
N-Arylation of Protected and Unprotected 5-Bromo-2-aminobenzimidazole as Organic Material: Non-Linear Optical (NLO) Properties and Structural Feature Determination through Computational Approach




Abstract
:1. Introduction
2. Results and Discussion
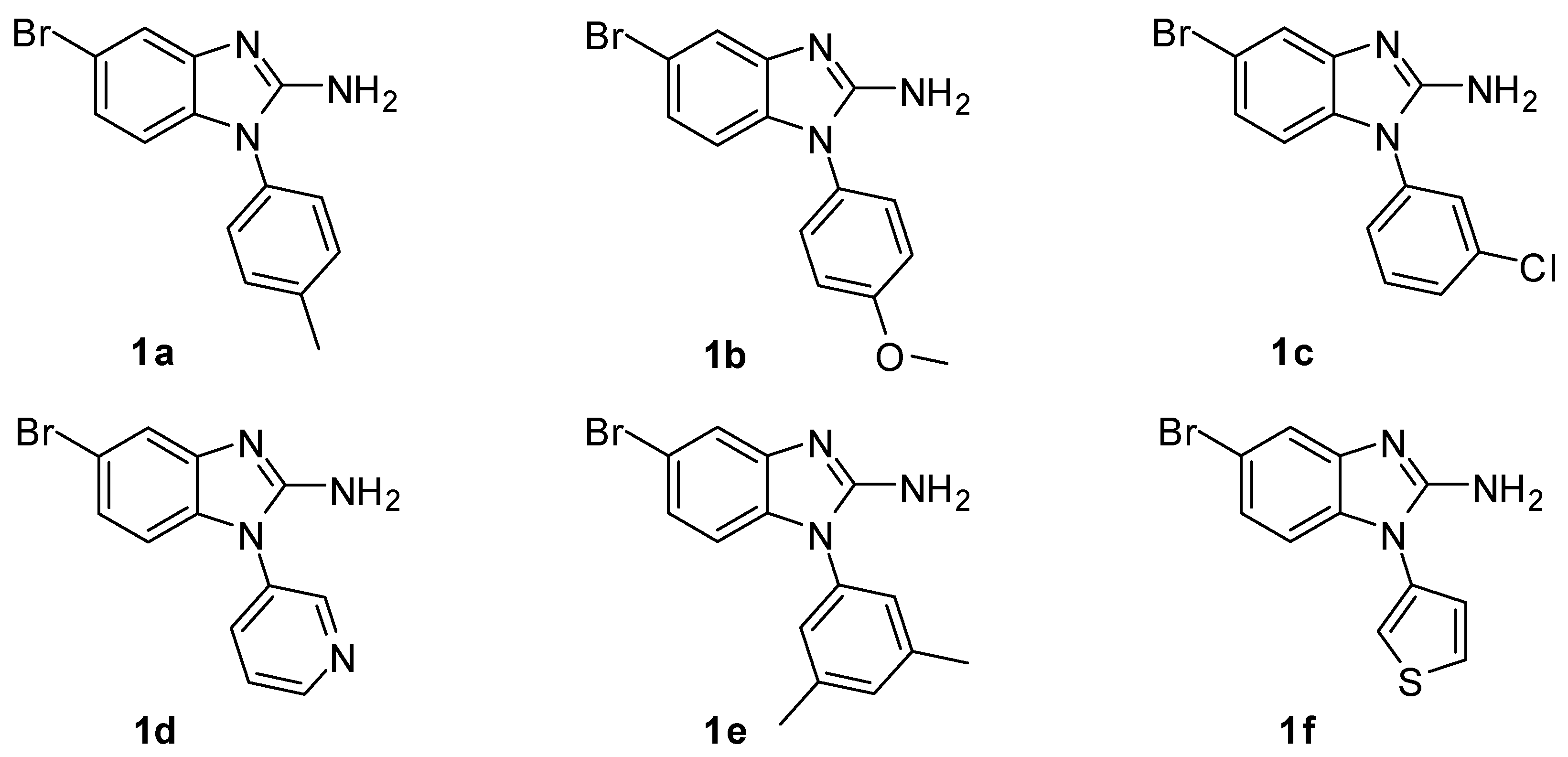
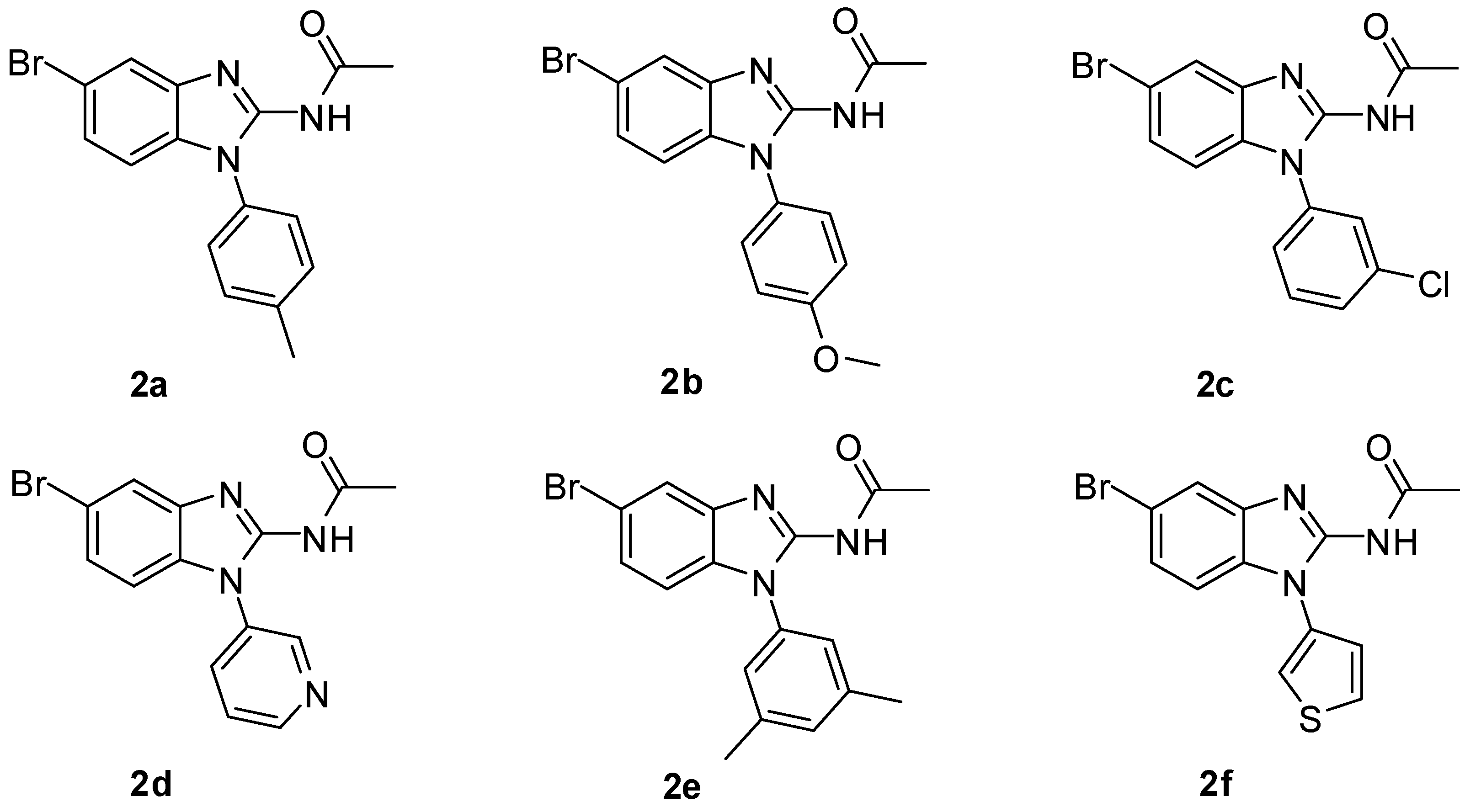
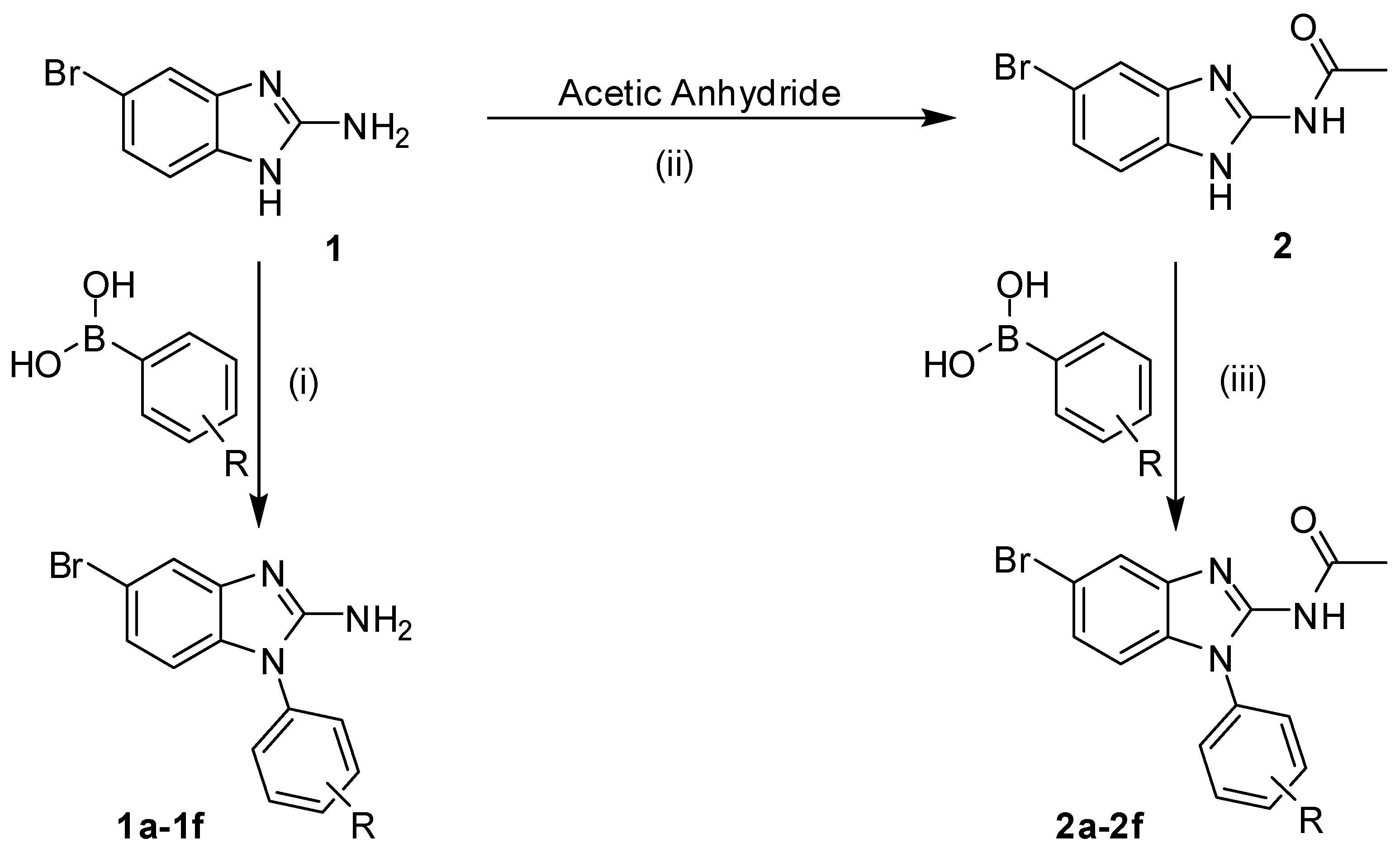
2.1. Chemistry
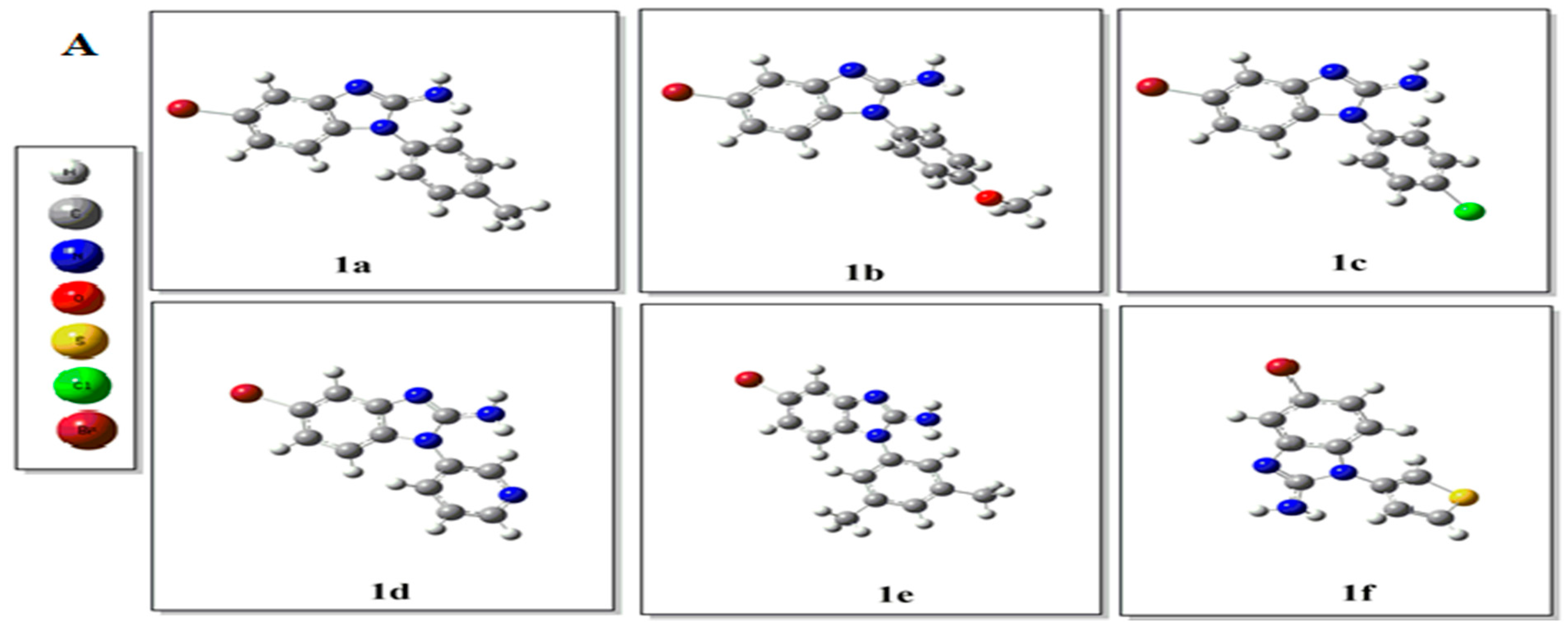
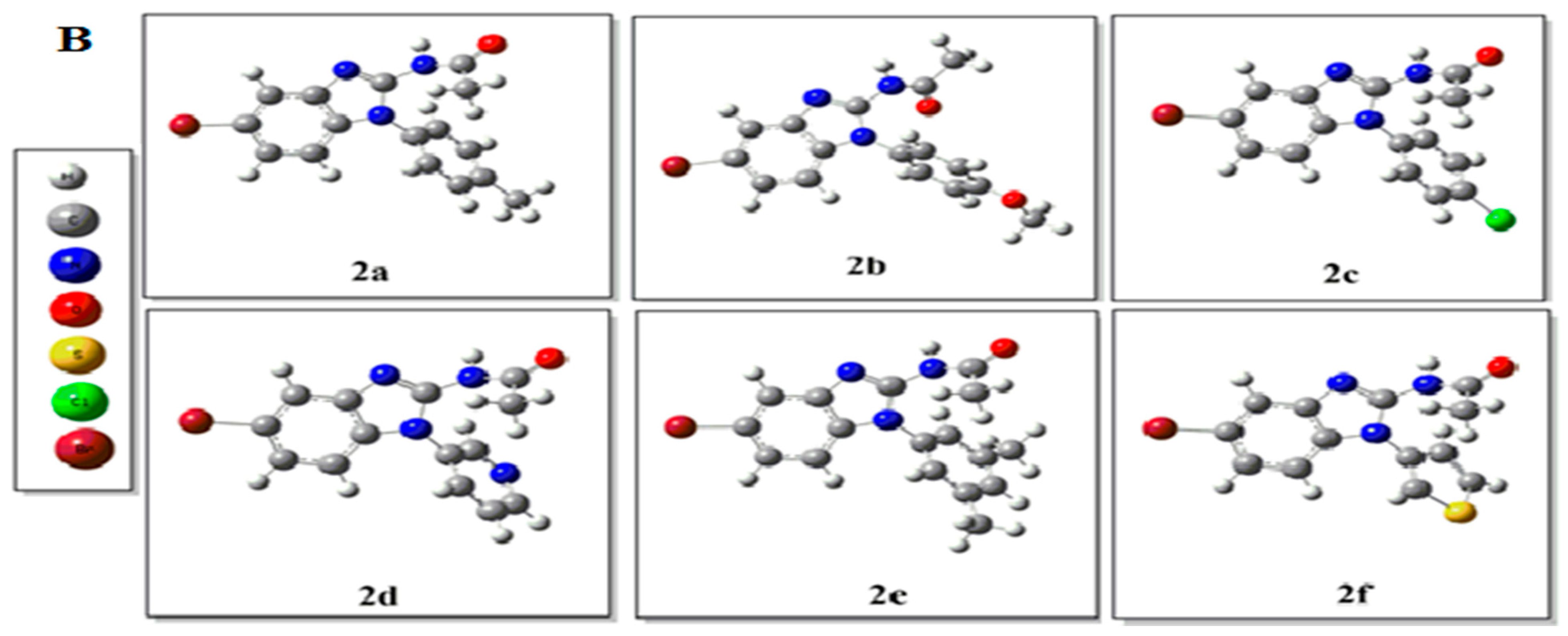
2.2. Density Functional Theory (DFT) Studies
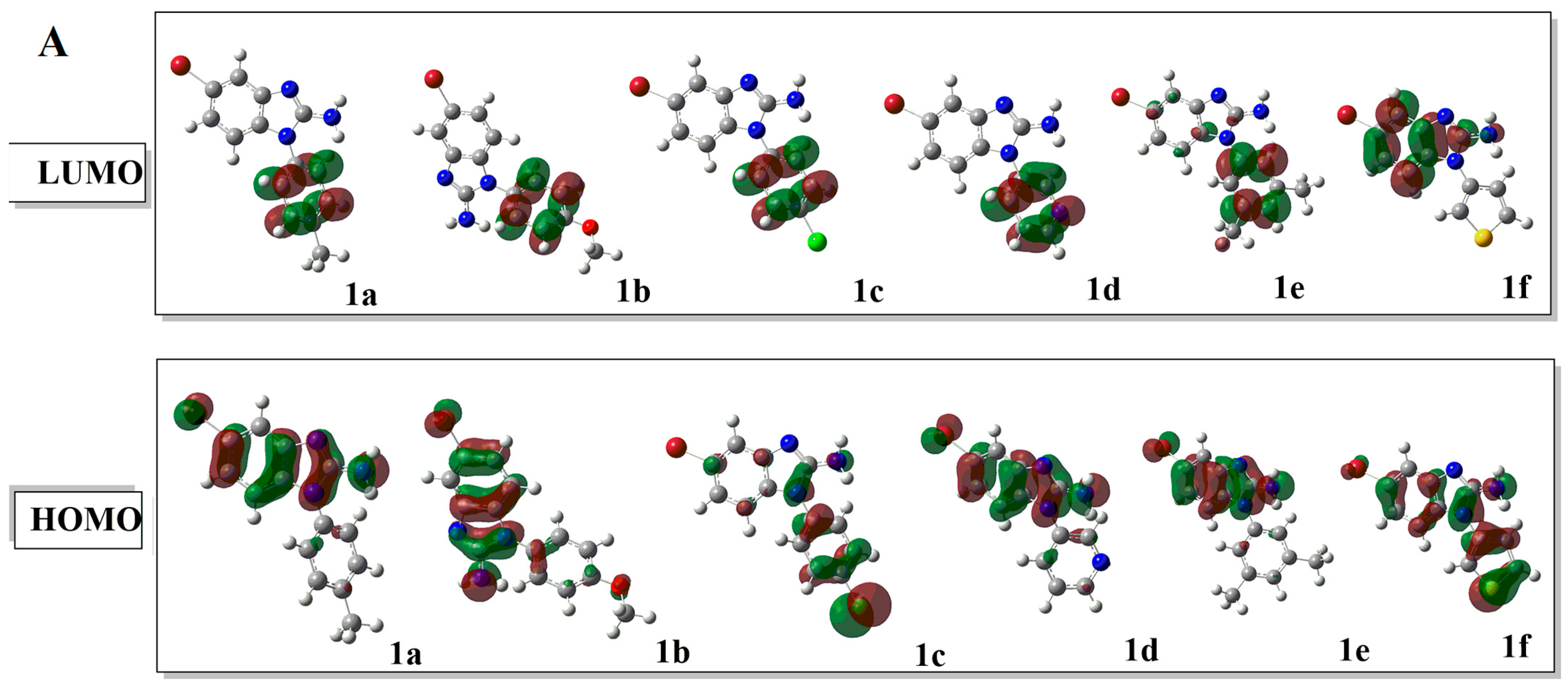
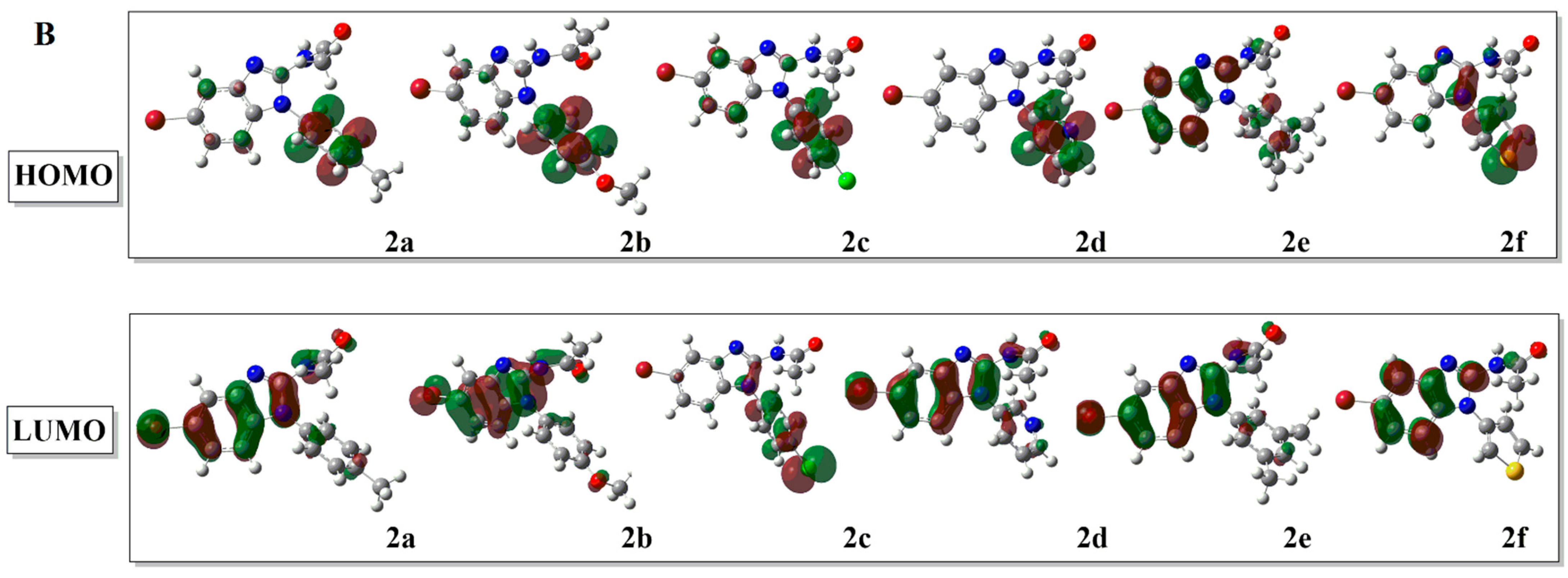
2.2.1. Frontier Molecular Orbital (FMO) Analysis
2.2.2. Reactivity Descriptor Parameters
2.3. Non-Linear Optical (NLO) Properties
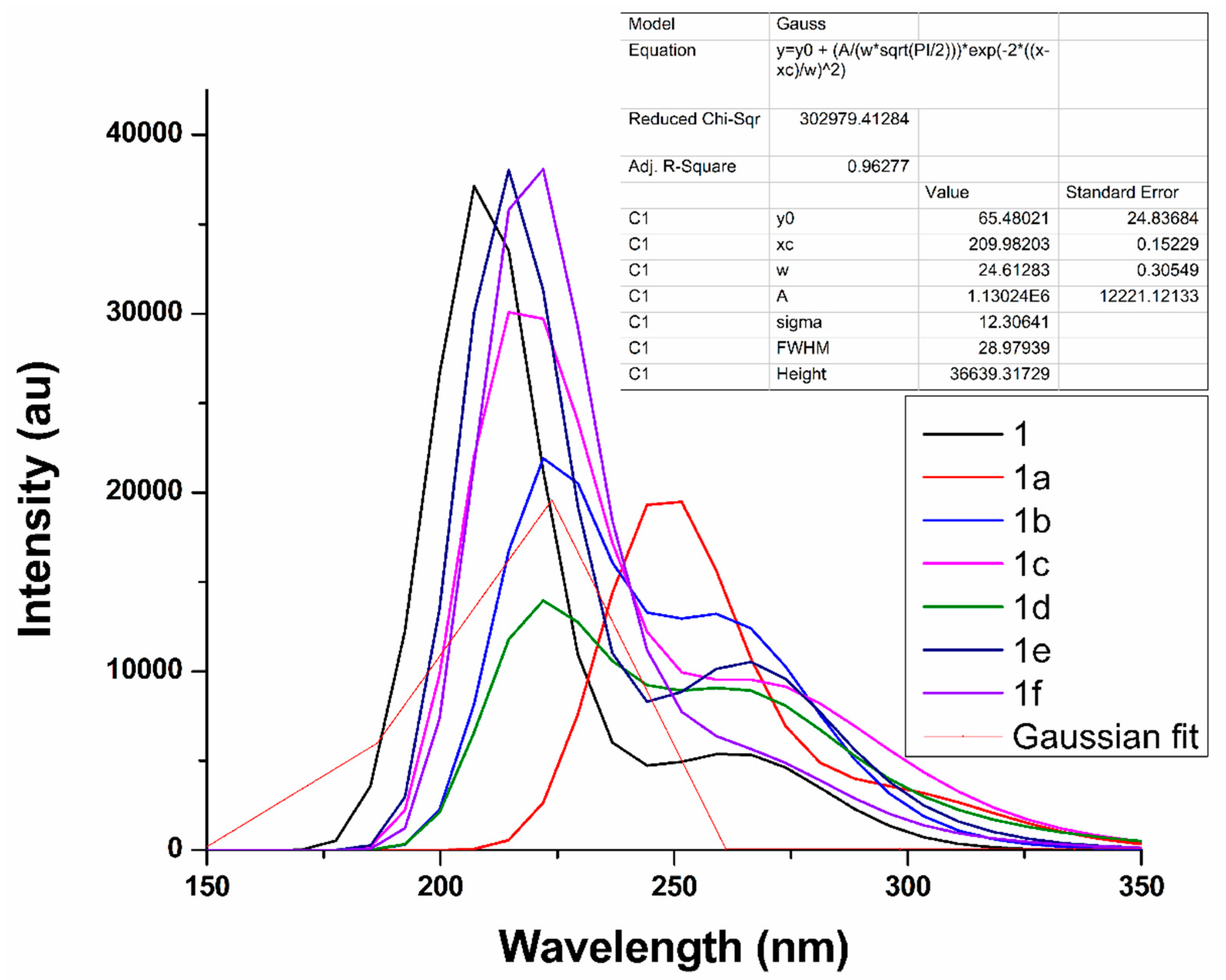
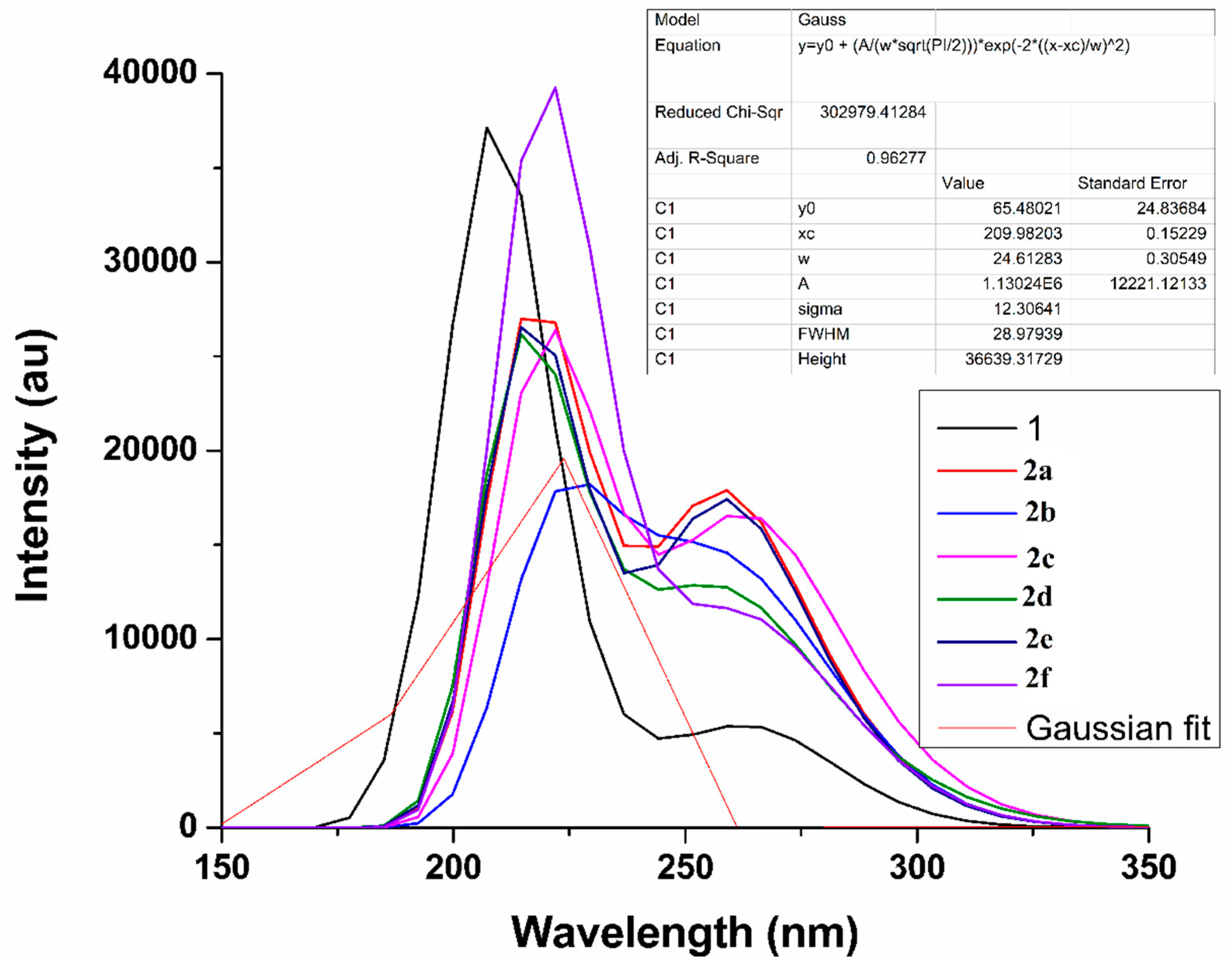
2.4. UV–VIS Absorption Analysis
3. Experimental
3.1. General Protocol for the Synthesis of Compounds
3.1.1. General Procedures for the Synthesis of 5-Bromo-1H-benzo[d]imidazol-2-amine Derivatives (1a–1f)
3.1.2. Synthesis of N-(5-Bromo-1H-benzo[d]imidazol-2-yl) Acetamide (2)
3.1.3. General Procedures for the Synthesis of N-(5-Bromo-1H-benzo[d]imidazol-2-yl) Acetamide Derivatives (2a–2f)
3.2. Characterization Data
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Karaffa, L.S. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; RSC Publishing: London, UK, 2013. [Google Scholar]
- Nawrocka, W.; Zimecki, M.; Kuznicki, T.; Kowalska, M.W. Immunotropic Properties of 2-Aminobenzimidazole Derivatives in Cultures of Human Peripheral Blood Cells, Part 5. Arch. Pharm. Int. J. Pharm. Med. Chem. 1999, 332, 85–90. [Google Scholar] [CrossRef]
- Mor, M.; Bordi, F.; Silva, C.; Rivara, S.; Zuliani, V.; Vacondio, F.; Rivara, M.; Barocelli, E.; Bertoni, S.; Ballabeni, V. Synthesis, biological activity, QSAR and QSPR study of 2-aminobenzimidazole derivatives as potent H3-antagonists. Bioorganic Med. Chem. 2004, 12, 663–674. [Google Scholar] [CrossRef]
