
The Glucocorticoid Receptor’s tau1c Activation Domain 35 Years on—Making Order out of Disorder

Division of Biomolecular and Cellular Medicine, Department of Laboratory Medicine, Karolinska Institutet, SE-141 57 Huddinge, Sweden
Receptors 2024, 3(1), 27-35; https://doi.org/10.3390/receptors3010003
Submission received: 3 November 2023
/
Revised: 6 December 2023
/
Accepted: 2 February 2024
/
Published: 5 February 2024
(This article belongs to the Special Issue Nuclear Receptors: Honorary Special Issue Commemorating the Work of Prof. Jan-Åke Gustafsson)
Abstract
:Almost exactly 35 years after starting to work with the human glucocorticoid receptor (hGR), it is interesting for me to re-evaluate the data and results obtained in the 1980s–1990s with the benefit of current knowledge. What was understood then and how can modern perspectives increase that understanding? The hGR’s tau1c activation domain that we delineated was an enigmatic protein domain. It was apparently devoid of secondary and tertiary protein structures but nonetheless maintained gene activation activity in the absence of other hGR domains, not only in human cells but also in yeast, which is evolutionarily very divergent from humans and which does not contain hGR or other nuclear receptors. We now know that the basic machinery of cells is much more conserved across evolution than was previously thought, so the hGR’s tau1c domain was able to utilise transcription machinery components that were conserved between humans and yeast. Further, we can now see that structure–function aspects of the tau1c domain conform to a general mechanistic framework, such as the acidic exposure model, that has been proposed for many activation domains. As for many transcription factor activation domains, it is now clear that tau1c activity requires regions of transient secondary structure. We now know that there is a tendency for positive Darwinian selection to target intrinsically disordered protein domains. It will be interesting to study the distribution and nature of the many single nucleotide variants of the hGR in this respect.
1. Introduction
Since this issue of Receptors is dedicated to Jan-Åke Gustafsson, it is appropriate to look back to 1988, the year I joined Jan-Åke’s group to study structure–function aspects of the human glucocorticoid receptor (hGR). Much of this work, and more, has been comprehensively reviewed elsewhere in this issue [1]. Equipped with current knowledge and understanding, I am reminded that the interpretation of measured empirical data is a changing entity that is at any time limited by the overall state of knowledge at that time. As frontiers of knowledge move forward, new understanding arising from older results becomes possible. It is with this consideration in mind that this perspectives article is written.
My introduction to the hGR came a couple of years after cloning of its cDNA [2]. Recent work on the purified rat glucocorticoid receptor protein had shown that the N-terminus containing the tau1 activation domain was more sensitive to proteolysis than the C-terminal domains and that it was the main recognition site for the mono-clonal antibodies that had been developed, thus providing circumstantial evidence for an open, less compact protein conformation for the N-terminal part of the receptor compared to other parts [3]. The C-terminal part of the receptor, containing the DNA-binding domain (DBD) and steroid-binding domain (SBD), was more highly conserved with other members of the nuclear receptor family and would later be the part of the protein for which tertiary structures could be determined [4,5,6,7]. Taken together with the fact that very little was known about the role of transcription factor activation domains in gene activation, these observations contributed to the hGR N-terminal domain being regarded as somewhat enigmatic.
2. The Human Glucocorticoid Receptor Can Work in Yeast
A second enigmatic aspect of our work was that the hGR functioned in yeast even though yeasts do not contain any nuclear receptors [8,9,10]. How could this be? The answer was not apparent until over a decade later when the genome sequences of yeast [11] and humans [12] could be compared, showing that many of the 30–40,000 human proteins manifested remarkable levels of homology to the 5000 or so yeast proteins. As more genomes were sequenced, it became apparent that very many protein families are conserved throughout eukaryotes and that a dominant mechanism of evolution appears to have occurred at the level of gene regulation, such that new functionality has arisen by differentially expressing different combinations of a relatively conserved group of protein designs. These conserved protein families include many components of the transcriptional machinery, and thus, yeast provided a simple model system that allowed us to map functional protein domains, select change-of-function mutants in the hGR, and identify functional partner proteins of the hGR [13,14,15,16].
3. Acidic and Hydrophobic Amino Acids Contribute to the hGR’s tau1c Domain Function
Using our model system, we identified a minimal 58-amino-acid region, the tau1 core (tau1c) [13], which maintained most of the activity of the previously identified larger tau1 activation domain [17]. Like the yeast Gal4p and viral VP16 activation domains, the tau1c domain was rich in acidic amino acids, and nuclear magnetic resonance (NMR) studies showed that like them, it lacked detectable secondary or tertiary structures at neutral pH [18]. Mutagenesis experiments showed that while most acidic residues were not important individually, activity was progressively lost as larger numbers of acidic residues were neutralised [19]. Thus, tau1c appeared to fulfil the criteria required to satisfy the acid blob or negative noodle hypothesis that was suggested at the time and whereby activators were thought to function by attracting important transcriptional co-factors through non-specific electrostatic interactions [20].
