
A Game with a Purpose: Designing Structural Modifications in Polymyxin B to Face Multi-Drug Resistant Bacteria †


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
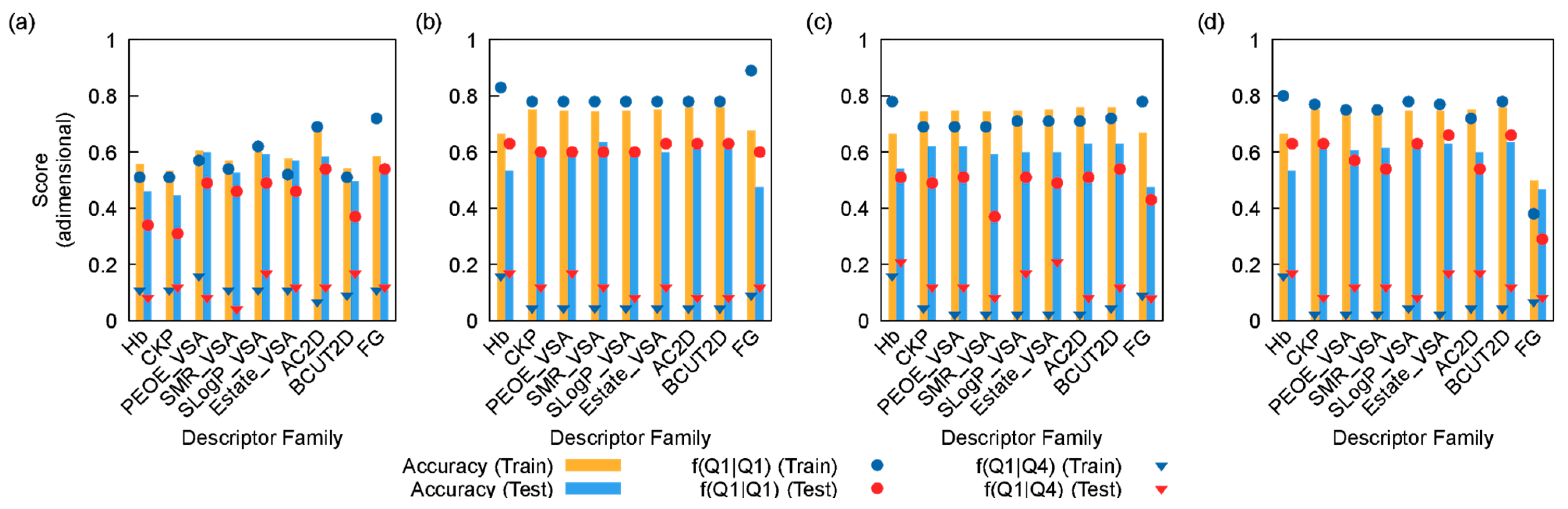
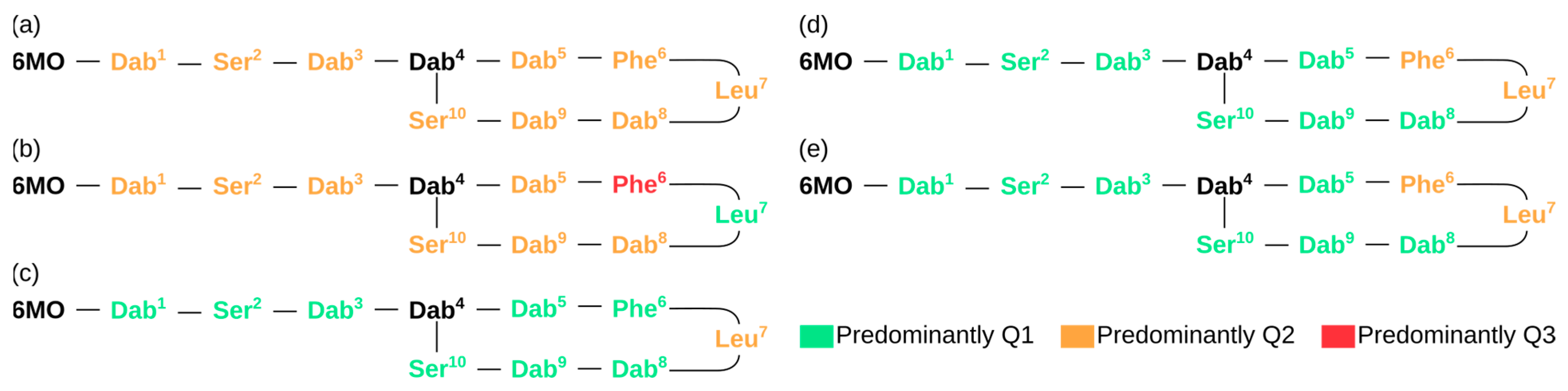
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garg, S.K.; Singh, O.; Juneja, D.; Tyagi, N.; Khurana, A.S.; Qamra, A.; Motlekar, S.; Barkate, H. Resurgence of Polymyxin B for MDR/XDR Gram-Negative Infections: An Overview of Current Evidence. Crit. Care Res. Pract. 2017, 2017, 3635609. [Google Scholar] [CrossRef] [PubMed]
- Manioglu, S.; Modaresi, S.M.; Ritzmann, N.; Thoma, J.; Overall, S.A.; Harms, A.; Upert, G.; Luther, A.; Barnes, A.B.; Obrecht, D.; et al. Antibiotic polymyxin arranges lipopolysaccharide into crystalline structures to solidify the bacterial membrane. Nat. Commun. 2022, 13, 6195. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-Activity Relationships of Polymyxin Antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.H.; Kier, L.B. The Molecular Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property Modeling. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Weinheim, Germany, 1991; Volume 2, pp. 367–422. [Google Scholar]
- Kier, L.B. An Index of Molecular Flexibility from Kappa Shape Attributes. Quant. Struct.-Act. Relat. 1989, 8, 221–224. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, I.; Inácio, J.; Jorge, P.; Teixeira, F. A Game with a Purpose: Designing Structural Modifications in Polymyxin B to Face Multi-Drug Resistant Bacteria. Med. Sci. Forum 2023, 23, 2. https://doi.org/10.3390/msf2023023002
Machado I, Inácio J, Jorge P, Teixeira F. A Game with a Purpose: Designing Structural Modifications in Polymyxin B to Face Multi-Drug Resistant Bacteria. Medical Sciences Forum. 2023; 23(1):2. https://doi.org/10.3390/msf2023023002
Chicago/Turabian StyleMachado, Inês, João Inácio, Paula Jorge, and Filipe Teixeira. 2023. "A Game with a Purpose: Designing Structural Modifications in Polymyxin B to Face Multi-Drug Resistant Bacteria" Medical Sciences Forum 23, no. 1: 2. https://doi.org/10.3390/msf2023023002