
Unusual Reactivities of ortho-Hydroxy-β-nitrostyrene


Abstract
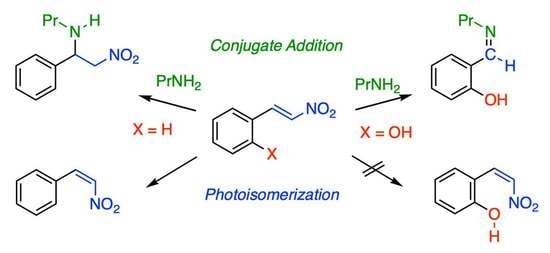
:
1. Introduction
2. Results and Discussion
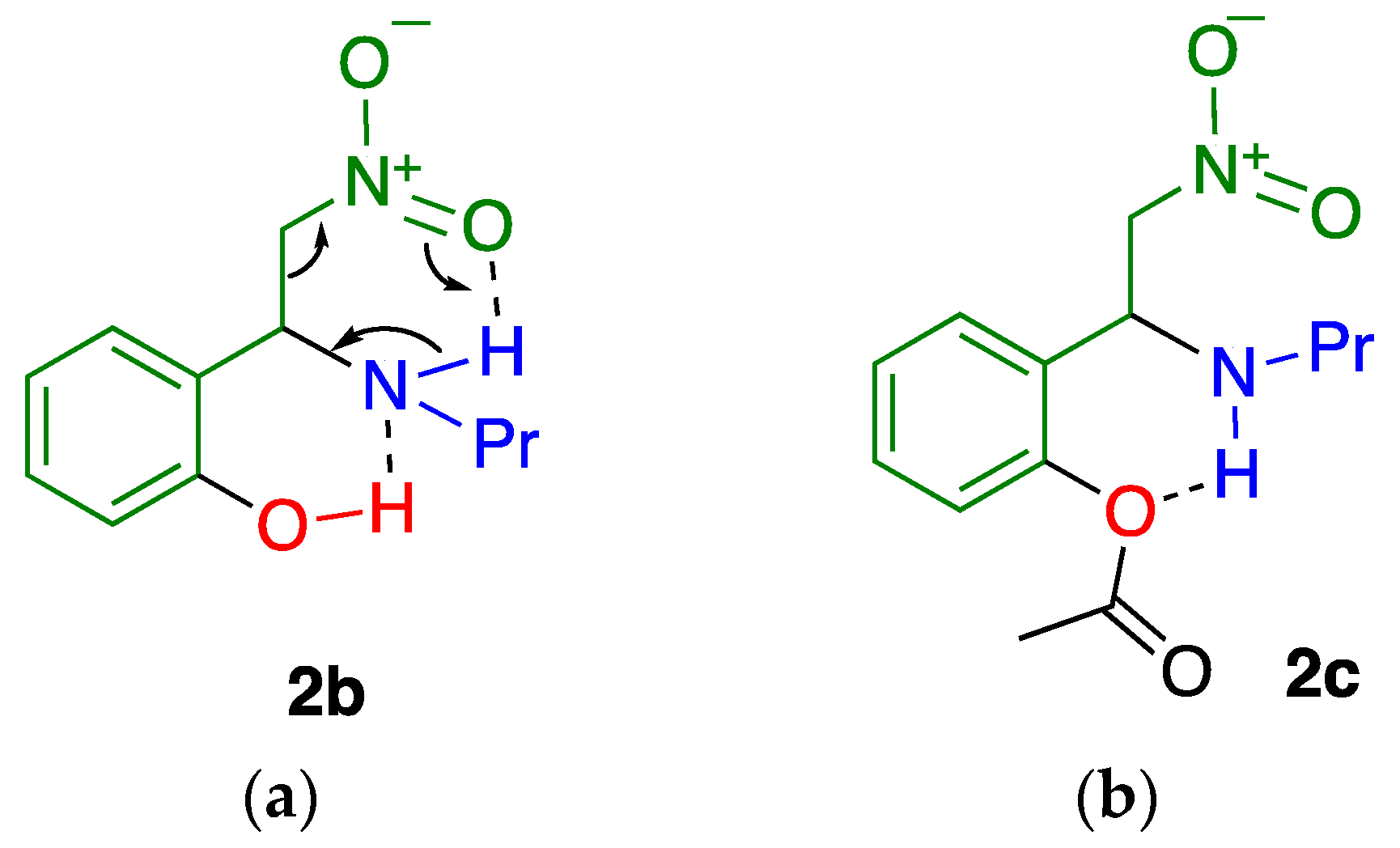
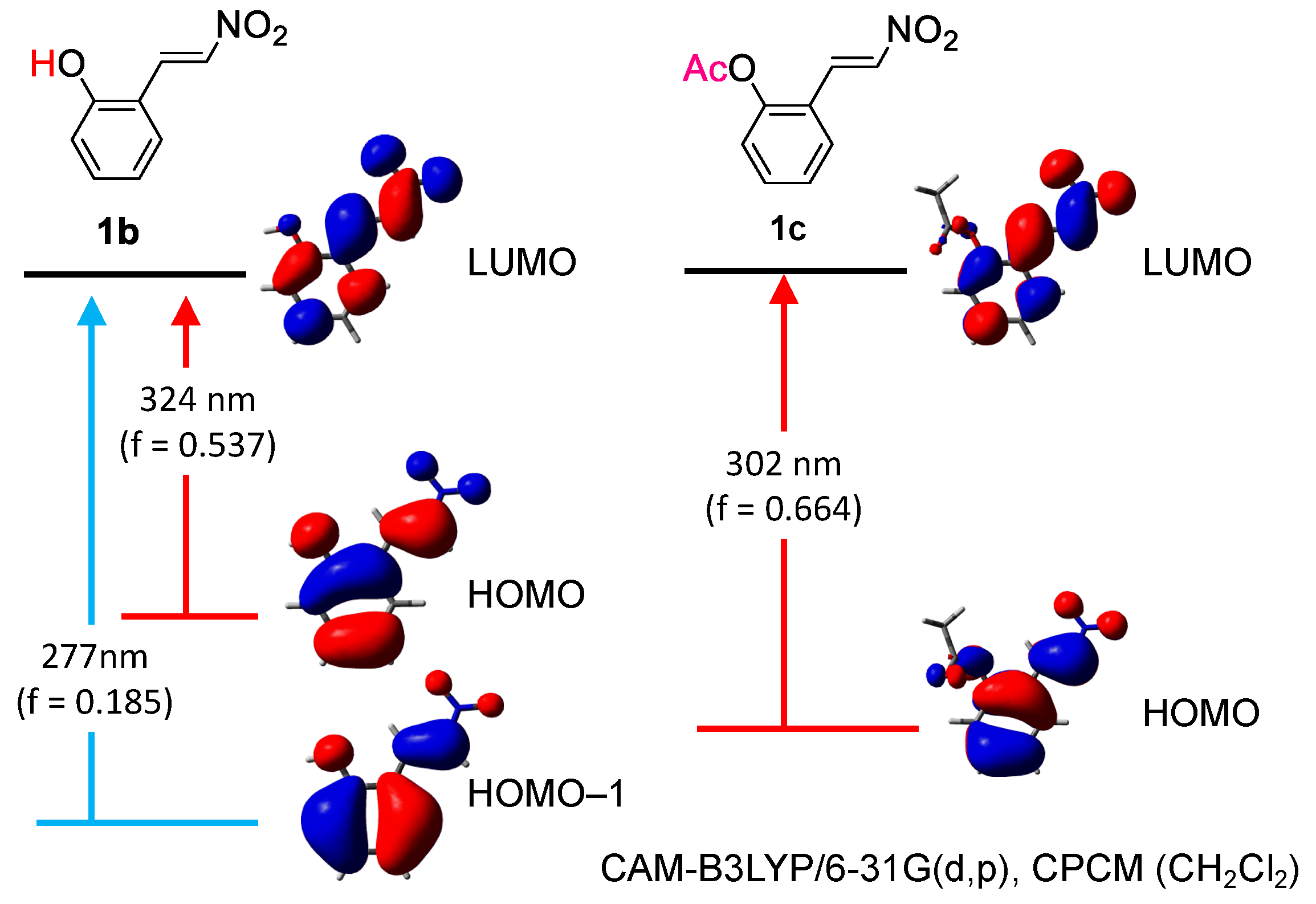
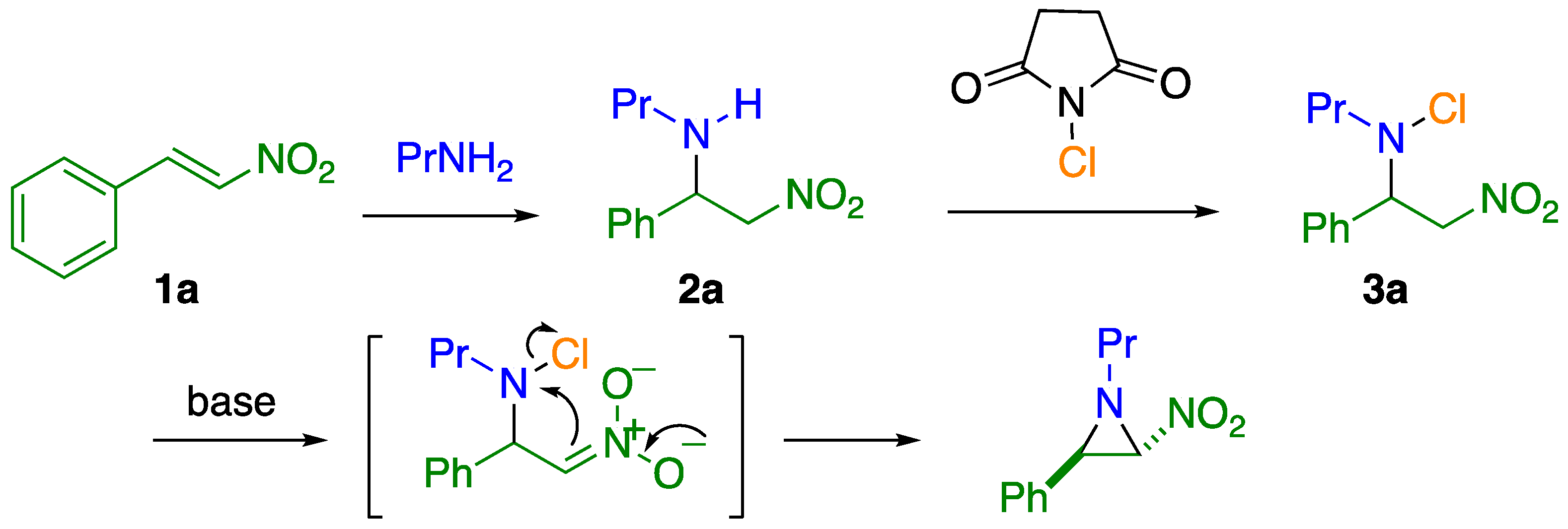
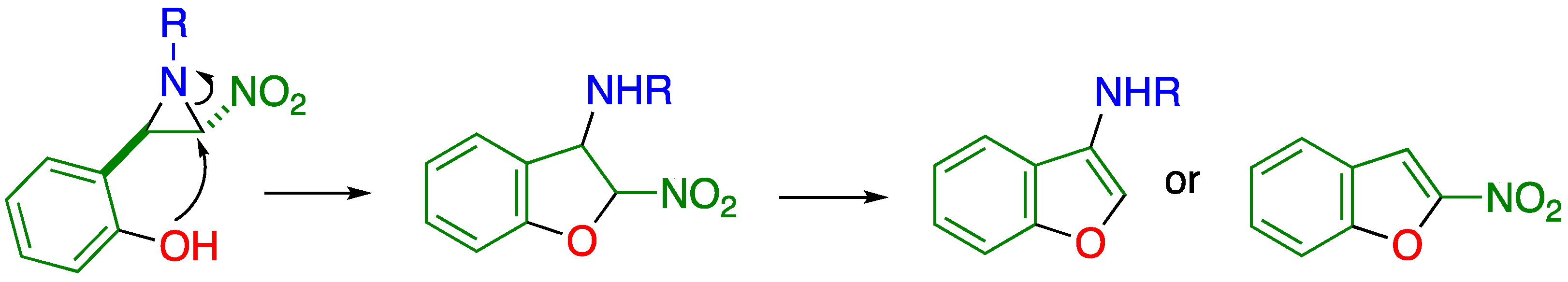
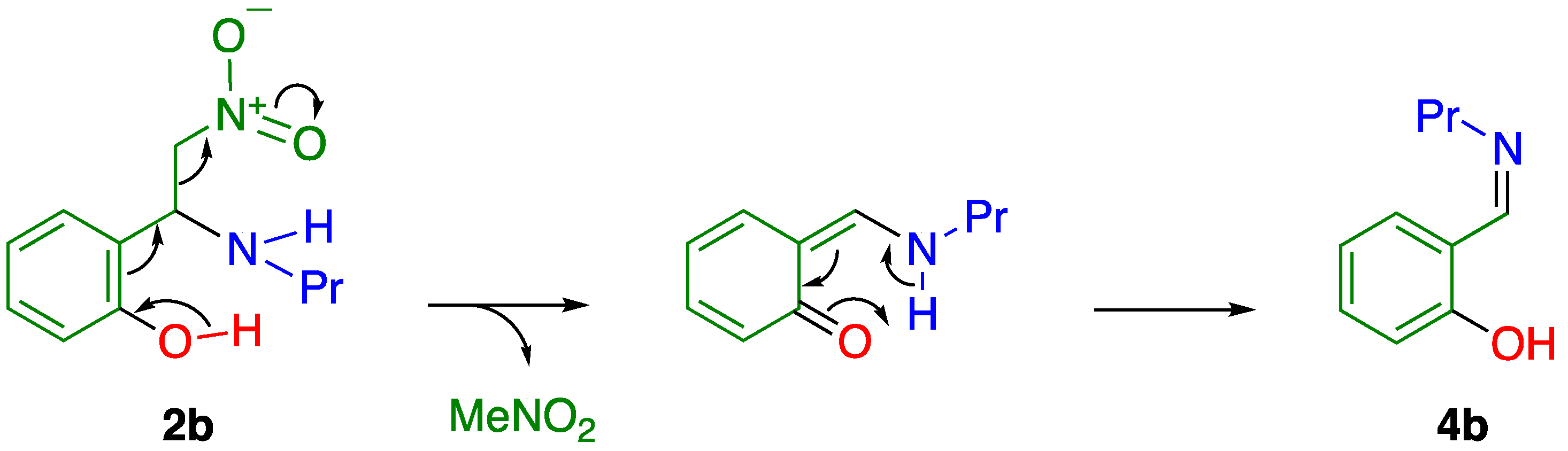
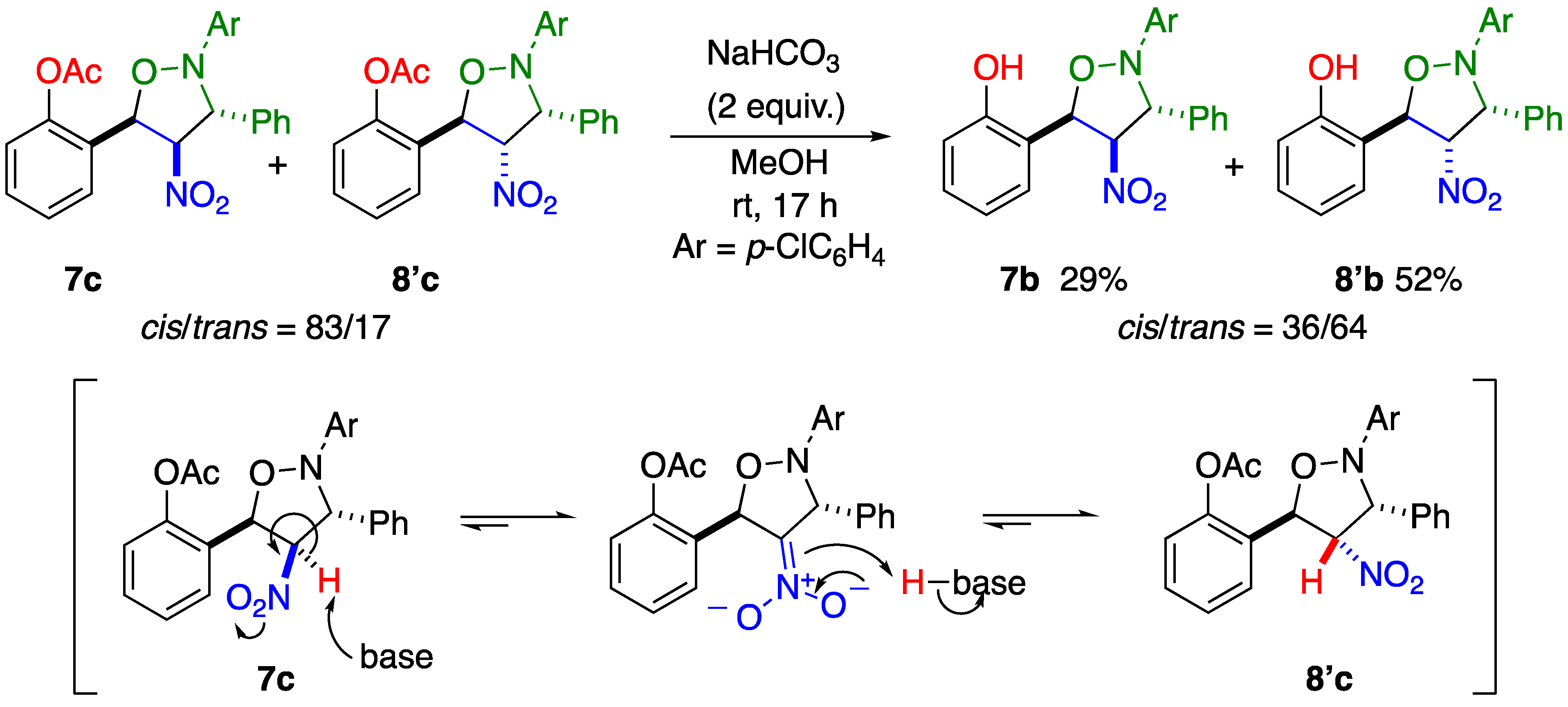
2.1. C–C Bond Cleavage
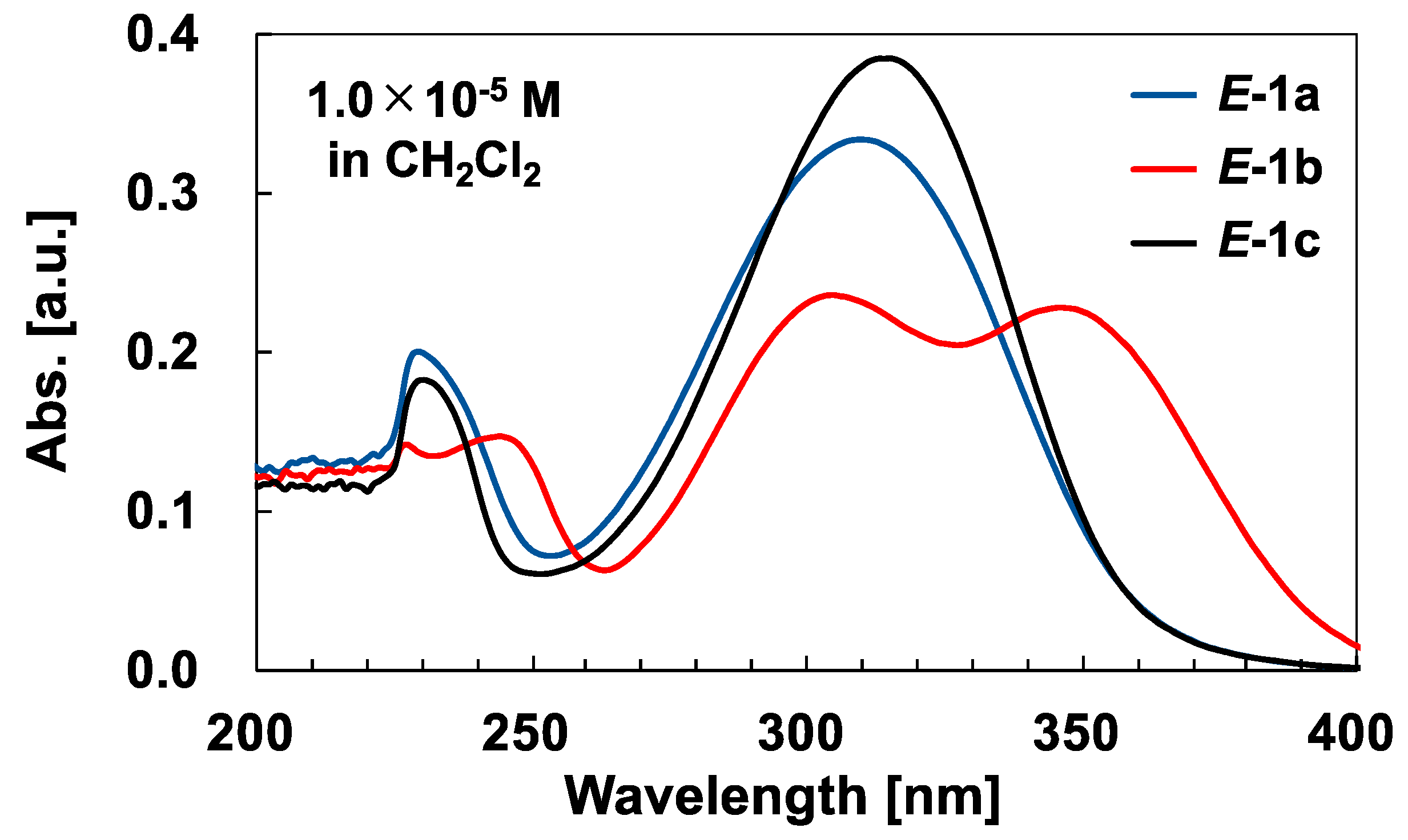
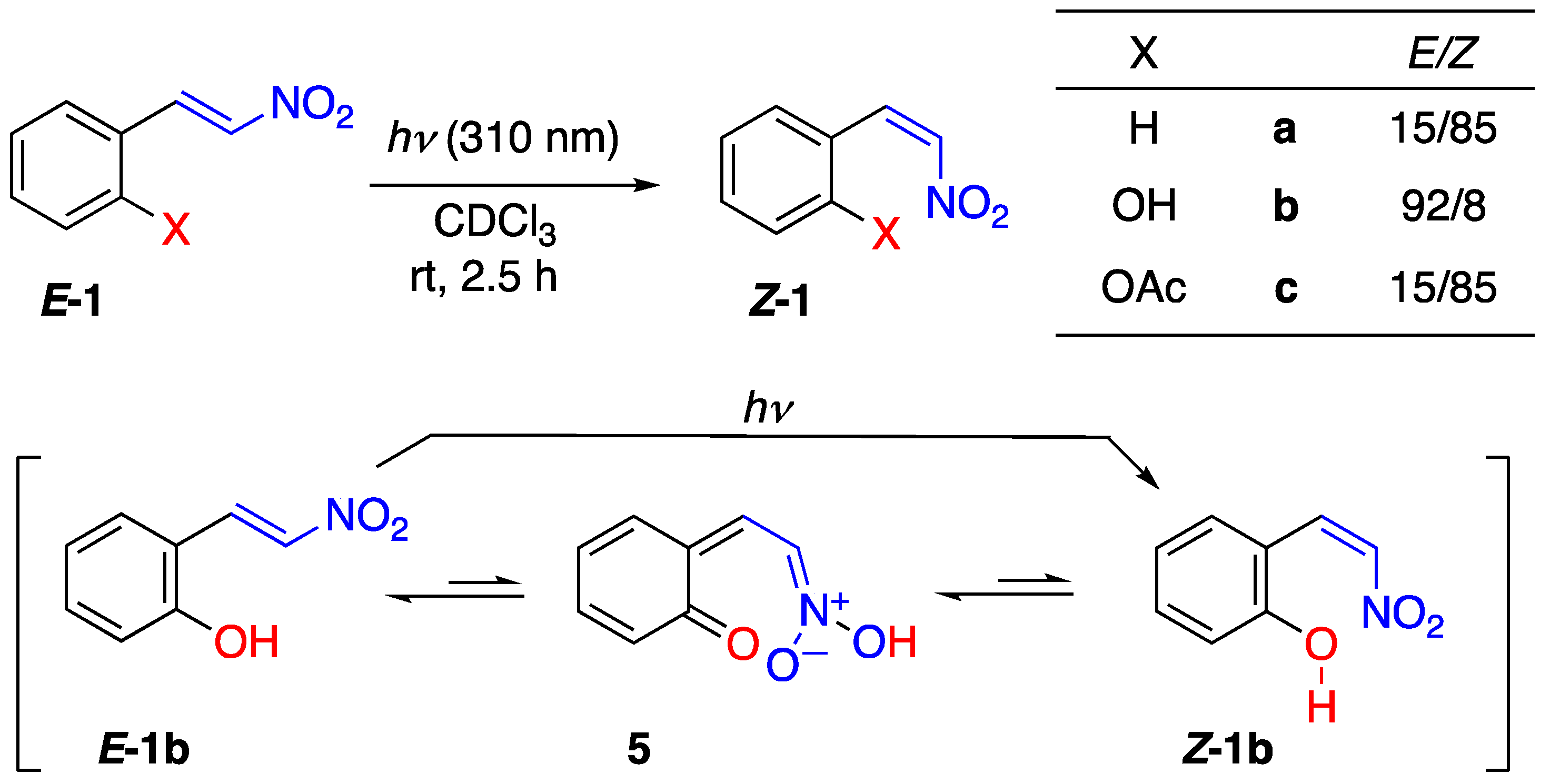
2.2. Photoisomerization
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Preparation of β-Nitrostyrenes 1
4.2.1. Synthesis of Nitrostyrene 1b
4.2.2. Acetylation of 1b
4.3. Conjugate Addition of Amine to β-Nitrostyrene
4.4. Photoisomerization of β-Nitrostyrene
4.5. 1,3-Dipolar Cycloaddition of β-Nitrostyrene of 1 with Nitrone 7
4.5.1. 2-(4-Chlorophenyl)-3,5-diphenyl-4-nitroisooxazolidine 8a and 9a
4.5.2. 5-(2-Acetoxyphenyl)-2-(4-chlorophenyl)-4-nitro-3-phenylisoxazolidine 8c and 9c
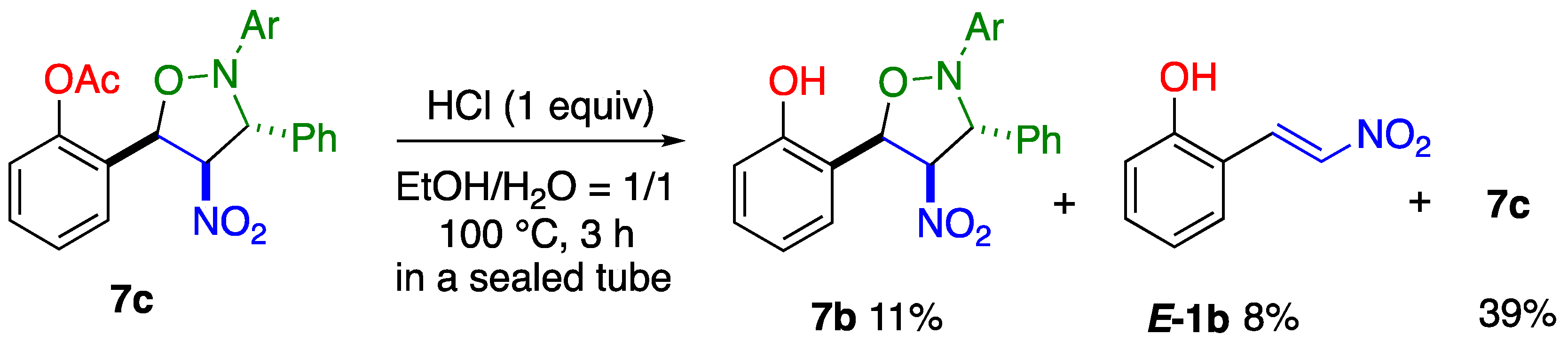
