

Exploring Inflammasome Complex as a Therapeutic Approach in Inflammatory Diseases


 , ,
, ,
Abstract
:1. Introduction
2. Methods
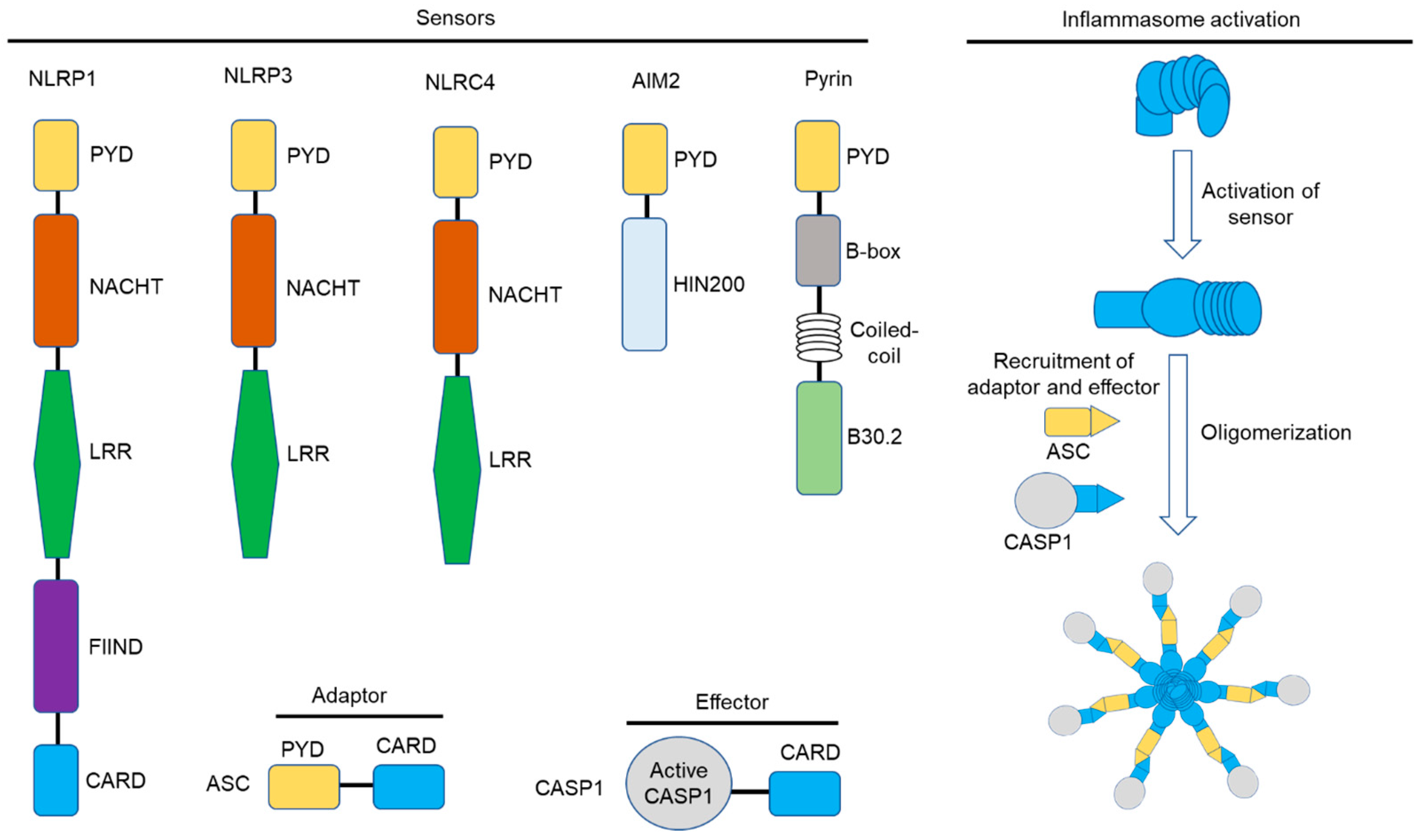
3. The Inflammasome: Mechanisms of Activation
3.1. The NLRP1 Inflammasome
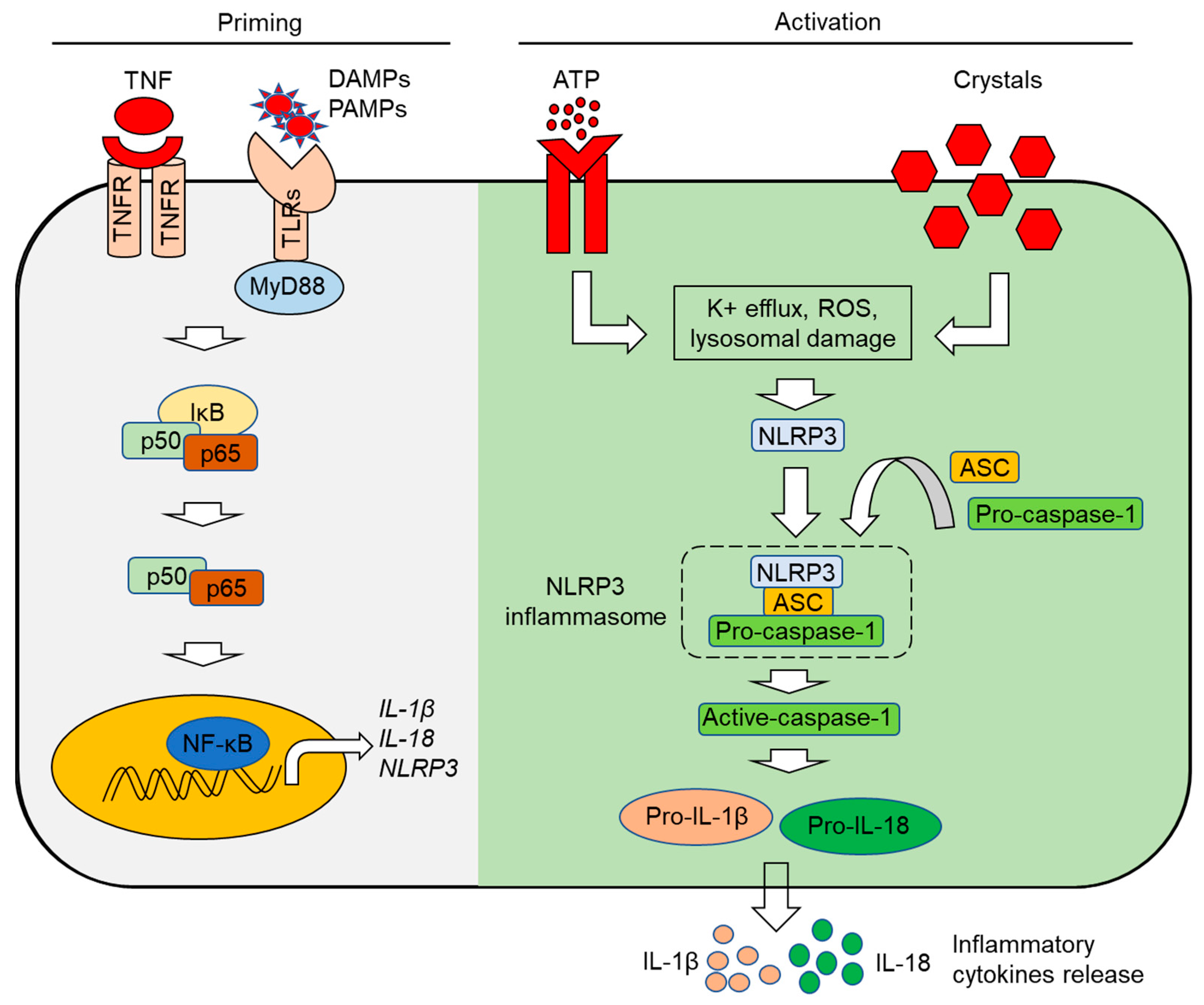
3.2. The NLRP3 Inflammasome
3.3. The NLRP6 Inflammasome
3.4. The NLRP7 Inflammasome
3.5. The NLRP12 Inflammasome
3.6. The NLRC4 Inflammasome
3.7. The AIM2 Inflammasome
3.8. The IFI16 Inflammasome
3.9. The Pyrin Inflammasome
3.10. Non-Canonical Inflammasomes
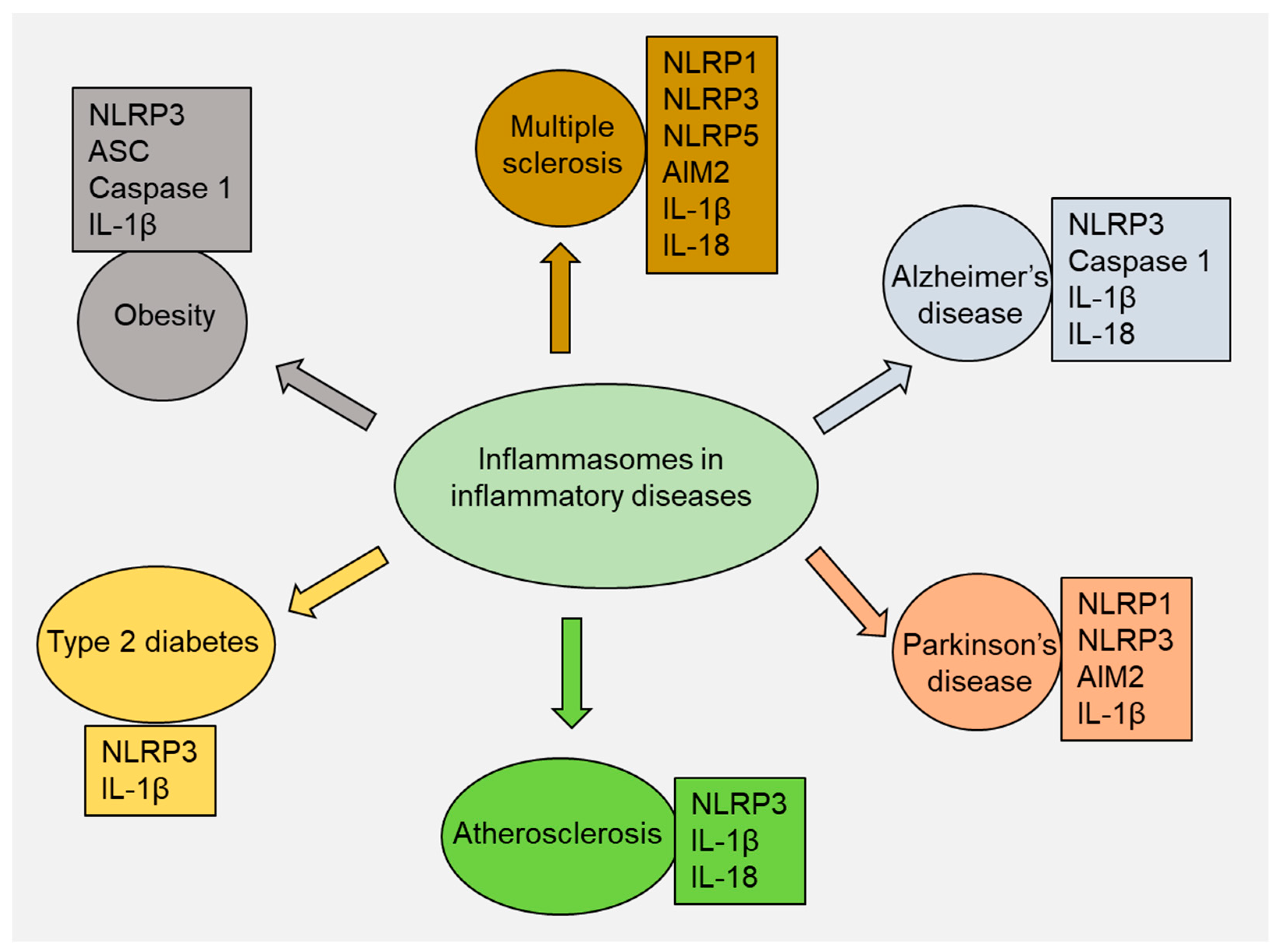
4. Role of Inflammasomes in Inflammatory Diseases
4.1. Multiple Sclerosis
4.2. Alzheimer’s Disease
4.3. Parkinson’s Disease
4.4. Atherosclerosis
4.5. Type 2 Diabetes
4.6. Obesity
4.7. Other Inflammatory Diseases
5. Inflammasomes as Therapeutic Targets in Inflammatory Diseases
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Rana, A.; Kaur, A.; Faraz, F.; Tandon, S. Targeting inflammasomes: A possible therapeutic approach for periodontal disease. World Acad. Sci. J. 2021, 3, 50. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Pedra, J.H.; Cassel, S.L.; Sutterwala, F.S. Sensing pathogens and danger signals by the inflammasome. Curr. Opin. Immunol. 2009, 21, 10–16. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Christgen, S.; Place, D.E.; Kanneganti, T.-D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Zeiser, R. NLRP3 inflammasome activation in cancer: A double-edged sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef]
- Bryant, C.; Fitzgerald, K.A. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 2009, 19, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.-L.; Cheung, S.W.-M.; Cheng, K.K.-Y. NLRP3 inflammasome activation in adipose tissues and its implications on metabolic diseases. Int. J. Mol. Sci. 2020, 21, 4184. