
Charge Transfer Complexes of 1,3,6-Trinitro-9,10-phenanthrenequinone with Polycyclic Aromatic Compounds

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Theoretical
2.2. Experimental
3. Materials and Methods
3.1. Synthesis
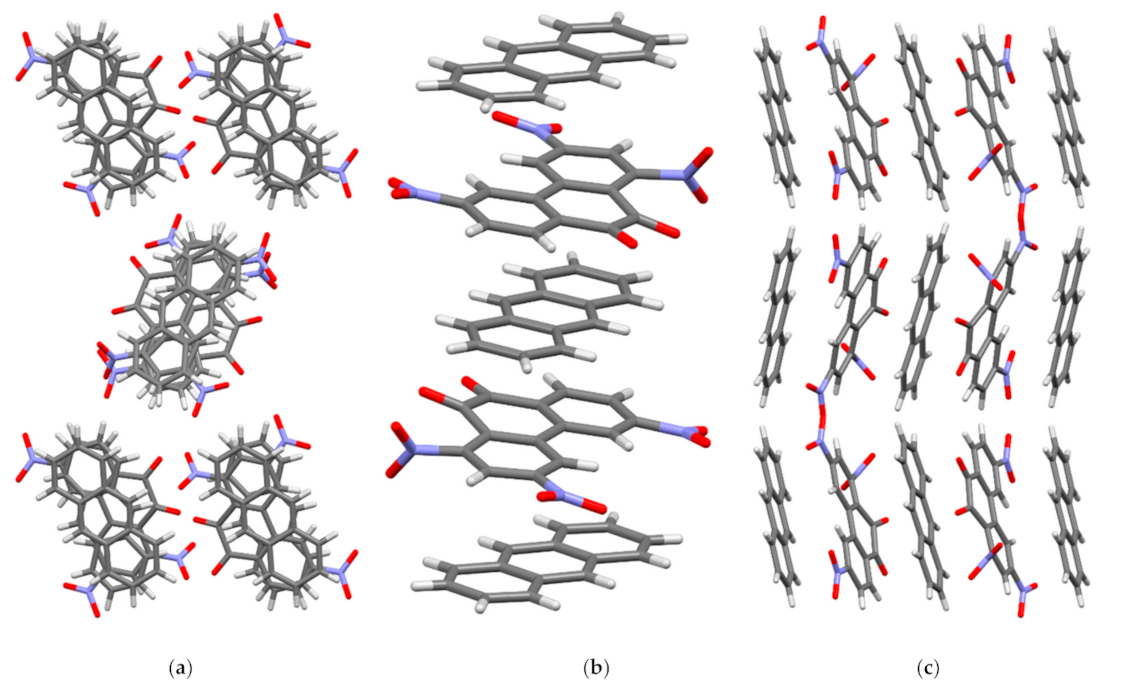
3.2. X-ray Crystallography and Structure Refinement
3.3. Quantum Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| ΔEMO | the energy difference between AELUMO and DEHOMO |
| DEHOMO | energy level of the HOMO of isolated donor molecule |
| AELUMO | energy level of the LUMO of isolated acceptor molecule |
| ΔCTCEMO | the energy difference between CTCELUMO and CTCEHOMO |
| CTCEHOMO | energy level of the HOMO of complex |
| CTCELUMO | energy level of the LUMO of complex |
| qNPA | partial NPA charges in the ground state |
| q*NPA | partial NPA charges in the first excited state |
| ΔEass | energy of association of donor and acceptor |
| A | acceptor |
| ACN | acenaphthene |
| ACR | acridine |
| AN | anthracene |
| AZU | azulene |
| BZ | benzene |
| COR | coronene |
| CRS | chrysene |
| CTCs | charge transfer complexes |
| D | donor |
| DBTTF | dibenzotetrathiafulvalene |
| DFT | density functional theory |
| DMPZ | 5,10-dimethylphenazine |
| HOMO | highest occupied molecular orbital |
| IQN | isoquinoline |
| LUMO | lowest unoccupied molecular orbital |
| MC | 9-methylcarbazole |
| NA | naphthalene |
| PA | phenanthrene |
| PD | pyridine |
| PEN | pentacene |
| POR | porphyrin |
| PQ | 1,3,6-trinitro-9,10-phenanthrenequinone |
| PYR | pyrene |
| QN | quinoline |
| TET | tetracene |
| TMDA | N,N,N’,N’-tetramethyl-p-phenylenediamine |
| TPH | tetraphene |
| TPL | triphenylene |
| TTF | tetrathiafulvalene |
References
- Pavelyev, V.G.; Parashchuk, O.D.; Krompiec, M.; Orekhova, T.V.; Perepichka, I.F.; Van Loosdrecht, P.H.M.; Paraschuk, D.Y.; Pshenichnikov, M.S. Charge transfer dynamics in donor-acceptor complexes between a conjugated polymer and fluorene acceptors. J. Phys. Chem. C 2014, 118, 30291–30301. [Google Scholar] [CrossRef] [Green Version]