- de Dios, A.; Shih, C.; de Uralde, B.L.; Sánchez, C.; del Prado, M.; Martín Cabrejas, L.M.; Pleite, S.; Blanco-Urgoiti, J.; Lorite, M.J.; Nevill, C.R. Design of potent and selective 2-aminobenzimidazole-based p38α MAP kinase inhibitors with excellent in vivo efficacy. J. Med. Chem. 2005, 48, 2270–2273. [Google Scholar] [CrossRef] [PubMed]
- Nawrocka, W.; Sztuba, B.; Kowalska, M.W.; Liszkiewicz, H.; Wietrzyk, J.; Nasulewicz, A.; Pełczyńska, M.; Opolski, A. Synthesis and antiproliferative activity in vitro of 2-aminobenzimidazole derivatives. II Farm. 2004, 59, 83–91. [Google Scholar]
- Iriepa, I.; Gil-Alberdi, B.; Gálvez, E.; Villasante, F.J.; Bellanato, J.; Carmona, P. Synthesis, structural, conformational and pharmacological study of some carbamates derived from 3-methyl-2, 4-diphenyl-3-azabicyclo [3.3. 1] nonan-9β-ol. J. Mol. Struct. 1999, 482, 437–442. [Google Scholar] [CrossRef]
- Vigorita, M.; Previtera, T.; Zappalà, C.; Trovato, A.; Monforte, M.; Barbera, R.; Pizzimenti, F. N-trifluoroacetyl derivatives as pharmacological agents. V. Evaluation of antiinflammatory and antimicrobial activities of some N-heterocyclic trifluoroacetamides. Farmaco 1990, 45, 223–235. [Google Scholar] [PubMed]
- Hu, Y.; Long, S.; Fu, H.; She, Y.; Xu, Z.; Yoon, J. Revisiting imidazolium receptors for the recognition of anions: Highlighted research during 2010–2019. Chem. Soc. Rev. 2021, 50, 589–618. [Google Scholar] [CrossRef] [PubMed]
- Wahe, H.; Asobo, P.F.; Cherkasov, R.A.; Nkengfack, A.E.; Folefoc, G.N.; Fomum, Z.T.; Doepp, D. Heterocycles of biological importance. Part 6. The formation of novel biologically active pyrimido [1,2-a] benzimid-azoles from electron deficient alkynes and 2-aminobenzimidazoles. Arkivoc 2003, 2003, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Nawrocka, W. Syntheses and pharmacological properties of new 2-aminobenzimidazole derivatives. Boll. Chim. Farm. 1996, 135, 18–23. [Google Scholar] [PubMed]
- Rossi, R.; Lessi, M.; Manzini, C.; Marianetti, G.; Bellina, F. Achievement of regioselectivity in transition metal-catalyzed direct C–H (hetero) arylation reactions of heteroarenes with one heteroatom through the use of removable protecting/blocking substituents or traceless directing groups. Tetrahedron 2016, 72, 1795–1837. [Google Scholar] [CrossRef] [Green Version]