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Ghani, A.; Mehal, W.Z. Inflammasome biology in fibrogenesis. Biochim. Biophys. Acta 2013, 1832, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.I.; McFadden, T.; Link, V.M.; Han, S.-J.; Karlsson, R.-M.; Stacy, A.; Farley, T.K.; Lima-Junior, D.S.; Harrison, O.J.; Desai, J.V. Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science 2021, 373, eabf3002. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Chavarría-Smith, J.; Vance, R.E. The NLRP1 inflammasomes. Immunol. Rev. 2015, 265, 22–34. [Google Scholar] [CrossRef]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, eabd0811. [Google Scholar] [CrossRef]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 2016, 167, 187–202.e17. [Google Scholar] [CrossRef] [PubMed]
- Fenini, G.; Karakaya, T.; Hennig, P.; Di Filippo, M.; Beer, H.-D. The NLRP1 inflammasome in human skin and beyond. Int. J. Mol. Sci. 2020, 21, 4788. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Lozada-Soto, K.M.; Russo, H.M.; Miller, B.A.; Dubyak, G.R. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: Roles for K+ efflux and Ca2+ influx. Am. J. Physiol. Cell Physiol. 2016, 311, C83–C100. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Amores-Iniesta, J.; Barberà-Cremades, M.; Martínez, C.M.; Pons, J.A.; Revilla-Nuin, B.; Martínez-Alarcón, L.; Di Virgilio, F.; Parrilla, P.; Baroja-Mazo, A.; Pelegrín, P. Extracellular ATP activates the NLRP3 inflammasome and is an early danger signal of skin allograft rejection. Cell Rep. 2017, 21, 3414–3426. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Latz, E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Pandey, A.; Shen, C.; Feng, S.; Man, S.M. Cell biology of inflammasome activation. Trends Cell Biol. 2021, 31, 924–939. [Google Scholar] [CrossRef]
- Clare, B. Inflammasome activation by Salmonella. Curr. Opin. Microbiol. 2021, 64, 27–32. [Google Scholar] [CrossRef]