- Al Kobaisi, M.; Bhosale, R.S.; El-Khouly, M.E.; La, D.D.; Padghan, S.D.; Bhosale, S.V.; Jones, L.A.; Antolasic, F.; Fukuzumi, S.; Bhosale, S.V. The sensitivity of donor – acceptor charge transfer to molecular geometry in DAN–NDI based supramolecular flower-like self-assemblies. Sci. Rep. 2017, 7, 16501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Du, K.; Wei, F.; Jiang, H.; Kloc, C. Crystal Growth, HOMO–LUMO Engineering, and Charge Transfer Degree in Perylene-Fx TCNQ (x = 1, 2, 4) Organic Charge Transfer Binary Compounds. Cryst. Growth Des. 2016, 16, 3019–3027. [Google Scholar] [CrossRef]
- Singh, M.; Chopra, D. Diversity in Mechanical Response in Donor–Acceptor Coupled Cocrystal Stoichiomorphs Based on Pyrene and 1,8-Dinitroanthraquinone Systems. Cryst. Growth Des. 2018, 18, 6670–6680. [Google Scholar] [CrossRef]
- Averkiev, B.; Isaac, R.; Jucov, E.V.; Khrustalev, V.N.; Kloc, C.; McNeil, L.E.; Timofeeva, T.V. Evidence of Low-Temperature Phase Transition in Tetracene–Tetracyanoquinodimethane Complex. Cryst. Growth Des. 2018, 18, 4095–4102. [Google Scholar] [CrossRef]
- Henderson, J.; Masino, M.; Hatcher, L.E.; Kociok-Köhn, G.; Salzillo, T.; Brillante, A.; Raithby, P.R.; Girlando, A.; Da Como, E. New Polymorphs of Perylene:Tetracyanoquinodimethane Charge Transfer Cocrystals. Cryst. Growth Des. 2018, 18, 2003–2009. [Google Scholar] [CrossRef]
- Starodub, V.A.; Starodub, T.N. Radical anion salts and charge transfer complexes based on tetracyanoquinodimethane and other strong π-electron acceptors. Russ. Chem. Rev. 2014, 83, 391. [Google Scholar] [CrossRef]
- Wang, W.; Luo, L.; Sheng, P.; Zhang, J.; Zhang, Q. Multifunctional Features of Organic Charge-Transfer Complexes: Advances and Perspectives. Chem.-A Eur. J. 2021, 27, 464–490. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Aquino, A.J.A.; Sue, A.C.-H.; Lischka, H. Analysis of charge transfer transitions in stacked π-electron donor–acceptor complexes. Phys. Chem. Chem. Phys. 2018, 20, 26957–26967. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.K. Electron affinities of polynuclear acceptors. Dinitro- and trinitrophenanthrenequinones. J. Phys. Chem. 1967, 71, 2277–2282. [Google Scholar] [CrossRef]
