
Liquids 2024, 4(2), 382-392; https://doi.org/10.3390/liquids4020019 (registering DOI) - 24 Apr 2024
Abstract
►
Show Figures
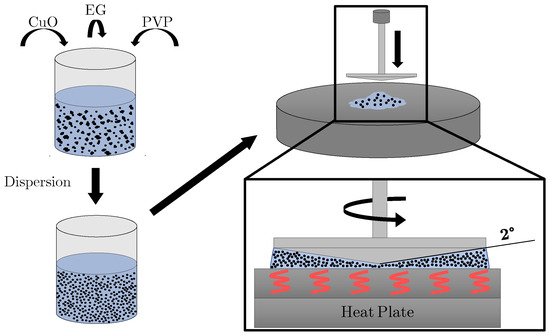
In this article, the particle concentration of finely dispersed copper(II) oxide nanosuspensions as precursors for reductive laser sintering (RLS) is optimized on the basis of rheological investigations. For this metallization process, a smooth, homogeneous and defect-free precursor layer is a prerequisite for adherent
[...] Read more.
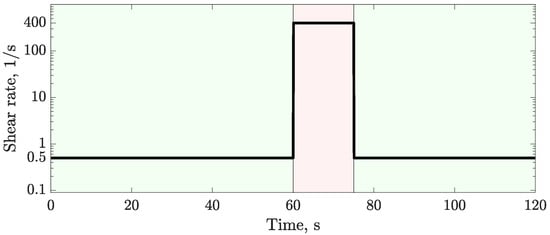
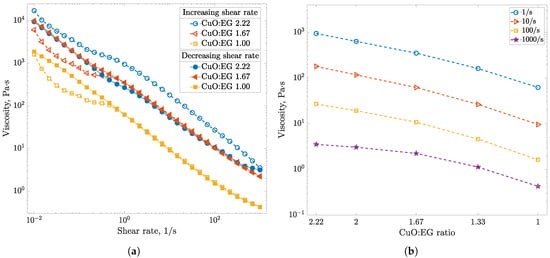