However, the characterisation of mutations that were selected for decreased or increased activity suggested a more complex structure–function relationship based on the hydrophobicity of key residues, such that reduced activity mutants had reduced hydrophobicity, and in a few cases, increased hydrophobicity increased activity [15]. These activity changes could be correlated with changes in the binding strength to partner proteins involved in gene activation [21]. The importance of hydrophobic residues had been reported earlier for the VP16 activation domain [22]. Notwithstanding the lack of detectable secondary structures in tau1c, these results could be rationalised in the context of hypothetical antipathic alpha helices, where key hydrophobic surfaces could be postulated to make inter- or intra-molecular interactions with other hydropathic surfaces in, for example, partner proteins bound by tau1c [15].
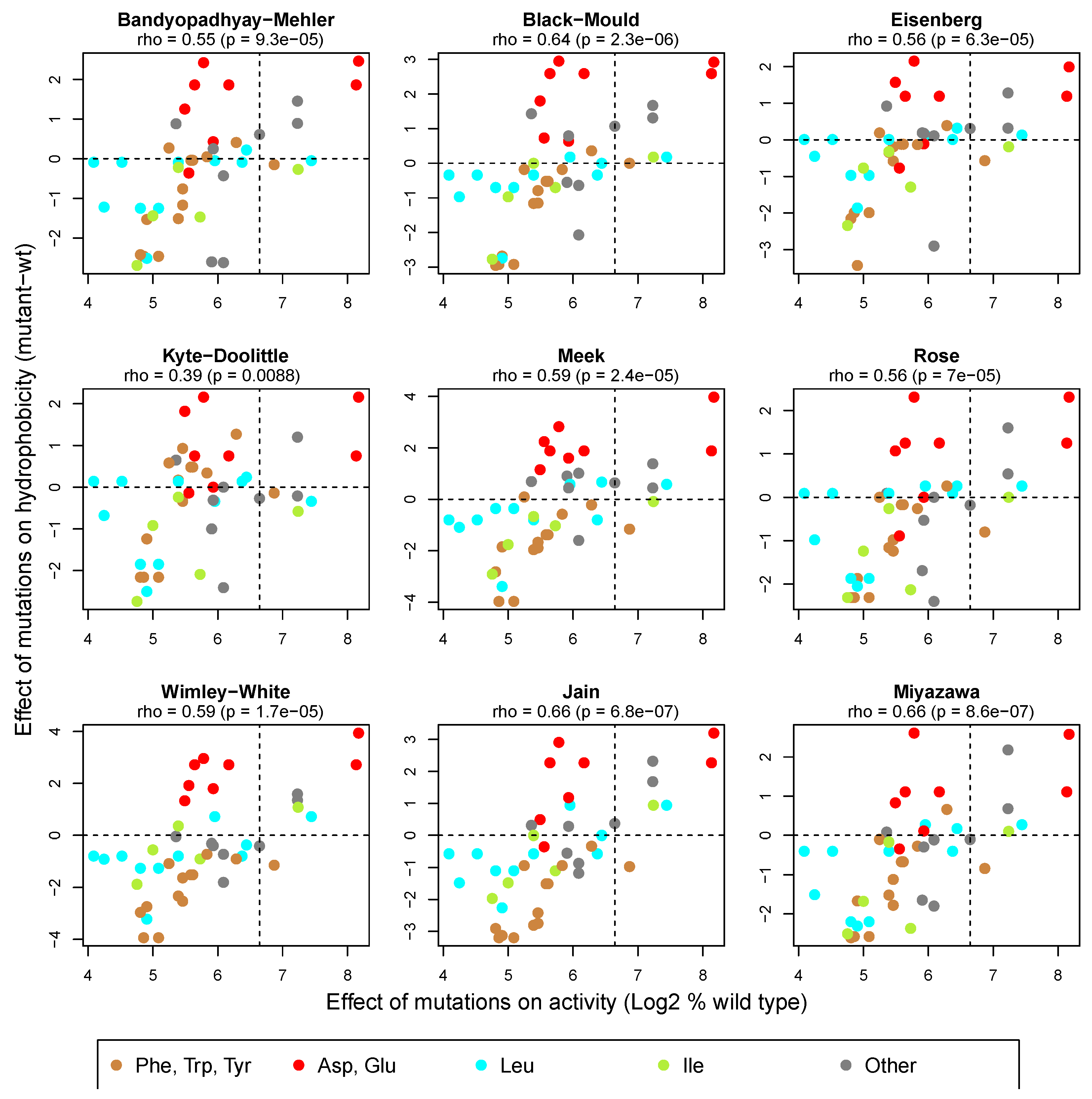
Retrospectively, it is interesting to note a recent genome-wide study of many human transcription factors that appears to establish the generality of the principles that we observed for the tau1c domain [23]. These principles include the importance both of hydrophobic amino acid clusters and acidic residues. An added insight from that systematic study is that the acidic residues are proposed to function by an “acidic exposure” process that presents hydrophobic residues as surface-exposed clusters with the capacity to bind to transcriptional co-activator proteins. These exposed protein conformations include, but are not restricted to, amphipathic helices. A further insight of the study was that leucine residues appear to play a more important “hydrophobic” role that valine and isoleucine, which are often considered to have similar hydrophobicity properties to leucine. Furthermore, it was concluded that some scales of estimated residue hydrophobicity (e.g., the Wimley–White scale [24]) may provide better estimates in this context than others (e.g., the commonly used Kyte–Doolittle scale [25]). Consistently, the effect of mutations on the activity of tau1c is not as well correlated with their effect on hydrophobicity estimated by the Kyte–Doolittle method (Rho = 0.39) compared to a range of other methods, including the Wimley–White method (Rho = 0.59, Figure 1). Residue classes where the effects of mutations on activity and hydrophobicity are not as well correlated include acidic residues and leucine residues (Figure 1, red and cyan points). Substitutions of most acidic residues reduce activity irrespective of their effect on hydrophobicity, consistent with the suggested importance of their acidity in the proposed acidic exposure process required to present hydrophobic surfaces suitable for partner protein interactions. In contrast, the D196Y and E221F substitutions cause a substantial increase in both activity (to 281% and 288%, respectively) and hydrophobicity (by 2.7 and 3.9 units, respectively, using the Wimley–White scale), perhaps because these acidic residues are not required to mediate the acidic exposure process but would increase the extent of the presented hydrophobic surface in their mutated form (see [15]).