- Neudorf, H.; Durrer, C.; Myette-Cote, E.; Makins, C.; O’Malley, T.; Little, J.P. Oral ketone supplementation acutely increases markers of NLRP3 inflammasome activation in human monocytes. Mol. Nutr. Food Res. 2019, 63, 1801171. [Google Scholar] [CrossRef]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.-Y. Antibiotics-driven gut microbiome perturbation alters immunity to vaccines in humans. Cell 2019, 178, 1313–1328.e13. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, T.; Huang, B.; Luo, M.; Chen, Z.; Zhao, Z.; Wang, J.; Leung, D.; Yang, X.; Chan, K.W. Excessive deubiquitination of NLRP3-R779C variant contributes to very-early-onset inflammatory bowel disease development. J. Allergy Clin. Immunol. 2021, 147, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Grenier, J.M.; Wang, L.; Manji, G.A.; Huang, W.-J.; Al-Garawi, A.; Kelly, R.; Carlson, A.; Merriam, S.; Lora, J.M.; Briskin, M. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-κB and caspase-1. FEBS Lett. 2002, 530, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Kern, L.; Elinav, E. The NLRP6 inflammasome. Immunology 2021, 162, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Angosto-Bazarra, D.; Molina-López, C.; Pelegrín, P. Physiological and pathophysiological functions of NLRP6: Pro-and anti-inflammatory roles. Commun. Biol. 2022, 5, 524. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zan, Y.; Sui, K.; Zhu, S. The latest breakthrough on NLRP6 inflammasome. Precis. Clin. Med. 2022, 5, pbac022. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef]
- Radian, A.D.; Khare, S.; Chu, L.H.; Dorfleutner, A.; Stehlik, C. ATP binding by NLRP7 is required for inflammasome activation in response to bacterial lipopeptides. Mol. Immunol. 2015, 67, 294–302. [Google Scholar] [CrossRef]
