
Validation of a Novel Double Control Quantitative Copy Number PCR Method to Quantify Off-Target Transgene Integration after CRISPR-Induced DNA Modification



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Standard Cell Culture Maintenance of SiMa and IMR90-4 Cell Lines
2.2. Stable Transfection of SiMa and IMR90-4 Cell Lines
2.3. Double-Control Quantitative Copy Number PCR
2.4. Insert Confirmation (PCR)
2.5. Gene-Expression Analysis
2.6. Statistical Analyses
3. Results
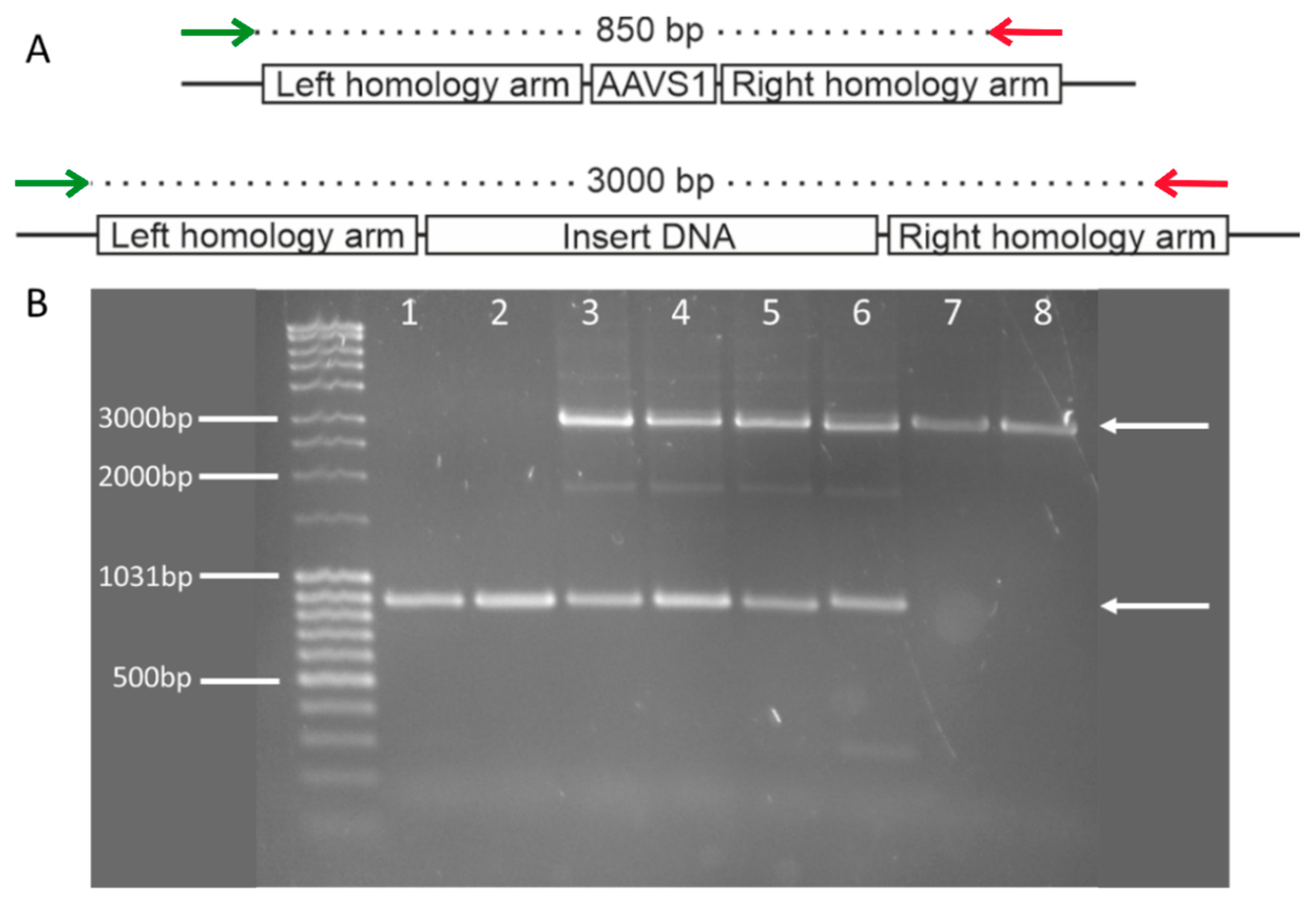
3.1. Insert Confirmation
3.2. Establishment of Technique for Copy-Number Quantification Using qPCR Analysis
3.3. Analysis of Donor DNA Copy Number by dc-qcnPCR
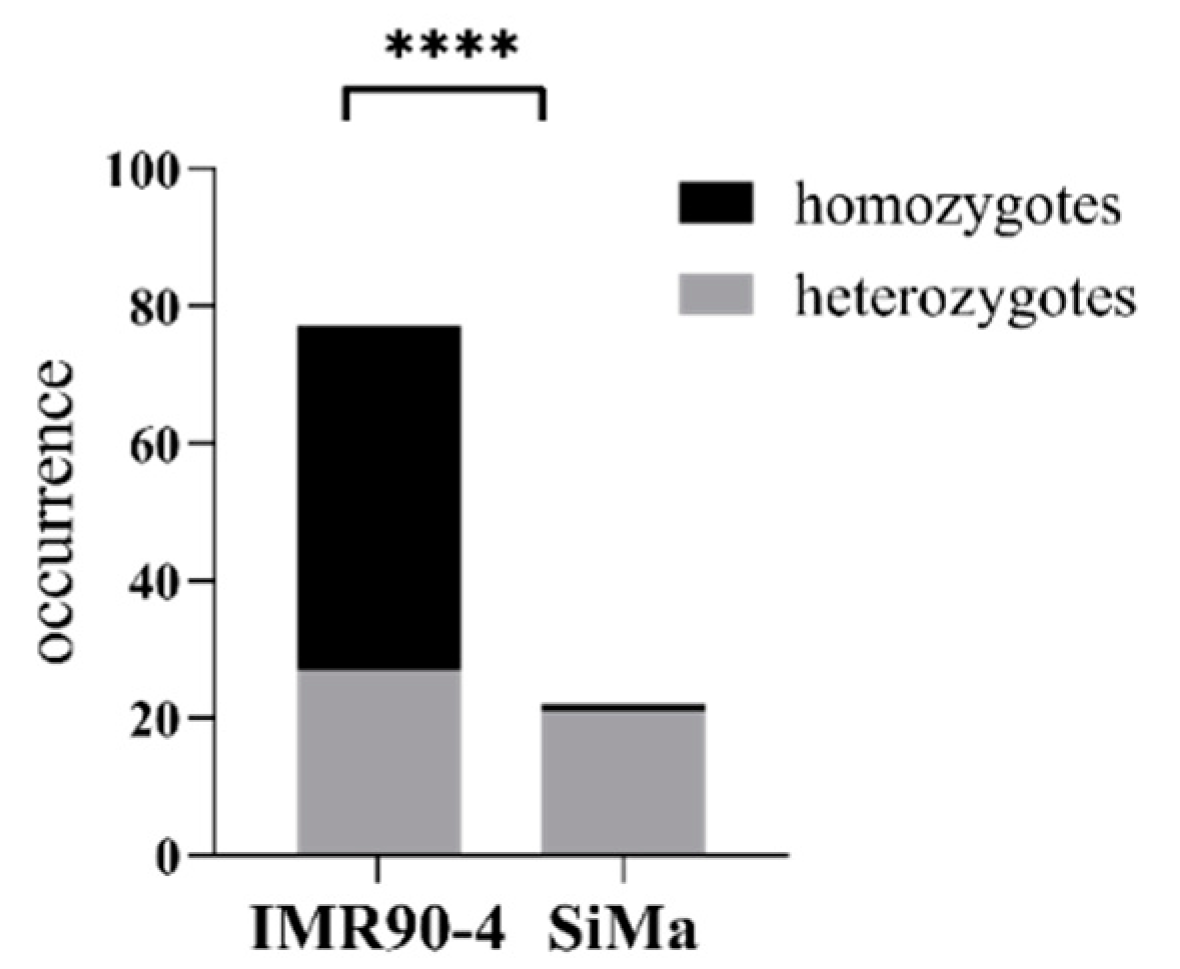
3.4. Defective Homology-Driven Double Strand Repair in SiMa Cells
4. Discussion
4.1. Analysis of Donor DNA Copy Number by dc-qcnPCR
4.2. Defective Homology-Driven Double Strand Repair in SiMa Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, R.; Dhaliwal, H.P.; Kukreja, R.V.; Singh, B.R. The Botulinum Toxin as a Therapeutic Agent: Molecular Structure and Mechanism of Action in Motor and Sensory Systems. Semin. Neurol. 2016, 36, 010–019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitz, S. The botulinum neurotoxin LD50 test—Problems and solutions. ALTEX Altern. Anim. Exp. 2010, 27, 114–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellett, S.; Tepp, W.H.; Johnson, E.A. Critical Analysis of Neuronal Cell and the Mouse Bioassay for Detection of Botulinum Neurotoxins. Toxins 2019, 11, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathe-Neuschäfer-Rube, A.; Neuschäfer-Rube, F.; Genz, L.; Püschel, G. Botulinum neurotoxin dose-dependently inhibits release of neurosecretory vesicle-targeted luciferase from neuronal cells. ALTEX Altern. Anim. Exp. 2015, 32, 201–210. Available online: http://www.altex.org/index.php/altex/article/view/211 (accessed on 7 October 2019).