4.6. Hydrolysis of 8c under Acidic Conditions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Nishiwaki, N. A Walk through Recent Nitro Chemistry Advances. Molecules 2020, 25, 3680. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Asymmetric Michael Addition Mediated by Chiral Ionic Liquids. Mini Rev. Org. Chem. 2018, 15, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Elkin, P.K.; Durfee, N.D.; Rawal, V.H. Diels-Alder Reactions of 1-Alkoxy-1-amino-1,3-butadienes: Direct Synthesis of 6-Substituted and 6,6-Disubstituted 2-Cyclohexenones and 6-Substituted 5,6-Dihydropyran-2-ones. Org. Lett. 2021, 23, 5288–5293. [Google Scholar] [CrossRef] [PubMed]
- Marcantonio, E.; Curti, C.; Battistini, L.; Sartori, A.; Cardinale, L.; Pelosi, G.; Zanardi, F. Direct, Asymmetric Synthesis of Carbocycle-Fused Uracils via [4+2] Cycloadditions: A Noncovalent Organocatalysis Approach. Adv. Synth. Catal. 2021, 363, 2625–2633. [Google Scholar] [CrossRef]
- Asahara, H.; Hiraishi, M.; Nishiwaki, N. One-Pot and Metal-Free Synthesis of 3-Arylated 4-Nitrophenols via Polyfunctionalized Cyclohexanones from β-Nitrostyrenes. Beistein. J. Org. Chem. 2020, 16, 1830–1836. [Google Scholar] [CrossRef]