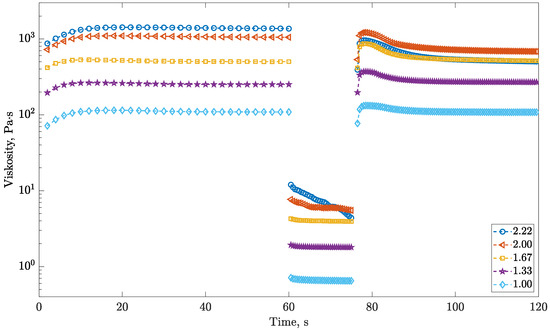
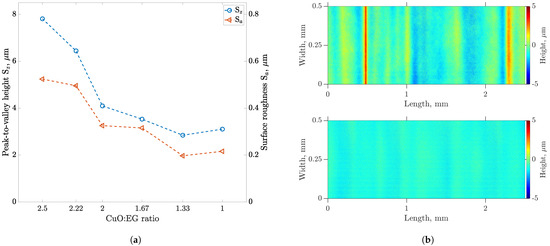
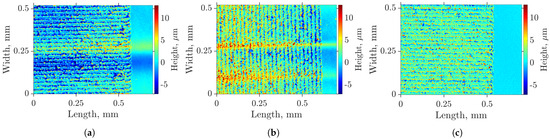
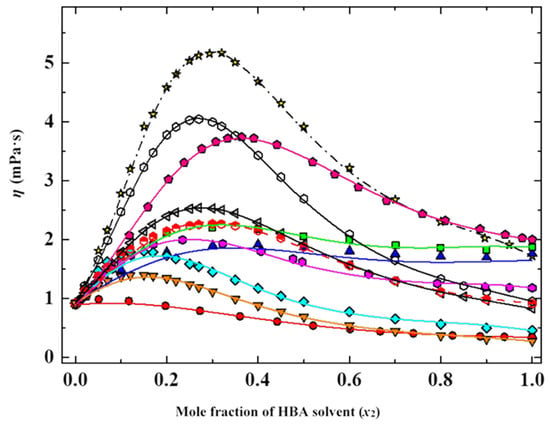
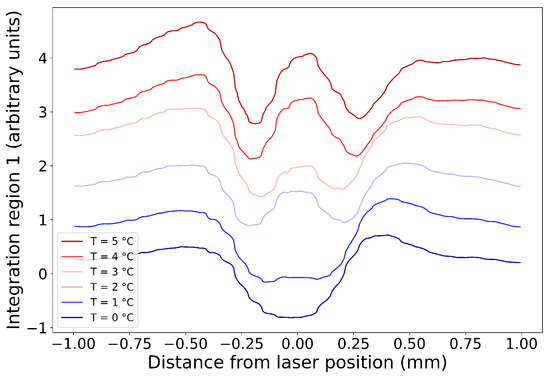
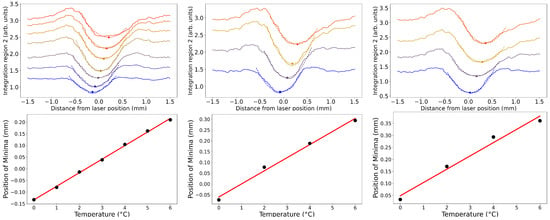
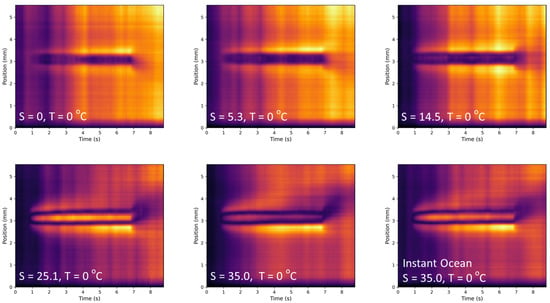
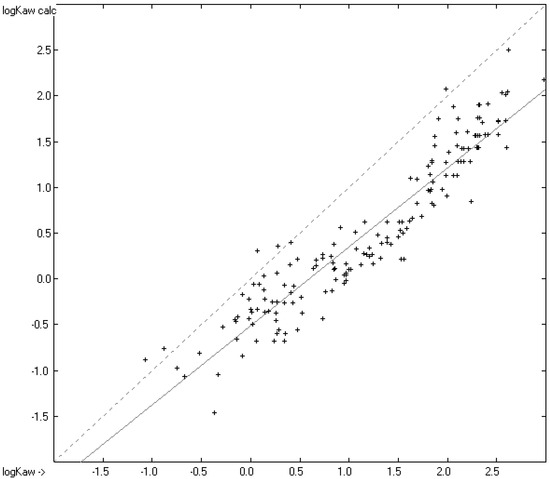
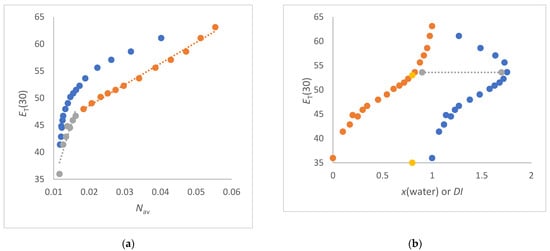
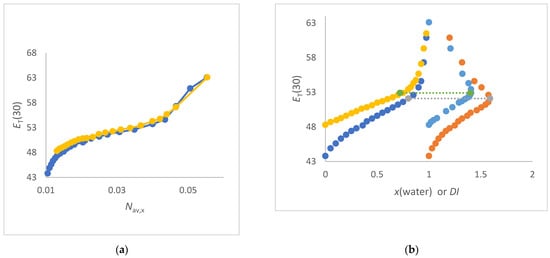
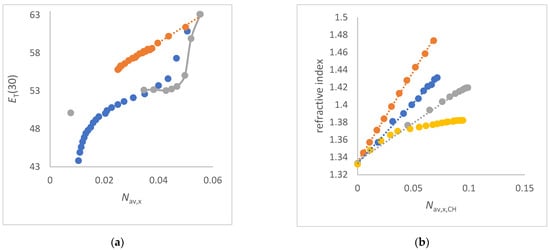
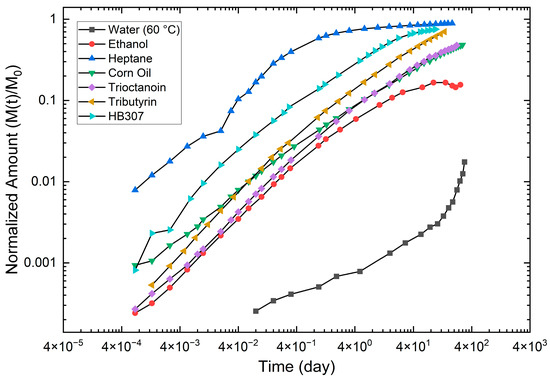
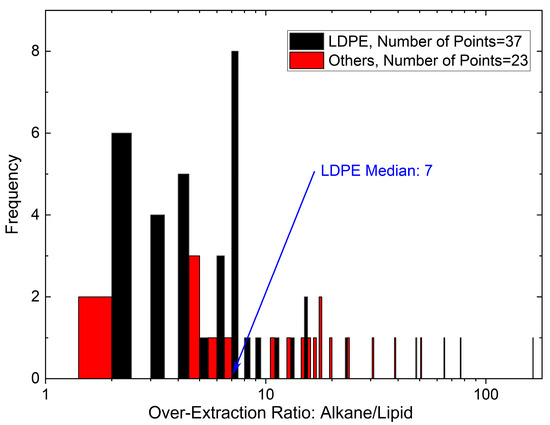
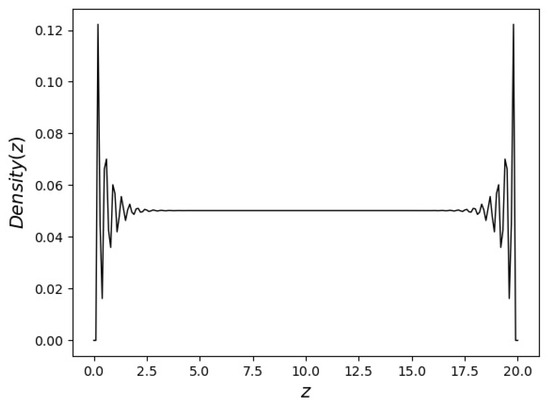
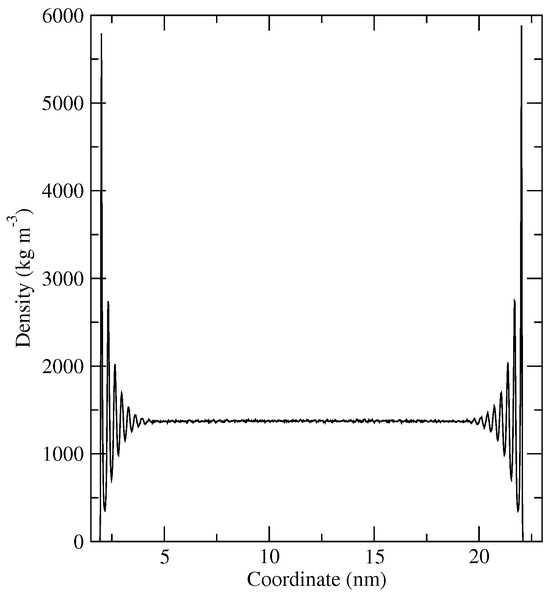