- Pathe-Neuschäfer-Rube, A.; Neuschäfer-Rube, F.; Haas, G.; Langoth-Fehringer, N.; Püschel, G. Cell-Based Reporter Release Assay to Determine the Potency of Proteolytic Bacterial Neurotoxins. Toxins 2018, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Neuschäfer-Rube, F.; Pathe-Neuschäfer-Rube, A.; Püschel, G.P. Discrimination of the Activity of Low-Affinity Wild-Type and High-Affinity Mutant Recombinant BoNT/B by a SIMA Cell-Based Reporter Release Assay. Toxins 2022, 14, 65. [Google Scholar] [CrossRef]
- Schenke, M.; Prause, H.C.; Bergforth, W.; Przykopanski, A.; Rummel, A.; Klawonn, F.; Seeger, B. Human-Relevant Sensitivity of iPSC-Derived Human Motor Neurons to BoNT/A1 and B1. Toxins 2021, 13, 585. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Oceguera-Yanez, F.; Kim, S.-I.; Matsumoto, T.; Tan, G.W.; Xiang, L.; Hatani, T.; Kondo, T.; Ikeya, M.; Yoshida, Y.; Inoue, H.; et al. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods 2016, 101, 43–55. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.; Chun-Jen, L.C.; Mo, W.; Dai, H.; Park, Y.Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014, 5, 3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherill-Rofe, D.; Rahat, D.; Findlay, S.; Mellul, A.; Guberman, I.; Braun, M.; Bloch, I.; Lalezari, A.; Samiei, A.; Sadreyev, R.; et al. Mapping global and local coevolution across 600 species to identify novel homologous recombination repair genes. Genome Res. 2019, 29, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Frey, M.K.; Pothuri, B. Homologous recombination deficiency (HRD) testing in ovarian cancer clinical practice: A review of the literature. Gynecol. Oncol. Res. Pract. 2017, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, O.; Knobloch, L.; Fornsaglio, J.; Varum, S.; Easley, C.; Schatten, G. DNA Damage Responses in Human Induced Pluripotent Stem Cells and Embryonic Stem Cells. PLoS ONE 2010, 5, e13410. [Google Scholar] [CrossRef] [Green Version]
- Marini, P.; MacLeod, R.A.F.; Treuner, C.; Bruchelt, G.; Böhm, W.; Wolburg, H.; Schweizer, P.; Girgert, R. SiMa, a New Neuroblastoma Cell Line Combining Poor Prognostic Cytogenetic Markers with High Adrenergic Differentiation. Cancer Genet. Cytogenet. 1999, 112, 161–164. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Schenke, M.; Schjeide, B.M.; Püschel, G.P.; Seeger, B. Analysis of Motor Neurons Differentiated from Human Induced Pluripotent Stem Cells for the Use in Cell-Based Botulinum Neurotoxin Activity Assays. Toxins 2020, 12, 276. [Google Scholar] [CrossRef]
- Southern, E.M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 1975, 98, 503–517. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wienert, B.; Wyman, S.K.; Richardson, C.D.; Yeh, C.D.; Akcakaya, P.; Porritt, M.J.; Morlock, M.; Vu, J.T.; Kazane, K.R.; Watry, H.L.; et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 2019, 364, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, H.G.; Penterman, J.; Whitton, H.J.; Spencer, S.J.; Flanagan, N.; Lei Zhang, M.C.; Huang, E.; Khedkar, A.S.; Toomey, J.M.; Shearer, C.A.; et al. Evaluation of Homology-Independent CRISPR-Cas9 Off-Target Assessment Methods. CRISPR J. 2020, 3, 440–453. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Mehrotra, M. PCR-Based Detection of DNA Copy Number Variation. In Clinical Applications of PCR; Methods in Molecular Biology; Luthra, R., Singh, R.R., Patel, K.P., Eds.; Springer: New York, NY, USA, 2016; Volume 1392, pp. 27–32. Available online: http://link.springer.com/10.1007/978-1-4939-3360-0_3 (accessed on 13 May 2022).