Contrary to the proposed special status of leucine residues in relation to isoleucine and valine in activation domains [23], many substitutions of leucine and isoleucine (cyan and green in Figure 1) seem to show a similarly high level of correlation between their effects on activity and hydrophobicity. However, a subset of leucine substitutions shows disproportionally large activity reductions in relation to the corresponding predicted reductions in hydrophobicity. These residues include L236V and L194V, for which activity is reduced to 17% and 23%, respectively, but where the hydrophobicity reduction (Wimley–White scale) is only 0.8 units. Interestingly, this subset is restricted to substitutions of only two of four analysed leucine residues (L194 and L236) in tau1c, suggesting that the special significance that has recently been attributed to leucine residues in activation domains may be restricted to specific molecular contexts.
4. Proteins without Apparent Structure Can Mediate Biological Function
In 1988, some of the most inciteful studies on activation domains had been performed on the yeast Gal4p and viral VP16 activation domains. These were shown to be generally small modular domains that had been referred to as “acid blobs” or “negative noodles” as a result of their predicted lack of ordered structure and the preponderance of acidic amino acid residues in their primary sequences [20]. Biophysical studies of recombinant Gal4p and VP16 activation domains confirmed the lack of ordered structure at neutral pH, although the Gal4p domain did adopt a β-sheet conformation under mildly acidic conditions [28,29]. However, the conventional view at the time was that protein functionality was coupled to tertiary structure formation and thus the idea that an unstructured protein could mediate biological function was not broadly accepted. It would take the bioinformatic discovery, some 15 years later, of many genomic regions, potentially encoding low complexity proteins, for the concept to gain traction that protein functionality could be mediated by so-called intrinsically disordered regions (IDRs) of proteins [30]. Understanding IDRs has since contributed to understanding of inter-protein interactions, membrane-less intra-cellular compartmentalisation involving phase transitions, and many other aspects of molecular biology [31,32].
As intimated above, our original studies showed no evidence of a structured conformation for the isolated tau1c domain in aqueous solution, but alpha helical conformation was detected in the presence of added tri-fluoro-ethanol (TFE), indicating a propensity of tau1c to form alpha helices under some conditions [18]. It is, however, unclear whether TFE conditions in vitro are relevant for physiological contexts in vivo. Importantly, similar effects were observed under more physiological in vitro conditions for larger regions of the hGR containing the tau1c domain [33,34]. Therefore, in light of the current knowledge about IDRs, we recently revisited the issue of structured conformation in the tau1c domain [35]. Analysis of chemical shifts in newly collected NMR data for the tau1c domain in aqueous buffer showed clear evidence of transient alpha-helical conformation in three segments that roughly corresponded to regions identified previously in the presence of TFE [18]. The transient alpha-helical regions have secondary structure propensity (SSP) values around 0.2–0.3, suggesting a dynamic ensemble of conformational forms with 20–30% helical conformation. The existence of transient secondary structure elements has been shown to be a general feature in many of the transcription factor activation domains that have been studied by NMR and which are otherwise considered to be intrinsically disordered [36].