- Linko, R.V.; Ryabov, M.A.; Strashnov, P.V.; Polyanskaya, N.A.; Davydov, V.V.; Dorovatovskii, P.V.; Lin’ko, I.V.; Khrustalev, V.N. Charge transfer complexes of nitro derivatives of 9,10-phenanthrenequinone with anthracene. crystal and molecular structures of the (1:1) complex of 2,4,7-trinitro- 9,10-phenanthrenequinone with anthracene. J. Struct. Chem. 2021, 62, 137–146. [Google Scholar] [CrossRef]
- Linko, R.V.; Ryabov, M.A.; Strashnov, P.V.; Polyanskaya, N.A.; Davydov, V.V.; Dorovatovskii, P.V.; Khrustalev, V.N. Quantum-Chemical Simulation of the Structure of Charge-Transfer Complexes of 9,10-Phenanthrenequinone Nitro-Derivatives with Phenanthrene. Crystal and Molecular Structure of 1:1 Complex of 2,4,7-Trinitro−9,10-phenanthrenequinone with Phenanthrene. Russ. J. Gen. Chem. 2020, 90, 1869–1877. [Google Scholar] [CrossRef]
- Winget, P.; Brédas, J.-L. Ground-State Electronic Structure in Charge-Transfer Complexes Based on Carbazole and Diarylamine Donors. J. Phys. Chem. C 2011, 115, 10823–10835. [Google Scholar] [CrossRef]
- Saito, G.; Murata, T. Mixed valency in organic charge transfer complexes. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2008, 366, 139–150. [Google Scholar] [CrossRef]
- Sinclair, V.C.; Robertson, J.M.; McL Mathieson, A. The crystal and molecular structure of anthracene. II. Structure investigation by the triple Fourier series method. Acta Crystallogr. 1950, 3, 251. [Google Scholar] [CrossRef]
- Sadova, N.I.; Vilkov, L.V. The Molecular Geometry of Nitro-compounds. Russ. Chem. Rev. 1982, 51, 87–105. [Google Scholar] [CrossRef]
- Britton, D. 2,3,5,6-Tetrachloro−1,4-dicyanobenzene–anthracene (1/1). Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, o1707–o1708. [Google Scholar] [CrossRef]
- Hu, P.; Li, H.; Li, Y.; Jiang, H.; Kloc, C. Single-crystal growth, structures, charge transfer and transport properties of anthracene-F4 TCNQ and tetracene-F4 TCNQ charge-transfer compounds. CrystEngComm 2017, 19, 618–624. [Google Scholar] [CrossRef]