- Maria Thomas, A.; Sujatha, A.; Anilkumar, G. Goldberg reaction: Development, mechanistic insights and applications. Mini Rev. Org. Chem. 2015, 12, 3–23. [Google Scholar] [CrossRef]
- Reddy, K.R.; Kumar, N.S.; Sreedhar, B.; Kantam, M.L. N-Arylation of nitrogen heterocycles with aryl halides and arylboronic acids catalyzed by cellulose supported copper (0). J. Mol. Catal. A Chem. 2006, 252, 136–141. [Google Scholar] [CrossRef]
- Guram, A.S.; Buchwald, S.L. Palladium-catalyzed aromatic aminations with in situ generated aminostannanes. J. Am. Chem. Soc. 1994, 116, 7901–7902. [Google Scholar] [CrossRef]
- Chan, D.M.; Monaco, K.L.; Wang, R.-P.; Winters, M.P. New N-and O-arylations with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar] [CrossRef]
- Rao, D.N.; Rasheed, S.; Kumar, K.A.; Reddy, A.S.; Das, P. Copper-Catalyzed C-NH2 Arylation of 2-Aminobenzimidazoles and Related C-Amino-NH-azoles. Adv. Synth. Catal. 2016, 358, 2126–2133. [Google Scholar] [CrossRef]
- Qiao, J.X.; Lam, P.Y. Recent Advances in Chan–Lam Coupling Reaction: Copper-Promoted C–Heteroatom Bond Cross-Coupling Reactions with Boronic Acids and Derivatives. In Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, 1&2, 2nd ed.; John Wiley & Sons GmbH: Weinheim, Germany, 2011; Volume 1, pp. 315–361. [Google Scholar]
- Cramer, C.J.; Bickelhaupt, F. Essentials of computational chemistry. Angew. Chem. Int. Ed. Engl. 2003, 42, 381. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Ledoux, I. New advances in molecular engineering for quadratic nonlinear optics. Synth. Met. 1993, 54, 123–137. [Google Scholar] [CrossRef]
- Duo-rong, Y.; Dong, X.; Nan, Z.; Ming-guo, L.; Min-hua, J. Organic nonlinear optical crystal MHBA for compact blue-violet laser. Chin. Phys. Lett. 1996, 13, 841. [Google Scholar]
- Muhammad, S.; Xu, H.; Janjua, M.R.S.A.; Su, Z.; Nadeem, M. Quantum chemical study of benzimidazole derivatives to tune the second-order nonlinear optical molecular switching by proton abstraction. PCCP 2010, 12, 4791–4799. [Google Scholar] [CrossRef]