To evaluate the possible relationship between the level of intrinsic protein disorder and tau1c activity, we recently re-evaluated the set of 60 change-of-function mutations affecting tau1c amino acids from our early work [15] in relation to whether their predicted effect on intrinsic disorder correlates to their previously measured effect on gene activation activity [27]. Several software were used to predict the effects of the mutations on intrinsic disorder, peptide-backbone stiffness and protein interaction propensity. Table 1 shows that there is a significant moderate negative correlation between the effects of mutations on tau1c activity and their effects on predicted disorder. In line with this, the effects of mutations on predicted stiffness of the tau1c peptide backbone and protein interaction propensity are positively correlated with their effect on activity. Consistently, molecular dynamics simulations predicted correlation between the effect of many mutations on the stability of a transient alpha-helical region and gene activation activity [27]. Thus, for the tau1 domain and perhaps most other activation domains, there is a correlation between ordered conformation and the level of their activity, perhaps due to the role of transient secondary structure elements in mediating interactions with partner proteins.
5. Coupled Binding and Folding of Activation Domains
It has become apparent that a major context for the formation of an ordered conformation in activation domains is the context of their interaction with partner proteins, a process referred to as coupled binding and folding [46]. We provided early evidence for this by showing the formation of helical conformation upon binding of the intrinsically disordered Myc activation domain to the TATA-binding protein (TBP) [47]. As for the tau1c domain, the cMyc activation domain has been shown to contain transient secondary structure elements and these are stabilised upon the binding of the domain to partner proteins [48].
To understand the mechanisms involved in the coupled binding and folding of the cMyc activation domain to TBP, we performed kinetic experiments [49]. The interaction takes place in two distinct temporal steps. The first step is rapid and sensitive to salt concentration, indicating the importance of ionic interactions. The second step is slower with the rate being correlated with temperature, thus suggesting an enthalpy-driven step involving protein folding. We later showed similar results using the VP16 and Gal4 activation domains binding to TBP, as well as the Swi1 and Snf5 subunits of the Swi/Snf chromatin remodelling complex [50]. We were never able to complete analogous studies with the tau1 domain due to technical difficulties, but others have shown changes in protein conformation upon binding of partner proteins by the hGR N-terminal domain [51,52], suggesting that similar binding mechanisms may apply. From our kinetic studies, we surmised that acidic residues in activation domains likely play an important role in the first interaction step. It is of course possible that they also play an important role in the recently proposed process of acidic exposure [23], which presents clusters of hydrophobic residues in a conformation favouring partner protein interaction during the second step. The acidic exposure process would thus be expected to favour transition from the first binding step to the second, enthalpy-driven step. Kinetic studies have subsequently been conducted for a variety of protein interactions, where different transactivation domains adopt “induced fit” and “preformed conformation capture” interaction strategies to different extents [53,54]. For the disordered N-terminus of the hGR, containing the tau1c domain, there is also evidence for allosteric regulation involving intra-molecular coupled binding and folding interactions [55].
6. Intrinsically Disordered Regions and Evolution
Our early work showed that missense mutations substituting amino acids in the tau1c region of the hGR could either increase or decrease the strength of the response to glucocorticoid signalling [15]. Thus, the intrinsically disordered tau1c domain would be expected to represent a facile target for adaptive mutations that modulate the strength of glucocorticoid response during evolution. We considered that perhaps the conformational flexibility of IDRs could also impart an intrinsic propensity for evolutionary adaptation because their spatial and dynamic flexibility could facilitate the ability to adopt new conformational forms, leading to new functionality. Interestingly, we later showed that yeast transcription factors, which contain a high content of IDRs, appear to have been preferred targets for adaptation processes during evolution [56]. Furthermore, in a genome-wide study of fully sequenced genomes from 64 wild and domesticated yeast strains (Saccharomyces cerevisiae or the very closely related Saccharomyces paradoxus), we showed that codon sites predicted to be under positive Darwinian selection were about three times more likely to occur in regions encoding predicted IDRs than in codon sites in regions encoding predicted secondary structure elements of proteins [57]. Similar findings have been reported in a broad range of other organism groups, including humans, as exemplified in [58,59,60,61]. Thus, the intrinsic evolvability of IDRs in transcription factors may provide part of the explanation for why evolution throughout eukaryotes has in part been driven by increased regulatory complexity rather than, e.g., the advent of new protein classes. A large number of single nucleotide variants have been identified for the hGR, with many occurring in the disordered N-terminal domain containing the tau1c activation domain, but the selective/functional significance of these variants remains to be systematically evaluated [62].
7. Conclusions and Future Perspectives
The main technical developments that are relevant for the re-evaluation of our early work on the tau1c activation domain of the hGR are large-scale sequencing of DNA allowing for the sequencing of whole genomes, extended and refined spectroscopic methods for studying protein conformation, and the development of software for the analysis of genomes and proteomes, as well as protein sequences and conformations. As described in this perspectives article, these developments have allowed for (i) the rationalisation of how human proteins are able to function in yeast, (ii) the understanding of structure–function aspects of tau1c within a generalised framework based on systematic studies of many transcription factors and (iii) the potential for understanding hGR variants within the scope of a better general understanding of the relationship between functional adaptation/conservation and protein conformation characteristics.