- Andrievskii, A.M.; Linko, R.V.; Grachev, M.K. Synthesis and reactions of trinitro−9,10-phenanthrenequinone derivatives. Russ. J. Org. Chem. 2013, 49, 1025–1030. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2004. [Google Scholar]
- Granovsky, A.A. FireFly Version 8.20. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 20 May 1997).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | DEHOMO | ΔEMO | CTCEHOMO | CTCELUMO | ΔCTCEMO | qNPA | q*NPA | ΔEass |
|---|---|---|---|---|---|---|---|---|
| [PQ-PD] | −6.97 | 2.40 | −7.43 | −3.92 | 3.51 | 0.138 | 0.080 | −59.4 |
| [PQ-Pd]’ | −6.97 | 2.40 | −7.61 | −4.46 | 3.14 | 0.015 | 0.001 | −46.6 |
| [PQ-BZ] | −6.96 | 2.39 | −7.50 | −4.40 | 3.10 | 0.026 | 0.008 | −47.0 |
| [PQ-QN] | −6.53 | 1.96 | −7.15 | −4.35 | 2.79 | 0.019 | 0.071 | −67.6 |
| [PQ-Qn]’ | −6.53 | 1.96 | −7.06 | −4.42 | 2.64 | 0.014 | 0.969 | −63.3 |
| [PQ-IQN] | −6.45 | 1.88 | −7.01 | −4.45 | 2.56 | −0.003 | 0.971 | −61.4 |
| [PQ-IQn]’ | −6.45 | 1.88 | −7.05 | −4.45 | 2.60 | 0.018 | 0.967 | −64.2 |
| [PQ-TPL] | −6.10 | 1.53 | −6.58 | −4.21 | 2.37 | 0.037 | 0.969 | −106.5 |
| [PQ-NA] | −6.04 | 1.47 | −6.62 | −4.43 | 2.19 | −0.001 | 0.976 | −63.0 |
| [PQ-PA] | −5.98 | 1.41 | −6.65 | −4.22 | 2.43 | 0.054 | 0.994 | −92.6 |
| [PQ-Pa] | −5.98 | 1.41 | −6.47 | −3.89 | 2.57 | 0.028 | 0.992 | −89.9 |
| [PQ-ACR] | −5.92 | 1.35 | −6.48 | −4.30 | 2.18 | 0.014 | 0.982 | −79.2 |
| [PQ-ACr]’ | −5.92 | 1.35 | −6.50 | −4.38 | 2.11 | 0.023 | 0.981 | −77.1 |
| [PQ-CRS] | −5.76 | 1.19 | −6.36 | −4.21 | 2.15 | 0.060 | 1.007 | −111.3 |
| [PQ-ACN] | −5.71 | 1.14 | −6.31 | −4.43 | 1.88 | 0.005 | 0.983 | −74.7 |
| [PQ-aCn]’ | −5.71 | 1.14 | −6.25 | −4.53 | 1.72 | 0.047 | 0.985 | −75.8 |
| [PQ-COR] | −5.70 | 1.13 | −6.13 | −4.21 | 1.92 | 0.031 | 1.010 | −115.3 |
| [PQ-PYR] | −5.58 | 1.01 | −6.20 | −4.20 | 2.00 | 0.068 | 0.992 | −97.5 |
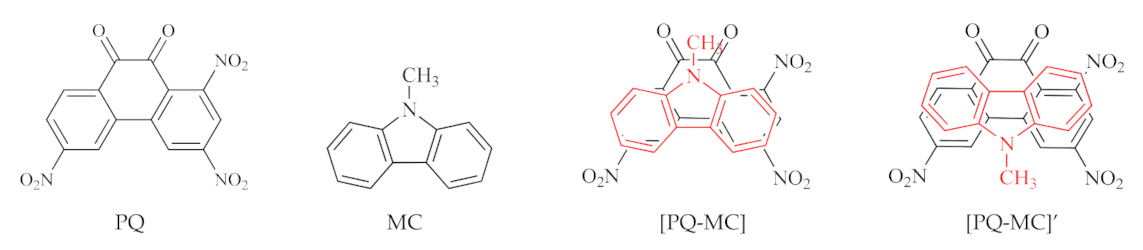
| [PQ-MC] | −5.57 | 1.00 | −6.27 | −4.31 | 1.96 | 0.070 | 1.012 | −94.0 |
| [PQ-Mc]’ | −5.57 | 1.00 | −6.16 | −4.23 | 1.92 | 0.071 | 1.008 | −87.0 |
| [PQ-TPH] | −5.57 | 1.00 | −6.17 | −4.18 | 1.99 | 0.066 | 1.006 | −104.8 |