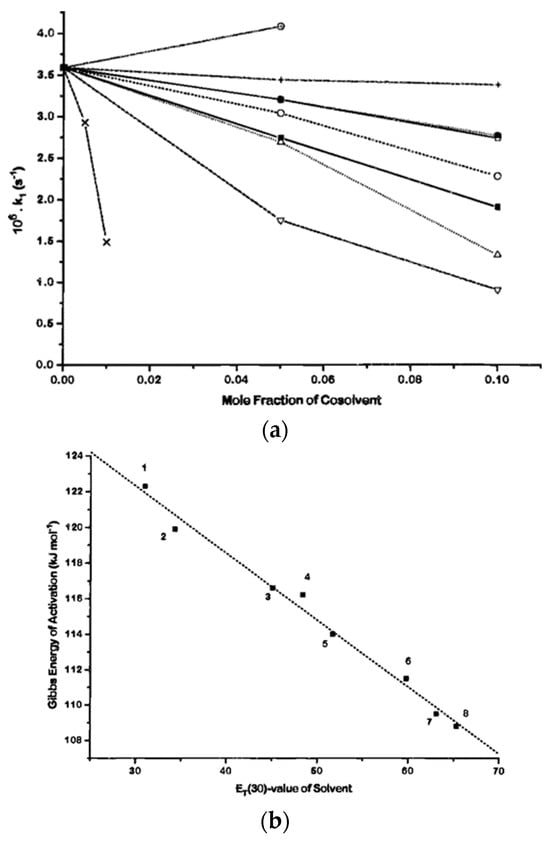
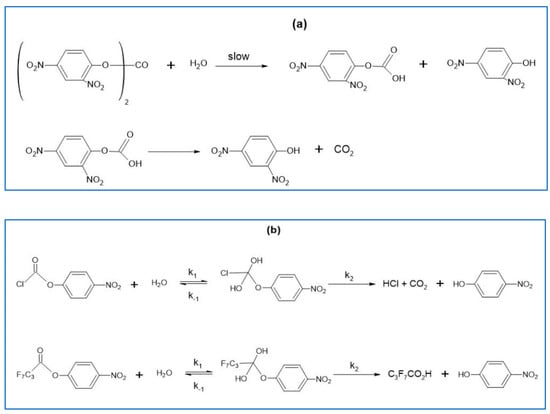
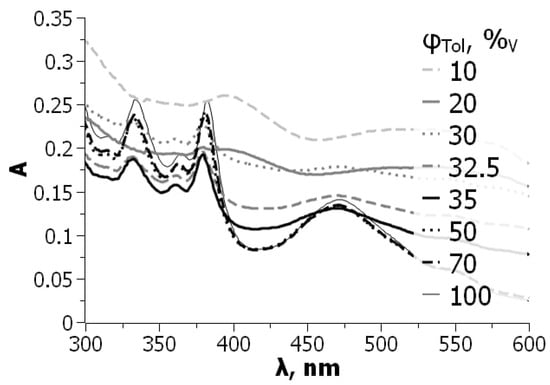
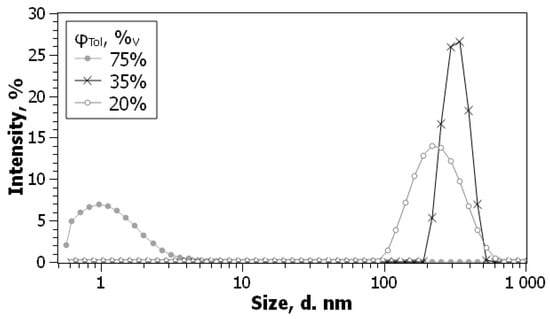
In this article, the particle concentration of finely dispersed copper(II) oxide nanosuspensions as precursors for reductive laser sintering (RLS) is optimized on the basis of rheological investigations. For this metallization process, a smooth, homogeneous and defect-free precursor layer is a prerequisite for adherent and reproducible copper structures. The knowledge of the rheological properties of an