The future will likely see the continued development of experimental and computer-based methods that will offer further understanding of IDRs, like the tau1c domain, in the context of the ensembles of conformational states that together define them. Such approaches will likely further the understanding of aspects such as the role of transient regions of secondary structure within IDRs, as well as the ability of individual IDRs to transition between different liquid or solid phases. Correlating the effects of natural or experimentally induced mutations on such biophysical aspects with their effects on biological function will continue to provide an important approach to link biophysical phenomena to protein functionality.
Funding
This research was funded by the Swedish Cancer Society, grant number: 21 1716 Pj 01 H.
Data Availability Statement
All analysed data have been published previously in sources as indicated.
Conflicts of Interest
The author declares no conflicts of interest.
References
- McEwan, I.J. From Antibodies to Crystals: Understanding the Structure of the Glucocorticoid Receptor and Related Proteins. Receptors 2023, 2, 166–175. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary Structure and Expression of a Functional Human Glucocorticoid Receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef]
- Gustafsson, J.-Å.; Carlstedt-Duke, J.; Okret, S.; Wikström, A.-C.; Wrange, Ö.; Payvar, F.; Yamamoto, K. Structure and Specific DNA Binding of the Rat Liver Glucocorticoid Receptor. J. Steroid Biochem. 1984, 20, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Härd, T.; Kellenbach, E.; Boelens, R.; Maler, B.A.; Dahlman, K.; Freedman, L.P.; Carlstedt-Duke, J.; Yamamoto, K.R.; Gustafsson, J.A.; Kaptein, R. Solution Structure of the Glucocorticoid Receptor DNA-Binding Domain. Science 1990, 249, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Baumann, H.; Paulsen, K.; Kovacs, H.; Berglund, H.; Wright, A.P.; Gustafsson, J.A.; Hard, T. Refined Solution Structure of the Glucocorticoid Receptor DNA-Binding Domain. Biochemistry 1993, 32, 13463–13471. [Google Scholar] [CrossRef]
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M.; et al. Crystal Structure of the Glucocorticoid Receptor Ligand Binding Domain Reveals a Novel Mode of Receptor Dimerization and Coactivator Recognition. Cell 2002, 110, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Kauppi, B.; Jakob, C.; Färnegårdh, M.; Yang, J.; Ahola, H.; Alarcon, M.; Calles, K.; Engström, O.; Harlan, J.; Muchmore, S.; et al. The Three-Dimensional Structures of Antagonistic and Agonistic Forms of the Glucocorticoid Receptor Ligand-Binding Domain: RU-486 Induces a Transconformation That Leads to Active Antagonism. J. Biol. Chem. 2003, 278, 22748–22754. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Yamamoto, K.R. Mammalian Glucocorticoid Receptor Derivatives Enhance Transcription in Yeast. Science 1988, 241, 965–967. [Google Scholar] [CrossRef]
- Wright, A.P.H.; Carlstedtduke, J.; Gustafsson, J.A. Ligand-Specific Transactivation of Gene-Expression by a Derivative of the Human Glucocorticoid Receptor Expressed in Yeast. J. Biol. Chem. 1990, 265, 14763–14769. [Google Scholar] [CrossRef]
- Wright, A.P.H.; Gustafsson, J.A. Glucocorticoid-Specific Gene Activation by the Intact Human Glucocorticoid Receptor Expressed in Yeast—Glucocorticoid Specificity Depends on Low-Level Receptor Expression. J. Biol. Chem. 1992, 267, 11191–11195. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 Genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Dahlman-Wright, K.; Almlof, T.; McEwan, I.J.; Gustafsson, J.A.; Wright, A.P. Delineation of a Small Region within the Major Transactivation Domain of the Human Glucocorticoid Receptor That Mediates Transactivation of Gene Expression. Proc. Natl. Acad. Sci. USA 1994, 91, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.P.; McEwan, I.J.; Dahlman-Wright, K.; Gustafsson, J.A. High Level Expression of the Major Transactivation Domain of the Human Glucocorticoid Receptor in Yeast Cells Inhibits Endogenous Gene Expression and Cell Growth. Mol. Endocrinol. 1991, 5, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Almlof, T.; Gustafsson, J.A.; Wright, A.P. Role of Hydrophobic Amino Acid Clusters in the Transactivation Activity of the Human Glucocorticoid Receptor. Mol. Cell Biol. 1997, 17, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, A.; Almlof, T.; Ford, J.; McEwan, I.J.; Gustafsson, J.A.; Wright, A.P. Role of the Ada Adaptor Complex in Gene Activation by the Glucocorticoid Receptor. Mol. Cell Biol. 1997, 17, 3065–3073. [Google Scholar] [CrossRef]
- Giguère, V.; Hollenberg, S.M.; Rosenfeld, M.G.; Evans, R.M. Functional Domains of the Human Glucocorticoid Receptor. Cell 1986, 46, 645–652. [Google Scholar] [CrossRef]
- Dahlman-Wright, K.; Baumann, H.; McEwan, I.J.; Almlof, T.; Wright, A.P.; Gustafsson, J.A.; Hard, T. Structural Characterization of a Minimal Functional Transactivation Domain from the Human Glucocorticoid Receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 1699–1703. [Google Scholar] [CrossRef]
