




Methods Protoc. 2024, 7(1), 12; https://doi.org/10.3390/mps7010012 - 26 Jan 2024
Viewed by 1361
Abstract
►
Show Figures
The rapid advancement of genetic technologies has made it possible to modify various plants through both genetic transformation and gene editing techniques. Poplar, with its rapid in vitro growth and regeneration enabling high rates of micropropagation, has emerged as a model system for
[...] Read more.
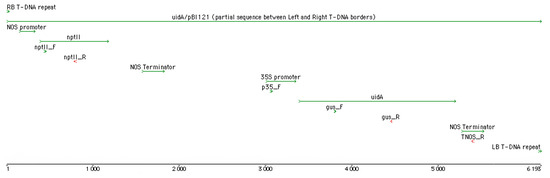
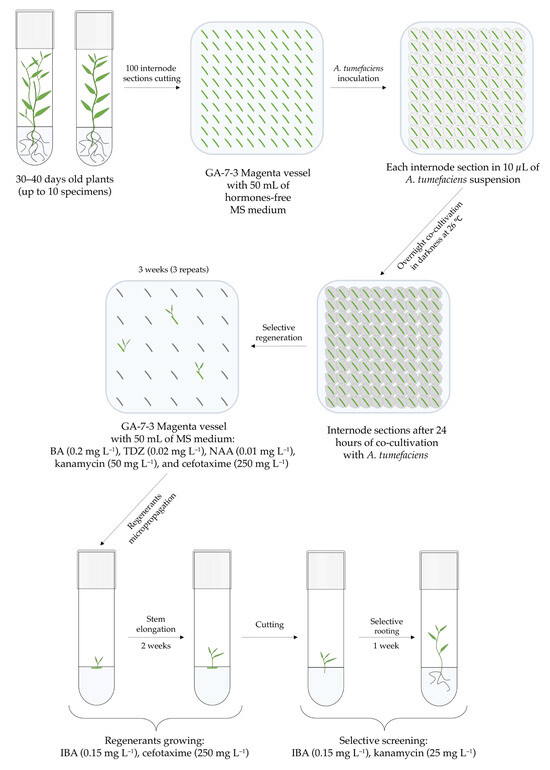
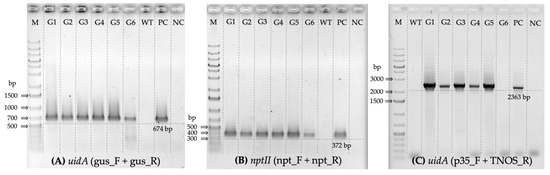
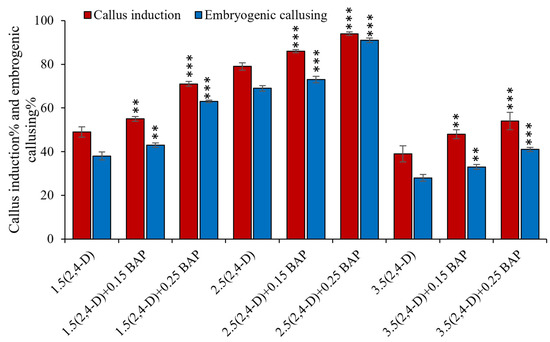
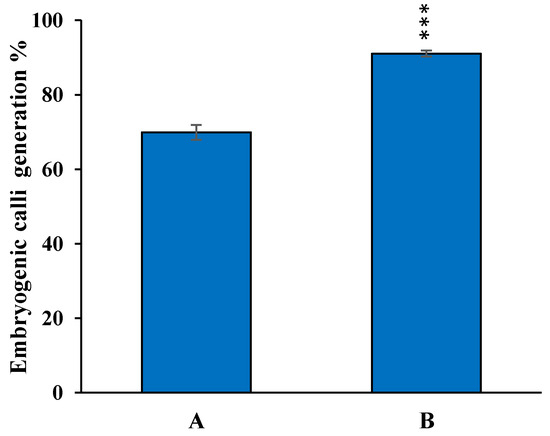
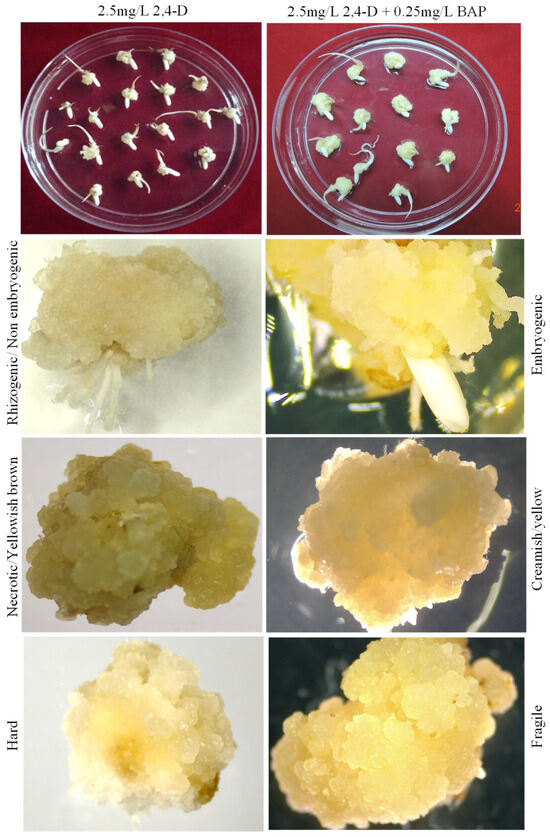
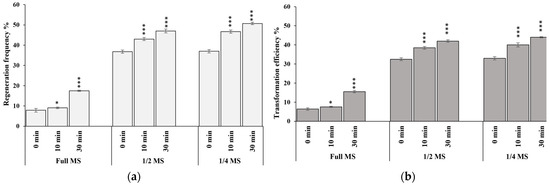
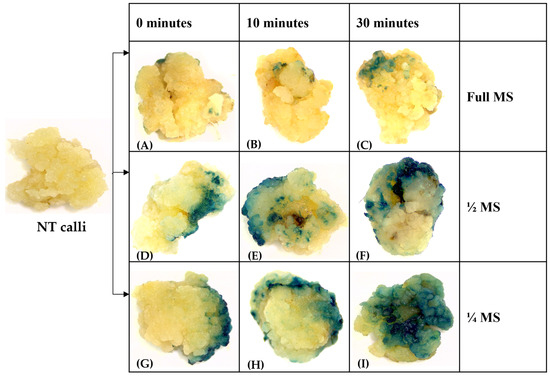
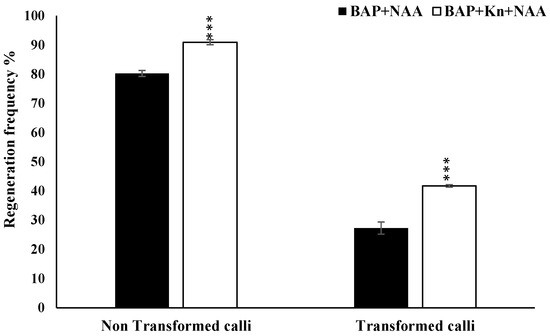
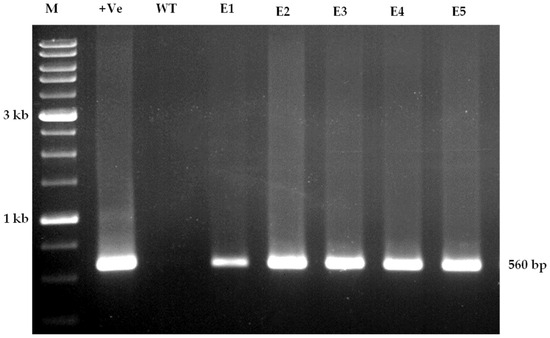