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R. Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef]
- Wei, W.; Cheng, Y.; Wang, B. Cancer and Genomic Instability. In Genome Stability; Elsevier: Amsterdam, The Netherlands, 2016; pp. 463–486. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780128033098000276 (accessed on 30 July 2021).
- Carter, M.; Shieh, J.C. Cell Culture Techniques. In Guide to Research Techniques in Neuroscience; Elsevier: Amsterdam, The Netherlands, 2010; pp. 281–296. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780123748492000136 (accessed on 21 May 2021).
- Li, H.; Zimmerman, S.E.; Weyemi, U. Genomic instability and metabolism in cancer. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 241–265. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1937644821000630 (accessed on 22 February 2022).
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Jin, M.H.; Oh, D.Y. ATM in DNA repair in cancer. Pharmacol. Ther. 2019, 203, 107391. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Tichy, E.D. Mechanisms maintaining genomic integrity in embryonic stem cells and induced pluripotent stem cells. Exp. Biol. Med. 2011, 236, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Haber, J.E. DNA Repair: The Search for Homology. BioEssays 2018, 40, 1700229. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int. J. Mol. Sci. 2020, 21, 6461. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [Green Version]
- Jachimowicz, R.D.; Beleggia, F.; Isensee, J.; Velpula, B.B.; Goergens, J.; Bustos, M.A.; Doll, M.A.; Shenoy, A.; Checa-Rodriguez, C.; Wiederstein, J.L.; et al. UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors. Cell 2019, 176, 505–519.e22. [Google Scholar] [CrossRef] [Green Version]
- Ceppi, I.; Howard, S.M.; Kasaciunaite, K.; Pinto, C.; Anand, R.; Seidel, R.; Cejka, P. CtIP promotes the motor activity of DNA2 to accelerate long-range DNA end resection. Proc. Natl. Acad. Sci. USA 2020, 117, 8859–8869. [Google Scholar] [CrossRef]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.; Yim, E.K.; Dai, H.; Jackson, A.P.; van der Burgt, I.; Pan, M.R.; Hu, R.; Li, K.; Lin, S.Y. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nat. Cell Biol. 2009, 11, 865–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Homma, A.; Sayadi, J.; Yang, S.; Ohashi, J.; Takumi, T. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci. Rep. 2016, 6, 19675. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.Z.; Gopalakrishna-Pillai, S.; Nay, S.L.; Park, S.W.; Bates, S.E.; Zeng, X.; Iverson, L.E.; O’Connor, T.R. DNA repair in human pluripotent stem cells is distinct from that in non-pluripotent human cells. PLoS ONE 2012, 7, e30541. [Google Scholar] [CrossRef]
- Fan, J.; Robert, C.; Jang, Y.Y.; Liu, H.; Sharkis, S.; Baylin, S.B.; Rassool, F.V. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutat. Res. Mol. Mech. Mutagen. 2011, 713, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Manghwar, H.; Li, B.; Ding, X.; Hussain, A.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv. Sci. 2020, 7, 1902312. [Google Scholar] [CrossRef]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schjeide, B.-M.M.; Schenke, M.; Seeger, B.; Püschel, G.P. Validation of a Novel Double Control Quantitative Copy Number PCR Method to Quantify Off-Target Transgene Integration after CRISPR-Induced DNA Modification. Methods Protoc. 2022, 5, 43. https://doi.org/10.3390/mps5030043
Schjeide B-MM, Schenke M, Seeger B, Püschel GP. Validation of a Novel Double Control Quantitative Copy Number PCR Method to Quantify Off-Target Transgene Integration after CRISPR-Induced DNA Modification. Methods and Protocols. 2022; 5(3):43. https://doi.org/10.3390/mps5030043
Chicago/Turabian StyleSchjeide, Brit-Maren Michaud, Maren Schenke, Bettina Seeger, and Gerhard Paul Püschel. 2022. "Validation of a Novel Double Control Quantitative Copy Number PCR Method to Quantify Off-Target Transgene Integration after CRISPR-Induced DNA Modification" Methods and Protocols 5, no. 3: 43. https://doi.org/10.3390/mps5030043