- Almlof, T.; Wright, A.P.; Gustafsson, J.A. Role of Acidic and Phosphorylated Residues in Gene Activation by the Glucocorticoid Receptor. J. Biol. Chem. 1995, 270, 17535–17540. [Google Scholar] [CrossRef]
- Sigler, P.B. Transcriptional Activation. Acid Blobs and Negative Noodles. Nature 1988, 333, 210–212. [Google Scholar] [CrossRef]
- Almlof, T.; Wallberg, A.E.; Gustafsson, J.A.; Wright, A.P. Role of Important Hydrophobic Amino Acids in the Interaction between the Glucocorticoid Receptor Tau 1-Core Activation Domain and Target Factors. Biochemistry 1998, 37, 9586–9594. [Google Scholar] [CrossRef]
- Cress, W.D.; Triezenberg, S.J. Critical Structural Elements of the VP16 Transcriptional Activation Domain. Science 1991, 251, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Staller, M.V.; Ramirez, E.; Kotha, S.R.; Holehouse, A.S.; Pappu, R.V.; Cohen, B.A. Directed Mutational Scanning Reveals a Balance between Acidic and Hydrophobic Residues in Strong Human Activation Domains. Cell Syst. 2022, 13, 334–345.e5. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S.H. Experimentally Determined Hydrophobicity Scale for Proteins at Membrane Interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Waibl, F.; Fernández-Quintero, M.L.; Wedl, F.S.; Kettenberger, H.; Georges, G.; Liedl, K.R. Comparison of Hydrophobicity Scales for Predicting Biophysical Properties of Antibodies. Front. Mol. Biosci. 2022, 9, 960194. [Google Scholar] [CrossRef] [PubMed]
- Salamanova, E.; Costeira-Paulo, J.; Han, K.-H.; Kim, D.-H.; Nilsson, L.; Wright, A.P.H. A Subset of Functional Adaptation Mutations Alter Propensity for α-Helical Conformation in the Intrinsically Disordered Glucocorticoid Receptor Tau1core Activation Domain. Biochim. Biophys. Acta BBA—Gen. Subj. 2018, 1862, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, P.; Williams, G. Structural Studies of the Acidic Transactivation Domain of the Vmw65 Protein of Herpes Simplex Virus Using 1H NMR. Biochemistry 1992, 31, 4150–4156. [Google Scholar] [CrossRef]
- Van Hoy, M.; Leuther, K.K.; Kodadek, T.; Johnston, S.A. The Acidic Activation Domains of the GCN4 and GAL4 Proteins Are Not Alpha Helical but Form Beta Sheets. Cell 1993, 72, 587–594. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and Combining Predictors of Mostly Disordered Proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Kulkarni, P.; Bhattacharya, S.; Achuthan, S.; Behal, A.; Jolly, M.K.; Kotnala, S.; Mohanty, A.; Rangarajan, G.; Salgia, R.; Uversky, V. Intrinsically Disordered Proteins: Critical Components of the Wetware. Chem. Rev. 2022, 122, 6614–6633. [Google Scholar] [CrossRef]
- Baskakov, I.V.; Kumar, R.; Srinivasan, G.; Ji, Y.S.; Bolen, D.W.; Thompson, E.B. Trimethylamine N-Oxide-Induced Cooperative Folding of an Intrinsically Unfolded Transcription-Activating Fragment of Human Glucocorticoid Receptor. J. Biol. Chem. 1999, 274, 10693–10696. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; Arnott, J.A.; Kumar, R. Naturally Occurring Osmolyte, Trehalose Induces Functional Conformation in an Intrinsically Disordered Activation Domain of Glucocorticoid Receptor. PLoS ONE 2011, 6, e19689. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Wright, A.; Han, K.H. An NMR Study on the Intrinsically Disordered Core Transactivation Domain of Human Glucocorticoid Receptor. BMB Rep. 2017, 50, 522–527. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, D.-H.; Han, J.J.; Cha, E.-J.; Lim, J.-E.; Cho, Y.-J.; Lee, C.; Han, K.-H. Understanding Pre-Structured Motifs (PreSMos) in Intrinsically Unfolded Proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web Server for the Prediction of Intrinsically Unstructured Regions of Proteins Based on Estimated Energy Content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K. Predicting Intrinsic Disorder from Amino Acid Sequence. Proteins Struct. Funct. Bioinforma. 2003, 53, 566–572. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence Complexity of Disordered Protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting Heterogeneous Sequence Properties Improves Prediction of Protein Disorder. Proteins Struct. Funct. Bioinforma. 2005, 61, 176–182. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A Meta-Predictor of Intrinsically Disordered Amino Acids. Biochim. Biophys. Acta BBA—Proteins Proteomics 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and Fast Prediction of Protein Disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. The DynaMine Webserver: Predicting Protein Dynamics from Sequence. Nucleic Acids Res. 2014, 42, W264–W270. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: Web Server for Predicting Protein Binding Regions in Disordered Proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Disfani, F.M.; Hsu, W.-L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a Computational Tool for Sequence-Based Prediction and Characterization of Short Disorder-to-Order Transitioning Binding Regions in Proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically Unstructured Proteins and Their Functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- McEwan, I.J.; Dahlman-Wright, K.; Ford, J.; Wright, A.P. Functional Interaction of the C-Myc Transactivation Domain with the TATA Binding Protein: Evidence for an Induced Fit Model of Transactivation Domain Folding. Biochemistry 1996, 35, 9584–9593. [Google Scholar] [CrossRef]