| [PQ-TPh]’ | −5.57 | 1.00 | −6.15 | −4.21 | 1.94 | 0.065 | 1.005 | −107.0 |
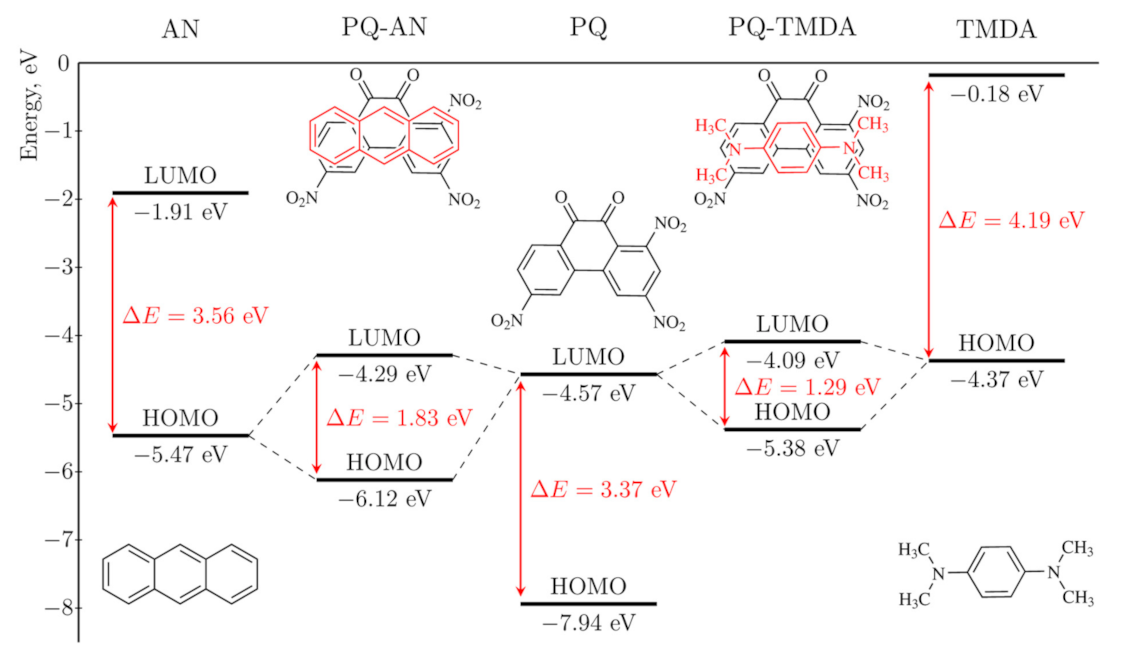
| [PQ-AN] | −5.47 | 0.90 | −6.12 | −4.29 | 1.83 | 0.046 | 0.990 | −82.3 |
| [PQ-AZU] | −5.44 | 0.87 | −6.02 | −4.45 | 1.57 | −0.004 | 0.969 | −63.5 |
| [PQ-AZu]’ | −5.44 | 0.87 | −6.27 | −4.18 | 2.10 | 0.114 | 0.971 | −73.3 |
| [PQ-POR] | −5.39 | 0.82 | −5.88 | −4.07 | 1.82 | 0.077 | 1.033 | −119.2 |
| [PQ-TET] | −5.10 | 0.53 | −5.70 | −4.24 | 1.45 | 0.066 | 1.018 | −106.1 |
| [PQ-DBTTF] | −4.89 | 0.32 | −5.53 | −4.00 | 1.53 | 0.216 | 1.081 | −124.0 |
| [PQ-PEN] | −4.85 | 0.28 | −5.45 | −4.08 | 1.37 | 0.134 | 1.042 | −118.0 |
| [PQ-TTF] | −4.52 | −0.05 | −5.48 | −4.03 | 1.45 | 0.240 | 1.089 | −102.3 |
| [PQ-DMPZ] | −4.39 | −0.18 | −5.26 | −4.22 | 1.04 | 0.135 | 1.058 | −107.5 |
| [PQ-TMDA] | −4.37 | −0.20 | −5.38 | −4.09 | 1.29 | 0.224 | 1.075 | −104.3 |
| Bond | d | Angle | ω | ||||
|---|---|---|---|---|---|---|---|
| I | [PQ-AN] | PQ, AN | I | [PQ-AN] | PQ, AN | ||
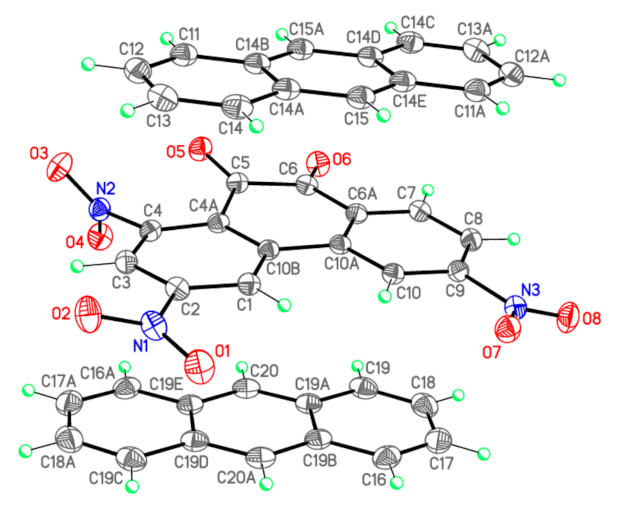
| O5–C5 | 1.2103(13) | 1.208 | 1.207 | O1–N1–O2 | 124.64(10) | 125.5 | 125.9 |
| O6–C6 | 1.2135(13) | 1.211 | 1.209 | O2–N1–C2 | 117.20(9) | 117.1 | 117.0 |
| O1–N1 | 1.2258(13) | 1.219 | 1.217 | O1–N1–C2 | 118.16(9) | 117.4 | 117.1 |
| O2–N1 | 1.2257(13) | 1.218 | 1.217 | O3–N2–O4 | 125.46(10) | 126.7 | 127.2 |
| O3–N2 | 1.2198(13) | 1.214 | 1.215 | O4–N2–C4 | 117.13(9) | 116.4 | 116.4 |
| O4–N2 | 1.2211(13) | 1.217 | 1.214 | O3–N2–C4 | 117.32(9) | 116.7 | 116.3 |
| O7–N3 | 1.2289(13) | 1.220 | 1.218 | O7–N3–O8 | 123.79(10) | 125.2 | 125.6 |
| O8–N3 | 1.2264(13) | 1.219 | 1.217 | O7–N3–C9 | 118.31(9) | 117.4 | 117.3 |
| N1–C2 | 1.4712(14) | 1.479 | 1.485 | O8–N3–C9 | 117.89(9) | 117.4 | 117.1 |
| N2–C4 | 1.4791(13) | 1.480 | 1.482 | O5–C5–C4A | 122.52(10) | 123.0 | 122.8 |
| N3–C9 | 1.4717(13) | 1.477 | 1.484 | O5–C5–C6 | 119.50(10) | 120.0 | 119.8 |
| C5–C6 | 1.5379(15) | 1.537 | 1.540 | C4A–C5–C6 | 117.91(9) | 117.0 | 117.3 |
| C10A–C10B | 1.4819(15) | 1.480 | 1.484 | O6–C6–C6A | 123.53(10) | 123.3 | 123.2 |