- Tayade, R.P.; Sekar, N. Benzimidazole-thiazole based NLOphoric styryl dyes with solid state emission—Synthesis, photophysical, hyperpolarizability and TD-DFT studies. Dye. Pigment. 2016, 128, 111–123. [Google Scholar] [CrossRef]
- Thakare, S.S.; Sreenath, M.C.; Chitrambalam, S.; Joe, I.H.; Sekar, N. Non-linear optical study of BODIPY-benzimidazole conjugate by solvatochromic, Z-scan and theoretical methods. Opt. Mater. 2017, 64, 453–460. [Google Scholar] [CrossRef]
- Tamer, Ö.; Gözüaçık, F.; Avcı, D.; Atalay, Y. Theoretical analysis on structural, spectroscopic, and electronic properties of some 2-aminobenzimidazole complexes by using PBE1PBE, B3LYP, and HF methods. Opt. Spectrosc. 2014, 116, 12–32. [Google Scholar] [CrossRef]
- Muthuraja, A.; Kalainathan, S. A study on growth, optical, mechanical, and NLO properties of 2-Mercaptobenzimidazole, 2-Phenylbenzimidazole and 2-Hydroxy benzimidazole single crystals: A comparative investigation. Mater. Technol. 2017, 32, 335–348. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Nichols, J.A.; Dixon, D.A. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: Molecular properties from density functional theory orbital energies. J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Liu, Z.; Yao, Q. Synthesis and characterization of 5-amino-1,3,6-trinitro-1H-benzo[d]imidazol-2(3H)-one as an energetic material. RSC Adv. 2014, 4, 42215–42219. [Google Scholar] [CrossRef]
- Yue, Y.; Zheng, Z.G.; Wu, B.; Xia, C.Q.; Yu, X.Q. Copper-Catalyzed Cross-Coupling Reactions of Nucleobases with Arylboronic Acids: An Efficient Access to N-Arylnucleobases. Eur. J. Org. Chem. 2005, 2005, 5154–5157. [Google Scholar] [CrossRef]
- Ikram, H.M.; Rasool, N.; Hashmi, M.A.; Anjum, M.A.; Ali, K.G.; Zubair, M.; Ahmad, G.; Mahmood, T. Density functional theory-supported studies of structural and electronic properties of substituted-phenol derivatives synthesized by efficient O-or C-arylation via Chan Lam or Suzuki cross-coupling reactions. Turk. J. Chem. 2019, 43, 1306–1321. [Google Scholar] [CrossRef]
- Inque, S.; Imanaka, Y. Reactions of organozinc coordination compounds II. Reactivity with secondary amines. J. Organomet. Chem. 1972, 35, 1–7. [Google Scholar] [CrossRef]