- Wu, M.-Y.; He, W.-W.; Liu, X.-Y.; Tan, B. Asymmetric Construction of Spirooxindoles by Organocatalytic Multicomponent Reactions Using Diazooxindoles. Angew. Chem. Int. Ed. 2015, 54, 9409–9413. [Google Scholar] [CrossRef]
- Banerji, A.; Gupta, M.; Biswas, P.K.; Prange, T.; Neuman, A. 1,3-Dipolar Cycloadditions. Part XII—Selective Cycloaddition Route to 4-Nitroisoxazolidine Ring Systems. J. Heterocycl. Chem. 2007, 44, 1045–1049. [Google Scholar] [CrossRef]
- Sridharan, V.; Muthusubramanian, S.; Sivasubramanian, S.; Polborn, K. Diastereoselective Synthesis of 2,3,4,5-Tetrasubstituted Isoxazolidines via 1,3-Dipolar Cycloaddition. Tetrahedron 2004, 60, 8881–8892. [Google Scholar] [CrossRef]
- Marčeková, M.; Ferko, B.; Detková, K.R.; Jakubec, P. Denitrative Cross-Couplings of Nitrostyrenes. Molecules 2020, 25, 3390. [Google Scholar] [CrossRef]
- Akhtar, R.; Zahoor, A.F.; Rasool, N.; Ahmad, M.; Ali, K.G. Recent Trends in the Chemistry of Sandmeyer Reaction: A Review. Mol. Divers. 2022, 26, 1837–1873. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Batra, S. Applications of Sodium Nitrite in Organic Synthesis. Eur. J. Org. Chem. 2019, 6424–6451. [Google Scholar] [CrossRef]