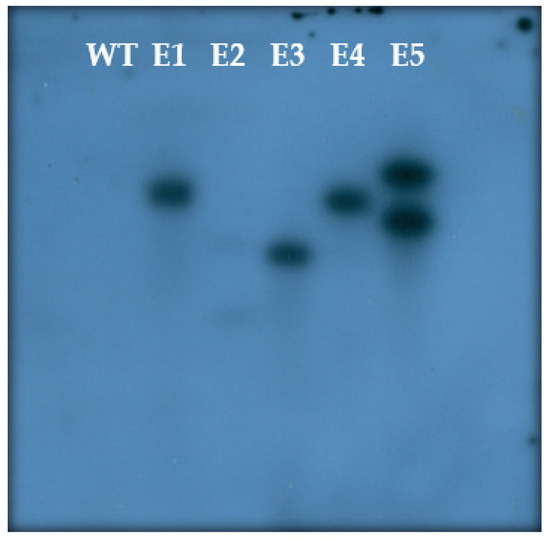
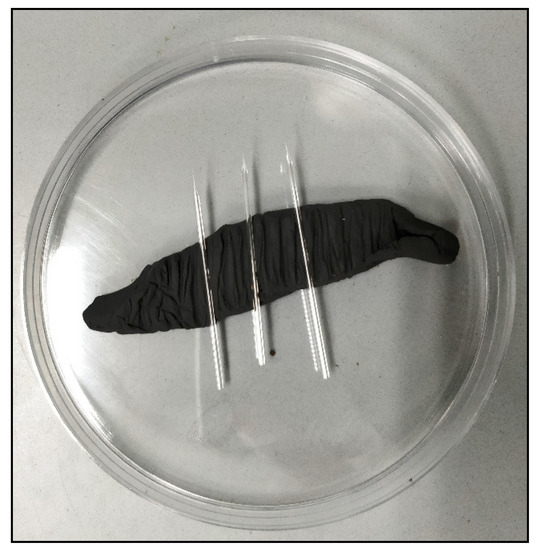
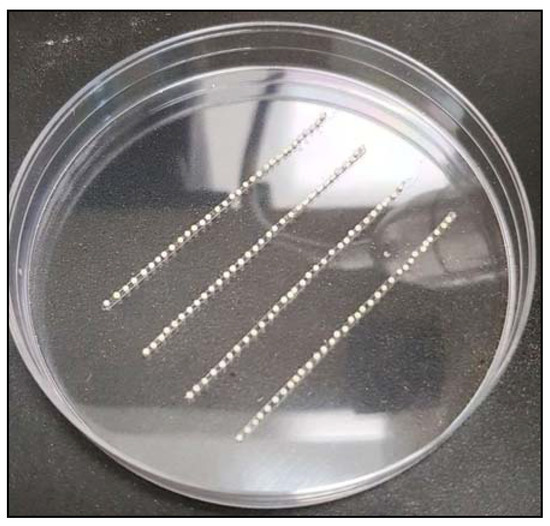
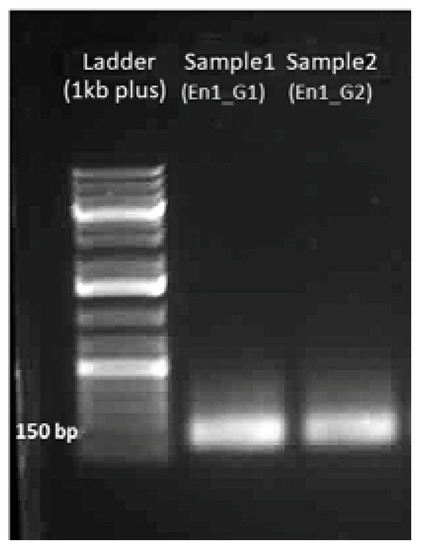
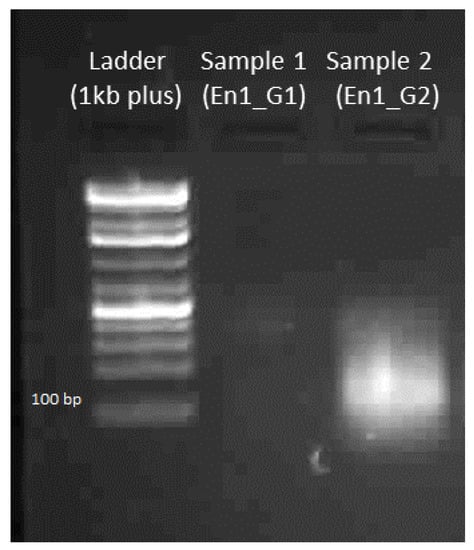
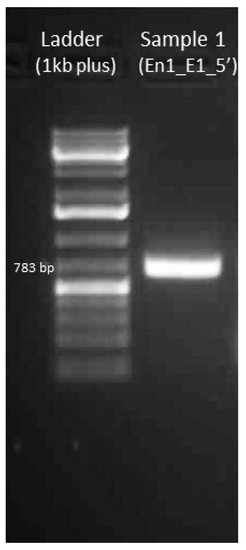
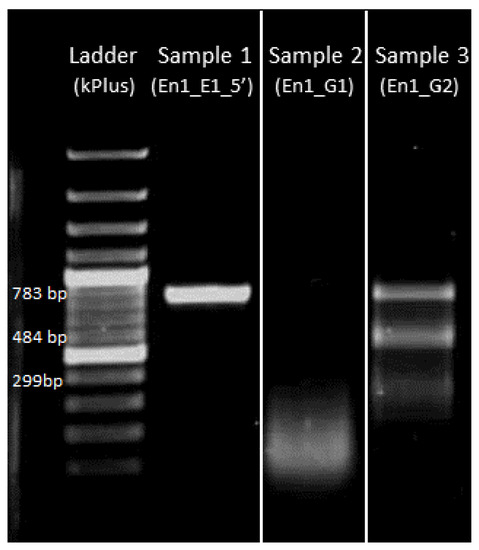
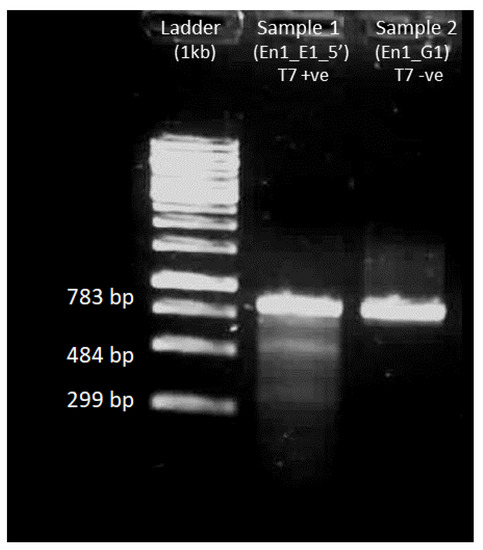
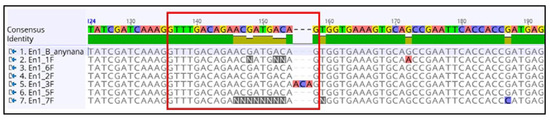
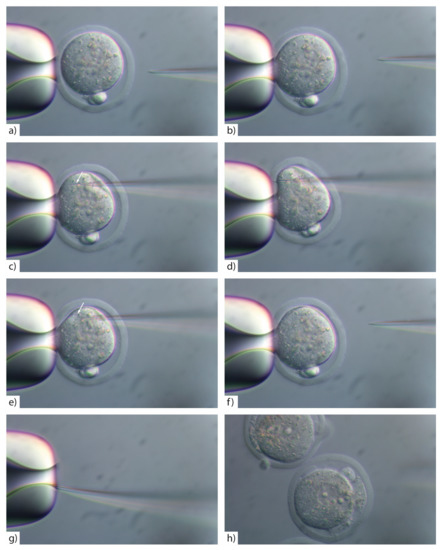
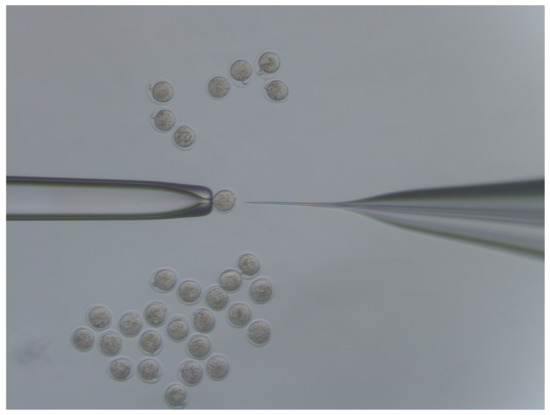
The rapid advancement of genetic technologies has made it possible to modify various plants through both genetic transformation and gene editing techniques. Poplar, with its rapid in vitro growth and regeneration enabling high rates of micropropagation, has emerged as a model system for the genetic transformation of woody plants. In this study, Populus × berolinensis