- Collum, D.B. Is N, N, N′, N′-tetramethylethylenediamine a good ligand for lithium? Acc. Chem. Res. 1992, 25, 448–454. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ochterski, J.W.; Petersson, G.A.; Montgomery, J.A., Jr. A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys. 1996, 104, 2598–2619. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Bannwarth, C.; Grimme, S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theor. Chem. 2014, 1040, 45–53. [Google Scholar] [CrossRef]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Russo, T.V.; Martin, R.L.; Hay, P.J. Density functional calculations on first-row transition metals. J. Chem. Phys. 1994, 101, 7729–7737. [Google Scholar] [CrossRef] [Green Version]
- Yoosefian, M.; Etminan, N. The role of solvent polarity in the electronic properties, stability and reactivity trend of a tryptophane/Pd doped SWCNT novel nanobiosensor from polar protic to non-polar solvents. RSC Adv. 2016, 6, 64818–64825. [Google Scholar] [CrossRef]
- Tayade, R.P.; Sekar, N. Novel thiazole based styryl dyes with benzimidazole unit-synthesis, photophysical and TD-DFT studies. J. Fluoresc. 2017, 27, 167–180. [Google Scholar] [CrossRef]
- Muhammad, S.; Kumar, S.; Koh, J.; Saravanabhavan, M.; Ayub, K.; Chaudhary, M. Synthesis, characterisation, optical and nonlinear optical properties of thiazole and benzothiazole derivatives: A dual approach. Mol. Simul. 2018, 44, 1191–1199. [Google Scholar] [CrossRef]
- Ayub, K. Are phosphide nano-cages better than nitride nano-cages? A kinetic, thermodynamic and non-linear optical properties study of alkali metal encapsulated X 12 Y 12 nano-cages. J. Mater. Chem. C 2016, 4, 10919–10934. [Google Scholar] [CrossRef]
- Hirata, S.; Nooijen, M.; Bartlett, R.J. High-order determinantal equation-of-motion coupled-cluster calculations for ionized and electron-attached states. Chem. Phys. Lett. 2000, 328, 459–468. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Kosar, N.; Mahmood, T.; Ayub, K.; Tabassum, S.; Arshad, M.; Gilani, M.A. Doping superalkali on Zn12O12 nanocage constitutes a superior approach to fabricate stable and high-performance nonlinear optical materials. Opt. Laser Technol. 2019, 120, 105753. [Google Scholar]