- Mukaijo, Y.; Yokoyama, S.; Nishiwaki, N. Comparison of Substitutiong Ability of Nitronate versus Enolate for Direct Substitution of a nitro Group. Molecules 2020, 25, 2048. [Google Scholar] [CrossRef] [PubMed]
- Gharui, C.; Pan, S.C. Employment of α-Nitroketones in Organic Synthesis. Org. Biomol. Chem. 2019, 17, 5190–5211. [Google Scholar] [CrossRef]
- Asahara, H.; Sofue, A.; Kuroda, Y.; Nishiwaki, N. Alkynylation and Cyanation of Alkenes Using Diverse Properties of a Nitro Group. J. Org. Chem. 2018, 83, 13691–13699. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Petrini, M. The Nitro to Carbonyl Conversion (Nef Reaction): New Perspectives for a Classical Transformation. Adv. Synth. Catal. 2015, 357, 2371–2402. [Google Scholar] [CrossRef]
- Hao, F.; Asahara, H.; Nishiwaki, N. Direct Aziridination of Nitroalkenes Affording N-Alkyl-C-nitroaziridines and the Subsequent Lewis Acid Mediated Isomerization to β-Nitroenamines. Org. Lett. 2017, 19, 5442–5445. [Google Scholar] [CrossRef]