K. Koch. (Berlin poplar) was chosen as the model organism due to its narrow leaves and spindle-shaped crown, which make it highly suitable for in vitro manipulations. Various protocols for the Agrobacterium-mediated transformation of poplar species have been developed to date. However, the genetic transformation procedures are often constrained by the complexity of the nutrient media used for plant regeneration and growth, which could potentially be simplified. Our study presents a cheaper, simplified, and relatively fast protocol for the Agrobacterium-mediated transformation of Berlin poplar. The protocol involved using internode sections without axillary buds as explants, which were co-cultivated in 10 µL droplets of bacterial suspension directly on the surface of a solid agar-based medium without rinsing and sterile paper drying after inoculation. We used only one regeneration Murashige and Skoogbased medium supplemented with BA (0.2 mg·L−1), TDZ (0.02 mg·L−1), and NAA (0.01 mg·L−1). Acetosyringone was not used as an induction agent for vir genes during the genetic transformation. Applying our protocol and using the binary plasmid pBI121 carrying the nptII selective and uidA reporter genes, we obtained the six transgenic lines of poplar. Transgenesis was confirmed through a PCR-based screening of kanamycin-selected regenerants for the presence of both mentioned genes, Sanger sequencing, and tests for detecting the maintained activity of both genes. The transformation efficiency, considering the 100 explants taken originally, was 6%.
Full article
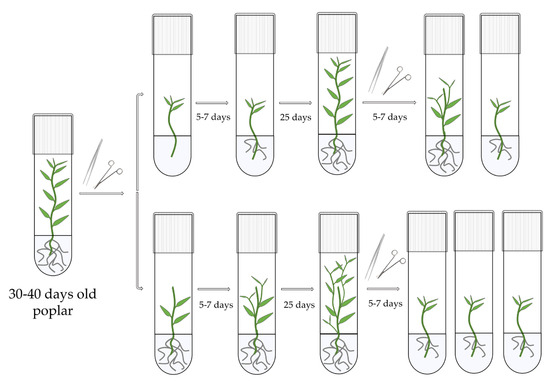
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}