- Lupfer, C.; Kanneganti, T.-D. Unsolved mysteries in NLR biology. Front. Immunol. 2013, 4, 285. [Google Scholar] [CrossRef]
- Duéñez-Guzmán, E.A.; Haig, D. The evolution of reproduction-related NLRP genes. J. Mol. Evol. 2014, 78, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Tweedell, R.E.; Kanneganti, T.D. NLRP12-PANoptosome in haemolytic, infectious and inflammatory diseases. Clin. Transl. Med. 2023, 13, e1409. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Lich, J.D.; Ye, Z.; Allen, I.C.; Gris, D.; Wilson, J.E.; Schneider, M.; Roney, K.E.; O’Connor, B.P.; Moore, C.B. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J. Immunol. 2010, 185, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tao, Y.; Wu, X.; Wu, J.; Shen, M.; Zheng, Z. The role of NLRP12 in inflammatory diseases. Eur. J. Pharmacol. 2023, 956, 175995. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, B.; Pandian, N.; Mall, R.; Wang, Y.; Sarkar, R.; Kim, H.J.; Malireddi, R.S.; Karki, R.; Janke, L.J.; Vogel, P. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell 2023, 186, 2783–2801. [Google Scholar] [CrossRef]
- Winsor, N.; Krustev, C.; Bruce, J.; Philpott, D.J.; Girardin, S.E. Canonical and noncanonical inflammasomes in intestinal epithelial cells. Cell. Microbiol. 2019, 21, e13079. [Google Scholar] [CrossRef]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.-Y.; Shen, Z.; Song, Y.-H. Inflammasomes as therapeutic targets in human diseases. Signal Transduct. Target. Ther. 2021, 6, 247. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Jones, J.W.; Kayagaki, N.; Broz, P.; Henry, T.; Newton, K.; O’Rourke, K.; Chan, S.; Dong, J.; Qu, Y.; Roose-Girma, M. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 2010, 107, 9771–9776. [Google Scholar] [CrossRef]
- Sauer, J.-D.; Witte, C.E.; Zemansky, J.; Hanson, B.; Lauer, P.; Portnoy, D.A. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 2010, 7, 412–419. [Google Scholar] [CrossRef]