- Iwai, K.; Wada, K.; Hao, F.; Asahara, H.; Nishiwaki, N. A Mechanistic Study for Aziridination of Nitroalkenes Mediated by N-Chlorosuccinimide. J. Oleo Sci. 2022, 71, 897–903. [Google Scholar] [CrossRef]
- Hao, F.; Nishiwaki, N. Chemistry of Nitroaziridines. Heterocycles 2019, 99, 54–72. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, A.; Srinivasulu, V.; Abu-Yousef, I.A.; Gorka, O.; Al-Tel, T.H. Domino Transformations of Ene/Yne Tethered Salicylaldehyde Derivatives: Pluripotent Platforms for the Construction of High sp3 Content and Privileged Architectures. Chem. Eur. J. 2019, 25, 15710–15735. [Google Scholar] [CrossRef]
- Nayar, C.R.; Ravikumar, R. Review: Second Order Nonlinearities of Schiff Bases Derived from Salicylaldehyde and Their Metal Complexes. J. Coord. Chem. 2014, 67, 1–16. [Google Scholar] [CrossRef]
- Masesane, I.B.; Desta, Z.Y. Reactions of Salicylaldehyde and Enolates or Their Equivalents: Versatile Synthetic Routes to Chromane Derivatives. Beilstein. J. Org. Chem. 2012, 8, 2166–2175. [Google Scholar] [CrossRef]
