isosteres at melatonin receptors as a function of their hydrogen bonding capability. Bioorg. Chem. 2004, 32, 1–12. [Google Scholar] [CrossRef]
- Barresi, E.; Nesi, G.; Citi, V.; Piragine, E.; Piano, I.; Taliani, S.; Da Settimo, F.; Rapposelli, S.; Testai, L.; Breschi, M.C.; et al. Iminothioethers as Hydrogen Sulfide Donors: From the Gasotransmitter Release to the Vascular Effects. J. Med. Chem. 2017, 60, 7512–7523. [Google Scholar] [CrossRef]
- Thomsen, I.; Clausen, K.; Scheibye, S.; Lawesson, S.-O. Thiation with 2,4-Bis(4-methoxyphenyl) -1,3,2,4-Dithiadiphosphetane 2,4-disulfide: N-Methylthiopyrrolidone. Org. Synth. 1984, 62, 158. [Google Scholar] [CrossRef]
- Mbaveng, A.; Ignat, A.G.; Ngameni, B.; Zaharia, V.; Ngadjui, B.T.; Kuete, V. In vitro antibacterial activities of p-toluenesulfonyl-hydrazinothiazoles and hydrazinoselenazoles against multi-drug resistant Gram-negative phenotypes. BMC Pharmacol. Toxicol. 2016, 17, 3. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Nishina, A.; Kimura, H.; Motoki, K.; Koketsu, M.; Ishihara, H. Tertiary selenoamide compounds are useful superoxide radical scavengers in vitro. Eur. J. Pharm. Sci. 2004, 23, 207–211. [Google Scholar] [CrossRef]
- Vishwanatha, T.M.; Narendra, N.; Chattopadhyay, B.; Mukherjee, M.; Sureshbabu, V.V. Synthesis of Selenoxo Peptides and Oligoselenoxo Peptides Employing LiAlHSeH. J. Org. Chem. 2012, 77, 2689–2702. [Google Scholar] [CrossRef]
- Bibelayi, D.; Lundemba, A.S.; Allen, F.H.; Galek, P.T.A.; Pradon, J.; Reilly, A.; Groom, C.R.; Yav, Z.G. Hydrogen bonding at C = Se acceptors in selenoureas, selenoamides and selones. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 317–325. [Google Scholar] [CrossRef]
- Lenardão, E.J.; Santi, C.; Sancineto, L. New Frontiers in Organoselenium Compounds; Springer International Publishing: Cham, Switzerland, 2018; ISBN 978-3-319-92404-5. [Google Scholar]
- Bhattacharyya, P.; Woollins, J. Selenocarbonyl synthesis using Woollins reagent. Tetrahedron Lett. 2001, 42, 5949–5951. [Google Scholar] [CrossRef] [Green Version]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A.M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef]
- Nourry, A.; Zambon, A.; Davies, L.; Niculescu-Duvaz, I.; Dijkstra, H.P.; Ménard, D.; Gaulon, C.; Niculescu-Duvaz, D.; Suijkerbuijk, B.M.J.M.; Friedlos, F.; et al. BRAF Inhibitors Based on an Imidazo[4,5]pyridin-2-one Scaffold and a Meta Substituted Middle Ring. J. Med. Chem. 2010, 53, 1964–1978. [Google Scholar] [CrossRef]
- Puszko, A.K.; Sosnowski, P.; Pułka-Ziach, K.; Hermine, O.; Hopfgartner, G.; Lepelletier, Y.; Misicka, A. Urea moiety as amide bond mimetic in peptide-like inhibitors of VEGF-A165/NRP-1 complex. Bioorg. Med. Chem. Lett. 2019, 29, 2493–2497. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Angell, Y.L.; Burgess, K. Peptidomimetics via copper-catalyzed azide-alkyne cycloadditions. Chem. Soc. Rev. 2007, 36, 1674–1689. [Google Scholar] [CrossRef]
- Brik, A.; Alexandratos, J.; Lin, Y.-C.; Elder, J.H.; Olson, A.J.; Wlodawer, A.; Goodsell, D.S.; Wong, C.-H. 1,2,3-Triazole as a Peptide Surrogate in the Rapid Synthesis of HIV-1 Protease Inhibitors. ChemBioChem 2005, 6, 1167–1169. [Google Scholar] [CrossRef]
- Bock, V.D.; Speijer, D.; Hiemstra, H.; van Maarseveen, J.H. 1,2,3-Triazoles as peptide bond isosteres: Synthesis and biological evaluation of cyclotetrapeptide mimics. Org. Biomol. Chem. 2007, 5, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Doiron, J.E.; Le, C.A.; Ody, B.K.; Brace, J.B.; Post, S.J.; Thacker, N.L.; Hill, H.M.; Breton, G.W.; Mulder, M.J.; Chang, S.; et al. Evaluation of 1,2,3-Triazoles as Amide Bioisosteres In Cystic Fibrosis Transmembrane Conductance Regulator Modulators VX-770 and VX-809. Chem. Eur. J. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Corredor, M.; Solà, J.; Alfonso, I. Disubstituted 1,2,3-triazoles as amide bond mimetics. Targets Heterocycl. Syst. 2017, 21, 1–22. [Google Scholar]
- Huisgen, R.; Szeimies, G.; Möbius, G.L. 1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen. Eur. J. Inorg. Chem. 1967, 100, 2494–2507. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Goddard-Borger, E.D.; Stick, R.V. An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar] [CrossRef]
- Whyte, A.; Olson, M.E.; Lautens, M. Palladium-Catalyzed, Norbornene-Mediated, ortho-Amination ipso-Amidation: Sequential C–N Bond Formation. Org. Lett. 2017, 20, 345–348. [Google Scholar] [CrossRef]
- Simonetta, S.H.; Golombek, D.A. An automated tracking system for Caenorhabditis elegans locomotor behavior and circadian studies application. J. Neurosci. Methods 2007, 161, 273–280. [Google Scholar] [CrossRef]
- Vairoletti, F.; Mahler, G.; Saiz, C.; Baron, A.; Salinas, G. Increased sensitivity of an infrared motility assay for nematicide discovery. microPubl. Biol. 2021, PMC8633989. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H.G.; Trautwein, A.; Willams, L.; Hink, M.; Lümmen, P.; Görgens, U.; Greul, J.N. Use of Aryl and Hetaryl Carboxamides as Endoparasiticides. U.S. Patent No. 9,422,276, 23 August 2016. [Google Scholar]
- Harris, T.W.; Antoshechkin, I.; Bieri, T.; Blasiar, D.; Chan, J.; Chen, W.J.; De La Cruz, N.; Davis, P.; Duesbury, M.; Fang, R.; et al. WormBase: A comprehensive resource for nematode research. Nucleic Acids Res. 2010, 38, D463–D467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE) 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2019.
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Otero, L.; Martínez-Rosales, C.; Barrera, E.; Pantano, S.; Salinas, G. Complex I and II Subunit Gene Duplications Provide Increased Fitness to Worms. Front. Genet. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Chassaing, S.; Sido, A.S.S.; Alix, A.; Kumarraja, M.; Pale, P.; Sommer, J. “Click Chemistry” in Zeolites: Copper(I) Zeolites as New Heterogeneous and Ligand-Free Catalysts for the Huisgen [3 + 2] Cycloaddition. Chem. Eur. J. 2008, 14, 6713–6721. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, C.; Aguirre, A. Palladium-catalyzed carbon–carbon coupling reactions using aryl Grignards. Tetrahedron Lett. 2002, 43, 7091–7094. [Google Scholar] [CrossRef]
- Song, W.; Zheng, N.; Li, M.; He, J.; Li, J.; Dong, K.; Ullah, K.; Zheng, Y. Rhodium(I)-Catalyzed Regioselective Azide-internal Alkynyl Trifluoromethyl Sulfide Cycloaddition and Azide-internal Thioalkyne Cycloaddition under Mild Conditions. Adv. Synth. Catal. 2019, 361, 469–475. [Google Scholar] [CrossRef]
- Meng, G.; Guo, T.; Ma, T.; Zhang, J.; Shen, Y.; Sharpless, K.B.; Dong, J. Modular click chemistry libraries for functional screens using a diazotizing reagent. Nature 2019, 574, 86–89. [Google Scholar] [CrossRef]
- Hu, M.; Li, J.; Yao, S.Q. In situ “click” Assembly of Small Molecule Matrix Metalloprotease Inhibitors Containing Zinc-Chelating Groups. Org. Lett. 2008, 10, 5529–5531. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Compound | X | Y | R | Yield (%) 1 |
|---|---|---|---|---|---|
| 1 | 5a | CF3 | O | CH3 | 40 |
| 2 | 5b | I | O | CH2CH3 | 76 |
| 3 | 5c | I | O | CH2CH(CH3)2 | 63 |
| 4 | 6 | I | S | CH3 | 47 |