- Ullah, F.; Kosar, N.; Ayub, K.; Mahmood, T. Superalkalis as a source of diffuse excess electrons in newly designed inorganic electrides with remarkable nonlinear response and deep ultraviolet transparency: A DFT study. Appl. Surf. Sci. 2019, 483, 1118–1128. [Google Scholar] [CrossRef]
- Mahmood, N.; Rasool, N.; Ikram, H.M.; Hashmi, M.A.; Mahmood, T.; Zubair, M.; Ahmad, G.; Rizwan, K.; Rashid, T.; Rashid, U. Synthesis of 3, 4-Biaryl-2, 5-Dichlorothiophene through Suzuki Cross-Coupling and Theoretical Exploration of Their Potential Applications as Nonlinear Optical Materials. Symmetry 2018, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Cao, R.; Wang, X.; Li, H.; Yuan, W.; Wang, G.; Wu, H.; Li, J. A multifunctional 3D ferroelectric and NLO-active porous metal− organic framework. J. Am. Chem. Soc. 2009, 131, 6894–6895. [Google Scholar] [CrossRef]
- Tahir, H.; Kosar, N.; Ayub, K.; Mahmood, T. Outstanding NLO response of thermodynamically stable single and multiple alkaline earth metals doped C20 fullerene. J. Mol. Liq. 2020, 305, 112875. [Google Scholar] [CrossRef]
- Lu, S.I. Computational study of static first hyperpolarizability of donor–acceptor substituted (E)-benzaldehyde phenylhydrazone. J. Comput. Chem. 2011, 32, 730–736. [Google Scholar] [CrossRef]
- Suman, G.; Bubbly, S.; Gudennavar, S.; Muthu, S.; Roopashree, B.; Gayatri, V.; Gowda, N.N. Structural investigation, spectroscopic and energy level studies of Schiff base: 2-[(3′-N-salicylidenephenyl) benzimidazole] using experimental and DFT methods. J. Mol. Struct. 2017, 1139, 247–254. [Google Scholar] [CrossRef]
- Wasim, F.; Kosar, N.; Mahmood, T.; Ayub, K. Sensor applications of polypyrrole for oxynitrogen analytes: A DFT study. J. Mol. Model. 2018, 24, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Arif, A.M.; Yousaf, A.; Xu, H.-L.; Su, Z.