- Kallitsakis, M.G.; Tancini, P.D.; Dixit, M.; Mpourmpakis, G.; Lykakis, I.N. Mechanistic Studies on the Michael Addition of Amines and Hydrazones to Nitrostyrenes: Nitroalkane Elimination via a Retro-aza-Henry-Type Process. J. Org. Chem. 2018, 83, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Yakura, T.; Nakazawa, M.; Takino, T.; Ikeda, M. Stereochemistry of 1,3-Dipolar Cycloadditions of Nitrones with (E)-1-Alkyl-2-nitroethenes. Chem. Pharm. Bull. 1992, 40, 2014–2018. [Google Scholar] [CrossRef] [Green Version]
- Wakita, K.; Kabuto, C.; Akine, S.; Nemoto, T.; Kwon, E.J. 2001, Release of Software (Yadokari-XG 2009) for Crystal Structure Analyses. Cryst. Soc. Jpn. 2009, 51, 218–224. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal Structure Determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Revision, C.; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; et al. Gaussian, 09; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Pérez, V.T.; de Arriba, F.Á.L.; Monleón, L.M.; Simón, L.; Rubio, O.H.; Sanz, F.; Morán, J.R. A High Yield Procedure for the Preparation of 2-Hydroxynitrostyrenes: Synthesis of Imines and Tetracyclic 1,3-Benzoxazines. Eur. J. Org. Chem. 2014, 2014, 3242–3248. [Google Scholar] [CrossRef]