ink is crucial for the selection of a suitable coating technology as well as for the adjustment of the ink formulation. Different dilutions of the nanosuspension were examined for their rheological behavior by recording flow curves. A strong shear thinning behavior was found and the viscosity decreases exponentially with increasing dilution. The viscoelastic behavior was investigated by a simulated doctor blade coating process using three-interval thixotropy tests. An overshoot in viscosity is observed, which decreases with increasing thinning of the precursor. As a comparison to these results, doctor blade coating of planar glass and polymer substrates was performed to prepare precursor layers for reductive laser sintering. Surface morphology measurements of the resulting coatings using laser scanning microscopy and rheological tests show that homogeneous precursor layers with constant thickness can be produced at a particle–solvent ratio of 1.33. A too-high particle content results in an irregular coating layer with deep grooves and a peak-to-valley height
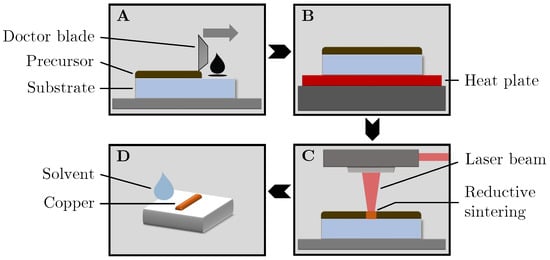
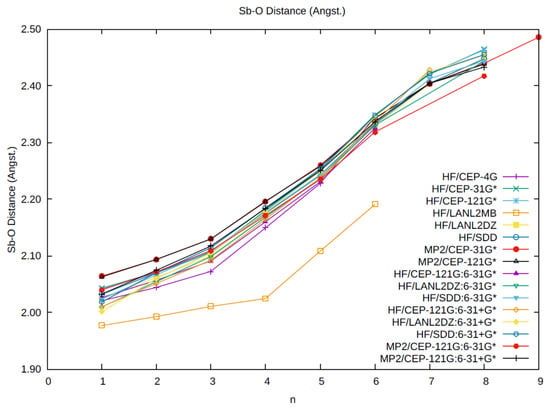
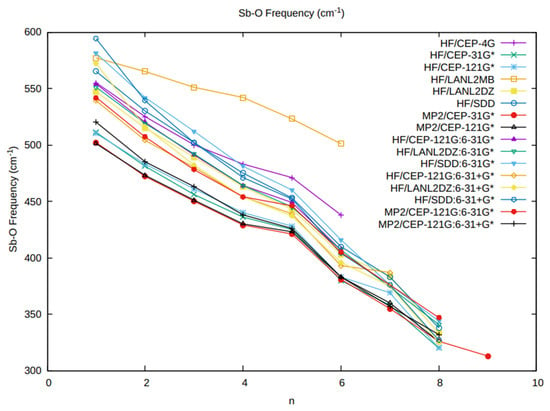