| C11–C12 | 1.3606(18) | 1.373 | 1.373 | O6–C6–C5 | 118.92(10) | 119.6 | 119.5 |
| C12–C13 | 1.4213(17) | 1.427 | 1.428 | C6A–C6–C5 | 117.54(9) | 117.1 | 117.2 |
| C11–C14B | 1.4303(17) | 1.431 | 1.431 | C3–C4–N2 | 115.79(9) | 115.7 | 115.9 |
| C14A–C15 | 1.3968(17) | 1.403 | 1.402 | C4A–C4–N2 | 120.83(9) | 121.4 | 121.4 |
| C14A–C14B | 1.4400(15) | 1.446 | 1.446 | C12–C11–C14B | 120.73(11) | 120.8 | 121.0 |
| Compound | I |
|---|---|
| CCDC | 2099997 |
| Formula | C14H5N3O8·C14H10 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Z | 4 |
| a,b,c, Å | 14.2721(14), 19.479(2), 8.1900(9) |
| α, β, γ, deg | 90, 99.041(8), 90 |
| V, Å3 | 2248.6(4) |
| Dx, g/cm3 | 1.540 |
| Radiation, λ, Å | Synchrotron, 0.79272 |
| μ, mm−1 | 0.147 |
| T, K | 100(2) |
| Specimen size, mm | 0.18 × 0.15 × 0.03 |
| Absorption correction | Semi-empirical |
| Tmin/Tmax | 0.966/0.987 |
| θmax, deg | 30.95 |
| Limits of h,k,l | −15<=h<=18; −25<=k<=18; −10<=l<=10; |
| Number of reflections: measured/independent (N1); | 13943/5033 |
| observed with I>2σ(I) (N2) | 4596 |
| Rint | 0.0276 |
| Number of parameters | 353 |
| Extinction coefficient | 0.029(3) |
| R1/wR2 by N1 | 0.0404/0.0975 |
| R1/wR2 by N2 | 0.0373/0.0950 |
| S | 1.023 |
| Δpmin/Δpmax, eÅ−3 | −0.220/0.261 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linko, R.; Ryabov, M.; Strashnov, P.; Dorovatovskii, P.; Khrustalev, V.; Davydov, V. Charge Transfer Complexes of 1,3,6-Trinitro-9,10-phenanthrenequinone with Polycyclic Aromatic Compounds. Molecules 2021, 26, 6391. https://doi.org/10.3390/molecules26216391
Linko R, Ryabov M, Strashnov P, Dorovatovskii P, Khrustalev V, Davydov V. Charge Transfer Complexes of 1,3,6-Trinitro-9,10-phenanthrenequinone with Polycyclic Aromatic Compounds. Molecules. 2021; 26(21):6391. https://doi.org/10.3390/molecules26216391
Chicago/Turabian StyleLinko, Roman, Michael Ryabov, Pavel Strashnov, Pavel Dorovatovskii, Victor Khrustalev, and Victor Davydov. 2021. "Charge Transfer Complexes of 1,3,6-Trinitro-9,10-phenanthrenequinone with Polycyclic Aromatic Compounds" Molecules 26, no. 21: 6391. https://doi.org/10.3390/molecules26216391