- Liu, Y.F.; Liu, S.N.; Zhao, P.H.; Li, X.H.; Liang, W.J.; Liu, Y.Q. Synthesis, Characterization and Crystal Structure of trans-2-(2-Hydroxyphenyl)-1-nitroethylene. Asian J. Chem. 2014, 26, 2475–2478. [Google Scholar] [CrossRef]
- Lewis, K.G.; Ghosh, S.K.; Bhuvanesh, N.; Gladysz, J.A. Cobalt(III) Werner Complexes with 1,2-Diphenylethylenediamine Ligands: Readily Available, Inexpensive, and Modular Chiral Hydrogen Bond Donor Catalysts for Enantioselective Organic Synthesis. ACS Cent. Sci. 2015, 1, 50–56. [Google Scholar] [PubMed]
- Vuagnoux-d’Augustin, M.; Alexakis, A. Influenece of the Double-Bond Geometry of the Michael Acceptor on Copper-Catalyzed Asymmetric Conjugate Addition. Eur. J. Org. Chem. 2007, 5852–5860. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | X | Yield of 2/% | Yield of 4/% | |
|---|---|---|---|---|
| 1 | H | a | 86 | 1 |
| 2 | OH | b | 35 | 59 |
| 3 | OAc | c | 91 | 9 |

| Entry | X | Time/min | Yield/% | Recovery/% | |||
|---|---|---|---|---|---|---|---|
| 3 | 4 | 1 | 2 | ||||
| 1 | H | A | 5 | 50 | 0 | 0 | 50 |
| 2 | OH | B | 5 | 0 | 0 | 0 | 100 |