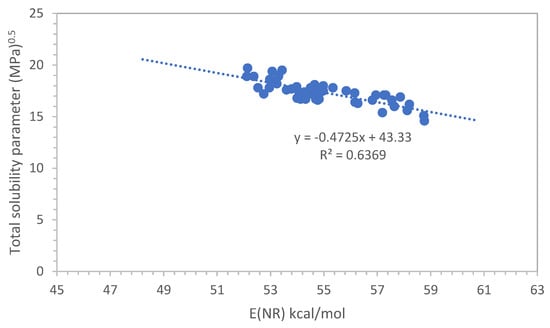
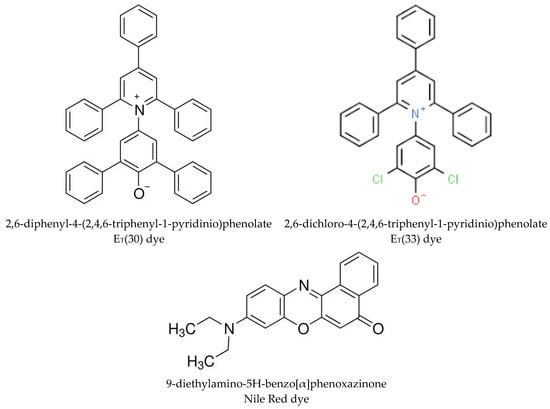
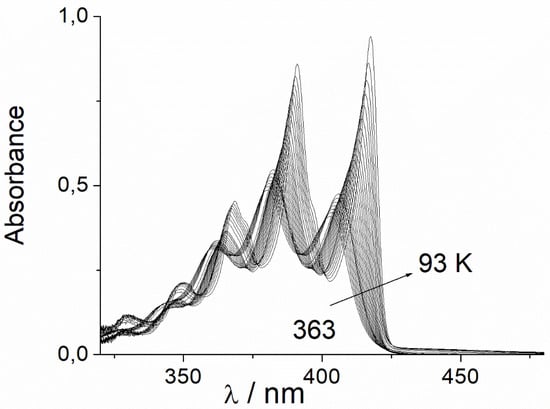
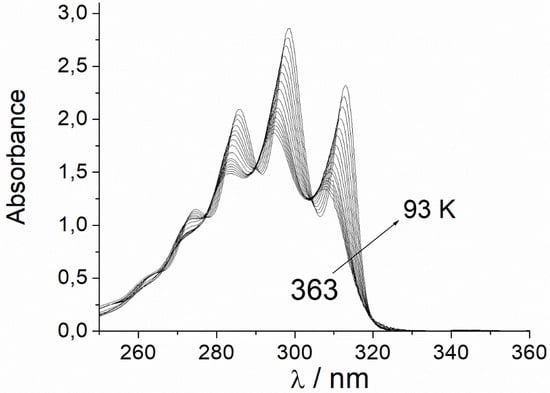
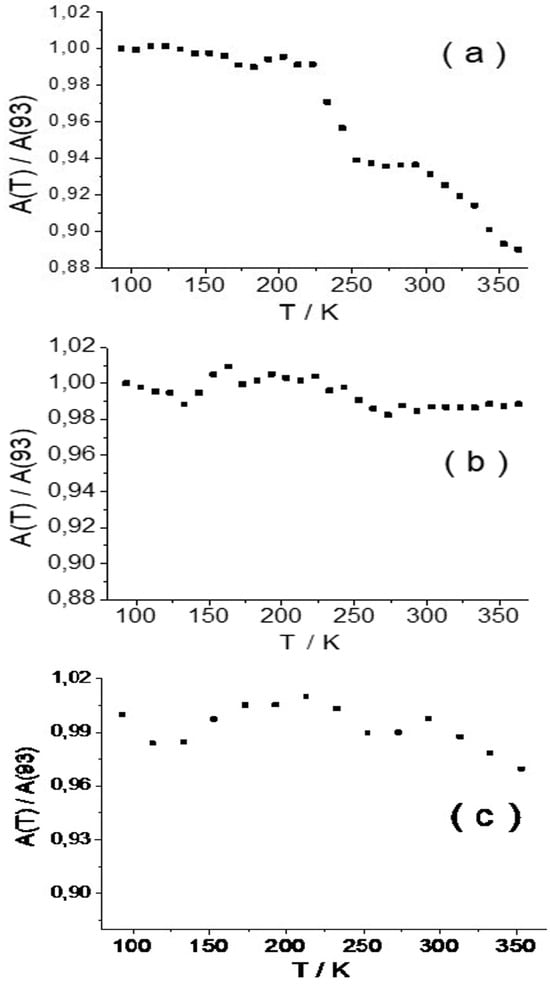
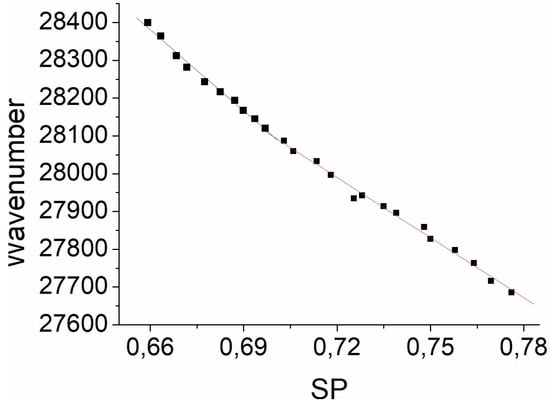
Figure 1
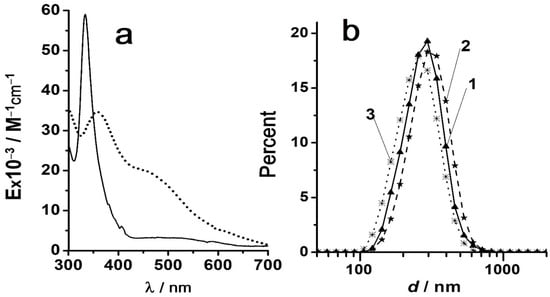


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
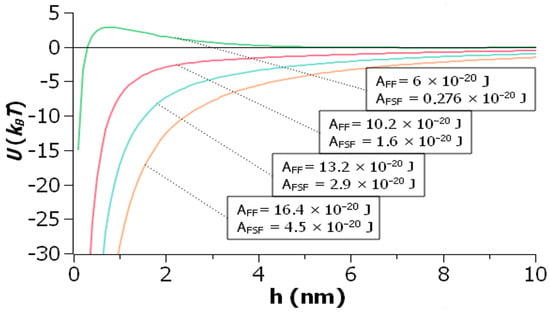{kind=link}
{kind=link}
{kind=link}
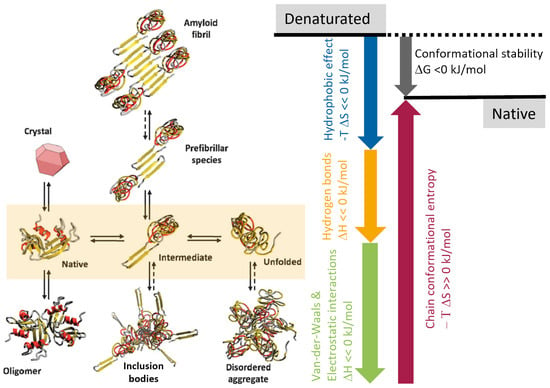{kind=link}
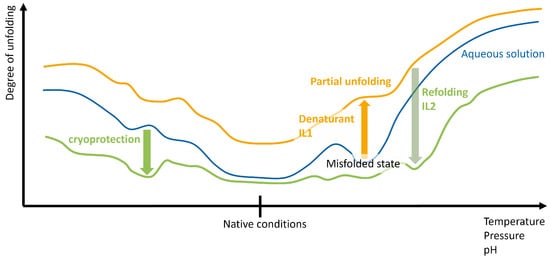{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}