-M. Spectroscopic behavior, FMO, NLO and substitution effect of 2-(1H-Benzo[d]imidazole-2-ylthio)-No-substituted-acetamides: Experimental and theoretical approach. Dye. Pigment. 2019, 171, 107742. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Kosar, N.; Mahmood, T.; Ayub, K. Role of dispersion corrected hybrid GGA class in accurately calculating the bond dissociation energy of carbon halogen bond: A benchmark study. J. Mol. Struct. 2017, 1150, 447–458. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Aryl Boronic Acid | Base | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 4-MeC6H4B(OH)2 | Et3N | 1a | trace |
| 2 | 4-MeC6H4B(OH)2 | TMEDA | 1a | 70 |
| 3 | 4-OMeC6H4B(OH)2 | TMEDA | 1b | 80 |
| 4 | 3-ClC6H4B(OH)2 | TMEDA | 1c | 78 |
| 5 | 3-NC5H4B(OH)2 | TMEDA | 1d | 73 |
| 6 | 3,5-Me2C6H3B(OH)2 | TMEDA | 1e | 71 |
| 7 | 3-SC4H3B(OH)2 | TMEDA | 1f | 69 |
| Entry | Aryl Boronic Acid | Base | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 4-MeC6H4B(OH)2 | Et3N | 2a | 40 |
| 2 | 4-MeC6H4B(OH)2 | Pyridine | 2a | 37 |
| 3 | 4-OMeC6H4B(OH)2 | Et3N | 2b | 42 |
| 4 | 3-ClC6H4B(OH)2 | Et3N | 2c | 41 |
| 5 | 3-NC5H4B(OH)2 | Et3N | 2d | 45 |
| 6 | 3,5-Me2C6H3B(OH)2 | Et3N | 2e | 49 |
| 7 | 3-SC4H3B(OH)2 | Et3N | 2f | 39 |
| Compounds | EHOMO (eV) | ELUMO (eV) | GH-L (eV) | μo (Debye) | αo (Au) | βo (Esu) |
|---|---|---|---|---|---|---|
| Density Functionals | B3LYP | CAM-B3LYP | ||||
| 1a | −5.81 | −1.12 | 4.69 | 8.46 | 228 | 4.61 × 10−30 |
| 1b | −5.78 | −1.11 | 4.66 | 7.95 | 215 | 5.66 × 10−30 |
| 1c | −5.99 | −1.50 | 4.49 | 4.91 | 210 | 3.81 × 10−30 |
| 1d | −6.02 | −1.77 | 4.25 | 5.63 | 190 | 4.40 × 10−30 |
| 1e | −5.79 | −1.10 | 4.69 | 7.56 | 222 | 4.00 × 10−30 |
| 1f | −5.91 | −1.39 | 4.52 | 6.32 | 188 | 4.30 × 10−30 |
| 2a | −6.44 | −1.40 | 5.04 | 6.05 | 235 | 2.02 × 10−30 |
| 2b | −6.19 | −1.11 | 5.08 | 5.93 | 240 | 3.54 × 10−30 |
| 2c | −6.59 | −1.68 | 4.91 | 3.74 | 236 | 2.00 × 10−30 |
| 2d | −6.66 | −1.87 | 4.79 | 5.35 | 215 | 2.40 × 10−30 |
| 2e | −6.42 | −1.39 | 5.03 | 6.03 | 247 | 2.01 × 10−30 |
| 2f | −6.52 | −1.52 | 5.00 | 4.69 | 215 | 2.37 × 10−30 |
| Compounds | I (eV) | EA (eV) | η (eV) | μ (eV) | ω (eV) |
|---|---|---|---|---|---|