| 3 | OH | B | 120 | 18 | 21 | 10 | 51 |
| 4 a | OH | B | 120 | 35 | 39 | 7 | 19 |
| 5 a | OH | B | 1000 | 21 | 47 | 24 | 8 |

| Entry | X | Ratio | Yield of cis-Isomer/% | Yield of trans-Isomer/% | Ratio | |||
|---|---|---|---|---|---|---|---|---|
| Z-1/E-1 | 7 | 7′ | 8 | 8′ | cis/trans | |||
| 1 a | H | a | 0/100 | 0 | 0 | 80 | 11 | 0/100 |
| 2 a | H | a | 96/4 | 45 | 30 | 15 | 3 | 81/19 |
| 3 a | OAc | c | 97/3 | 18 | 12 | 8 | 2 | 75/25 |
| 4 b | OAc | c | 97/3 | 23 | 15 | 7 | 1 | 83/17 |
| 5 c | OAc | c | 99/1 | 43 | 10 | 18 | 3 | 72/28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwai, K.; Wada, K.; Nishiwaki, N. Unusual Reactivities of ortho-Hydroxy-β-nitrostyrene. Molecules 2022, 27, 4804. https://doi.org/10.3390/molecules27154804
Iwai K, Wada K, Nishiwaki N. Unusual Reactivities of ortho-Hydroxy-β-nitrostyrene. Molecules. 2022; 27(15):4804. https://doi.org/10.3390/molecules27154804
Chicago/Turabian StyleIwai, Kento, Khimiya Wada, and Nagatoshi Nishiwaki. 2022. "Unusual Reactivities of ortho-Hydroxy-β-nitrostyrene" Molecules 27, no. 15: 4804. https://doi.org/10.3390/molecules27154804