- Andresen, C.; Helander, S.; Lemak, A.; Farès, C.; Csizmok, V.; Carlsson, J.; Penn, L.Z.; Forman-Kay, J.D.; Arrowsmith, C.H.; Lundström, P.; et al. Transient Structure and Dynamics in the Disordered C-Myc Transactivation Domain Affect Bin1 Binding. Nucleic Acids Res. 2012, 40, 6353–6366. [Google Scholar] [CrossRef]
- Hermann, S.; Berndt, K.D.; Wright, A.P. How Transcriptional Activators Bind Target Proteins. J. Biol. Chem. 2001, 276, 40127–40132. [Google Scholar] [CrossRef]
- Ferreira, M.E.; Hermann, S.; Prochasson, P.; Workman, J.L.; Berndt, K.D.; Wright, A.P. Mechanism of Transcription Factor Recruitment by Acidic Activators. J. Biol. Chem. 2005, 280, 21779–21784. [Google Scholar] [CrossRef]
- Kumar, R.; Volk, D.E.; Li, J.; Lee, J.C.; Gorenstein, D.G.; Thompson, E.B. TATA Box Binding Protein Induces Structure in the Recombinant Glucocorticoid Receptor AF1 Domain. Proc. Natl. Acad. Sci. USA 2004, 101, 16425–16430. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; Awasthi, S.; Guo, C.; Goswami, D.; Ling, J.; Griffin, P.R.; Simons, S.S.; Kumar, R. Binding of the N-Terminal Region of Coactivator TIF2 to the Intrinsically Disordered AF1 Domain of the Glucocorticoid Receptor Is Accompanied by Conformational Reorganizations. J. Biol. Chem. 2012, 287, 44546–44560. [Google Scholar] [CrossRef] [PubMed]
- Shammas, S.L.; Crabtree, M.D.; Dahal, L.; Wicky, B.I.M.; Clarke, J. Insights into Coupled Folding and Binding Mechanisms from Kinetic Studies*. J. Biol. Chem. 2016, 291, 6689–6695. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.C.; Kuravsky, M.; Shammas, S.L.; Matthews, J.M. Binding and Folding in Transcriptional Complexes. Curr. Opin. Struct. Biol. 2021, 66, 156–162. [Google Scholar] [CrossRef] [PubMed]
- White, J.T.; Li, J.; Grasso, E.; Wrabl, J.O.; Hilser, V.J. Ensemble Allosteric Model: Energetic Frustration within the Intrinsically Disordered Glucocorticoid Receptor. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170175. [Google Scholar] [CrossRef] [PubMed]
- Beskow, A.; Wright, A.P. Comparative Analysis of Regulatory Transcription Factors in Schizosaccharomyces Pombe and Budding Yeasts. Yeast 2006, 23, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Grahn, M.; Wright, A.P. Proteome-Wide Evidence for Enhanced Positive Darwinian Selection within Intrinsically Disordered Regions in Proteins. Genome Biol. 2011, 12, R65. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, A.; Bockwoldt, M.; Cooney, C.R.; Heiland, I.; Gossmann, T.I. Human Long Intrinsically Disordered Protein Regions Are Frequent Targets of Positive Selection. Genome Res. 2018, 28, 975–982. [Google Scholar] [CrossRef]
- Ridout, K.E.; Dixon, C.J.; Filatov, D.A. Positive Selection Differs between Protein Secondary Structure Elements in Drosophila. Genome Biol. Evol. 2010, 2, 166–179. [Google Scholar] [CrossRef]
- Abrams, A.J.; Cannatella, D.C.; Hillis, D.M.; Sawyer, S.L. Recent Host-Shifts in Ranaviruses: Signatures of Positive Selection in the Viral Genome. J. Gen. Virol. 2013, 94, 2082–2093. [Google Scholar] [CrossRef]
- Dunning, L.T.; Dennis, A.B.; Thomson, G.; Sinclair, B.J.; Newcomb, R.D.; Buckley, T.R. Positive Selection in Glycolysis among Australasian Stick Insects. BMC Evol. Biol. 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Leventhal, S.M.; Lim, D.; Green, T.L.; Cantrell, A.E.; Cho, K.; Greenhalgh, D.G. Uncovering a Multitude of Human Glucocorticoid Receptor Variants: An Expansive Survey of a Single Gene. BMC Genet. 2019, 20, 16. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Extent of correlation between the effect of tau1c substitution mutations on gene activation activity and their effect on hydrophobicity using different scales to estimate the hydrophobicity of amino acid residues. Scatter plots showing the effect of 45 single residue substitution mutants on hydrophobicity (wild-type value subtracted from the mutant residue value) as a function of their effect on activity (mutant activity as a percentage of wild type). The logarithmic scale is used to improve data point separation. Points are coloured according to the identity of the substituted residue in the wild-type tau1c protein. Dotted lines divide the plot into quadrants based on X and Y values for the wild-type tau1c domain. Rho correlation coefficients were calculated using Spearman’s rank correlation tests (two-sided), and p-values testing the null hypothesis that Rho = 0 are shown. Amino acid hydrophobicity scores for the different methods were taken from Table 2 in [26]. Values for the effect of mutations on tau1c activity were taken from the previously published Supplementary Table 1 in [27].