| 1a | 5.81 | 1.12 | 2.34 | −3.47 | 2.56 |
| 1b | 5.78 | 1.11 | 2.33 | −3.45 | 2.55 |
| 1c | 5.99 | 1.50 | 2.24 | −3.75 | 3.12 |
| 1d | 6.02 | 1.77 | 2.13 | −3.89 | 3.56 |
| 1e | 5.79 | 1.10 | 2.35 | −3.44 | 2.53 |
| 1f | 5.91 | 1.39 | 2.26 | −3.65 | 2.94 |
| 2a | 6.44 | 1.40 | 2.52 | −3.92 | 3.04 |
| 2b | 6.19 | 1.11 | 2.54 | −3.65 | 2.63 |
| 2c | 6.59 | 1.68 | 2.45 | −4.14 | 3.49 |
| 2d | 6.66 | 1.87 | 2.39 | −4.26 | 3.80 |
| 2e | 6.42 | 1.39 | 2.52 | −3.90 | 3.03 |
| 2f | 6.52 | 1.52 | 2.50 | −4.02 | 3.23 |
| Compounds | Hyperpolarizability (βo) |
|---|---|
| 1b | 5.66 × 10−30 |
| Spbzl | 1.27 × 10−29 |
| Spbzl | 8.50 × 10−30 |
| Pbzlb | 1.48 × 10−29 |
| Compounds | fo | ∆E | λmax |
|---|---|---|---|
| 1 | 0.71 | 5.94 | 209 |
| 1a | 0.16 | 5.00 | 248 |
| 1b | 0.32 | 5.49 | 226 |
| 1c | 0.48 | 5.85 | 212 |
| 1d | 0.18 | 5.62 | 221 |
| 1e | 0.65 | 5.82 | 213 |
| 1f | 0.45 | 5.71 | 217 |
| 2a | 0.40 | 5.69 | 218 |
| 2b | 0.14 | 5.56 | 223 |
| 2c | 0.27 | 5.70 | 218 |
| 2d | 0.48 | 5.78 | 214 |
| 2e | 0.37 | 5.74 | 216 |
| 2f | 0.31 | 5.67 | 219 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mumtaz, M.; Rasool, N.; Ahmad, G.; Kosar, N.; Rashid, U. N-Arylation of Protected and Unprotected 5-Bromo-2-aminobenzimidazole as Organic Material: Non-Linear Optical (NLO) Properties and Structural Feature Determination through Computational Approach. Molecules 2021, 26, 6920. https://doi.org/10.3390/molecules26226920
Mumtaz M, Rasool N, Ahmad G, Kosar N, Rashid U. N-Arylation of Protected and Unprotected 5-Bromo-2-aminobenzimidazole as Organic Material: Non-Linear Optical (NLO) Properties and Structural Feature Determination through Computational Approach. Molecules. 2021; 26(22):6920. https://doi.org/10.3390/molecules26226920
Chicago/Turabian StyleMumtaz, Mubeen, Nasir Rasool, Gulraiz Ahmad, Naveen Kosar, and Umer Rashid. 2021. "N-Arylation of Protected and Unprotected 5-Bromo-2-aminobenzimidazole as Organic Material: Non-Linear Optical (NLO) Properties and Structural Feature Determination through Computational Approach" Molecules 26, no. 22: 6920. https://doi.org/10.3390/molecules26226920