| Entry | Compound | R | Yield (%) 1 |
|---|---|---|---|
| 1 | 7 |  | 23 |
| 2 | 8a |  | 99 |
| 3 | 8b |  | 99 |
| 4 | 8c |  | 84 |

| Entry | X | R | Azide | Compound, (Yield %) 1 |
|---|---|---|---|---|
| 1 | H |  | 11a | 9a (93) |
| 3 | H |  | 11b | 9b (56) |
| 4 | CF3 |  | 11b | 9c (73) |

| Entry | Comp | R1 | R2 | % C. elegans Motility Reduction (10 μM) 1 |
|---|---|---|---|---|
| 1 | 1a | CF3 |  | 100 |
| 2 | 1b | I |  | 100 |
| 3 | 2 | I |  | 6 |
| 4 | 3 | I |  | 92 |
| 5 | 4 | I |  | 100 |
| 6 | 5a | CF3 |  | 17 |
| 7 | 5b | I |  | 31 |
| 8 | 5c | I |  | 27 |
| 9 | 6 | I |  | 59 |
| 10 | 7 | I |  | 47 |
| 11 | 8c | CF3 |  | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vairoletti, F.; Paulino, M.; Mahler, G.; Salinas, G.; Saiz, C. Structure-Based Bioisosterism Design, Synthesis, Biological Evaluation and In Silico Studies of Benzamide Analogs as Potential Anthelmintics. Molecules 2022, 27, 2659. https://doi.org/10.3390/molecules27092659
Vairoletti F, Paulino M, Mahler G, Salinas G, Saiz C. Structure-Based Bioisosterism Design, Synthesis, Biological Evaluation and In Silico Studies of Benzamide Analogs as Potential Anthelmintics. Molecules. 2022; 27(9):2659. https://doi.org/10.3390/molecules27092659
Chicago/Turabian StyleVairoletti, Franco, Margot Paulino, Graciela Mahler, Gustavo Salinas, and Cecilia Saiz. 2022. "Structure-Based Bioisosterism Design, Synthesis, Biological Evaluation and In Silico Studies of Benzamide Analogs as Potential Anthelmintics" Molecules 27, no. 9: 2659. https://doi.org/10.3390/molecules27092659