Figure 1.
Extent of correlation between the effect of tau1c substitution mutations on gene activation activity and their effect on hydrophobicity using different scales to estimate the hydrophobicity of amino acid residues. Scatter plots showing the effect of 45 single residue substitution mutants on hydrophobicity (wild-type value subtracted from the mutant residue value) as a function of their effect on activity (mutant activity as a percentage of wild type). The logarithmic scale is used to improve data point separation. Points are coloured according to the identity of the substituted residue in the wild-type tau1c protein. Dotted lines divide the plot into quadrants based on X and Y values for the wild-type tau1c domain. Rho correlation coefficients were calculated using Spearman’s rank correlation tests (two-sided), and p-values testing the null hypothesis that Rho = 0 are shown. Amino acid hydrophobicity scores for the different methods were taken from Table 2 in [26]. Values for the effect of mutations on tau1c activity were taken from the previously published Supplementary Table 1 in [27].


{kind=link}
Table 1.
Correlation between the effect of tau1c change-of-activity mutations on gene activation activity and their effect on predicted intrinsic disorder, peptide backbone rigidity and protein interaction propensity.
Table 1.
Correlation between the effect of tau1c change-of-activity mutations on gene activation activity and their effect on predicted intrinsic disorder, peptide backbone rigidity and protein interaction propensity.
| Predictor | Spearman’s Rho Value 10 | p-Value 11 |
|---|---|---|
| Intrinsic disorder | ||
| IUPred (long) 1 | −0.53 | 1.24 × 10−5 |
| IUPred (short) 1 | −0.50 | 4.81 × 10−5 |
| VSL2 2 | −0.42 | 8.07 × 10−4 |
| VL_XT 3 | −0.42 | 9.37 × 10−4 |
| VSL3 4 | −0.41 | 1.07 × 10−3 |
| Pondr_Fit 5 | −0.43 | 6.32 × 10−4 |
| Espritz (xray) 6 | −0.42 | 9.41 × 10−4 |
| Espritz (nmr) 6 | −0.44 | 5.12 × 10−4 |
| Petide backbone rigidity | ||
| Dynamine 7 | 0.39 | 2.08 × 10−3 |
| Protein interaction propensity | ||
| ANCHOR 8 | 0.62 | 1.50 × 10−7 |
| MoRFpred 9 | 0.44 | 2.08 × 10−3 |
1. Dosztanyi et al. [37]; 2. Obradovic et al. [38]; 3. Romero et al. [39]; 4. Obradovic et al. [40]; 5. Xue et al. [41]; 6. Walsh et al. [42]; 7. Cilia et al. [43]; 8. Dosztanyi et al. [44]; 9. Disfani et al. [45]; 10. derived from Spearman’s rank correlation analysis, two-sided; 11. null hypothesis—Rho = 0.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wright, A.P.H. The Glucocorticoid Receptor’s tau1c Activation Domain 35 Years on—Making Order out of Disorder. Receptors 2024, 3, 27-35. https://doi.org/10.3390/receptors3010003
AMA Style
Wright APH. The Glucocorticoid Receptor’s tau1c Activation Domain 35 Years on—Making Order out of Disorder. Receptors. 2024; 3(1):27-35. https://doi.org/10.3390/receptors3010003
Chicago/Turabian StyleWright, Anthony P. H. 2024. "The Glucocorticoid Receptor’s tau1c Activation Domain 35 Years on—Making Order out of Disorder" Receptors 3, no. 1: 27-35. https://doi.org/10.3390/receptors3010003