- Dihlmann, S.; Erhart, P.; Mehrabi, A.; Nickkholgh, A.; Lasitschka, F.; Böckler, D.; Hakimi, M. Increased expression and activation of absent in melanoma 2 inflammasome components in lymphocytic infiltrates of abdominal aortic aneurysms. Mol. Med. 2014, 20, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göß, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef]
- Javierre, B.M.; Fernandez, A.F.; Richter, J.; Al-Shahrour, F.; Martin-Subero, J.I.; Rodriguez-Ubreva, J.; Berdasco, M.; Fraga, M.F.; O’Hanlon, T.P.; Rider, L.G. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Dihlmann, S.; Tao, S.; Echterdiek, F.; Herpel, E.; Jansen, L.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M.; Kloor, M. Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int. J. Cancer 2014, 135, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, L.; Liu, H.; Duan, X.; Dickerson, E.; Shen, H.; Panchanathan, R.; Choubey, D. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol. Cancer Res. 2013, 11, 1193–1202. [Google Scholar] [CrossRef]
- Liu, D.; Lum, K.K.; Treen, N.; Núñez, C.T.; Yang, J.; Howard, T.R.; Levine, M.; Cristea, I.M. IFI16 phase separation via multi-phosphorylation drives innate immune signaling. Nucleic Acids Res. 2023, 51, 6819–6840. [Google Scholar] [CrossRef]
- Fan, Z.; Chen, R.; Yin, W.; Xie, X.; Wang, S.; Hao, C. Effects of AIM2 and IFI16 on Infectious Diseases and Inflammation. Viral Immunol. 2023, 36, 438–448. [Google Scholar] [CrossRef]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef]
- Gray, E.E.; Winship, D.; Snyder, J.M.; Child, S.J.; Geballe, A.P.; Stetson, D.B. The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 2016, 45, 255–266. [Google Scholar] [CrossRef]
- Heilig, R.; Broz, P. Function and mechanism of the pyrin inflammasome. Eur. J. Immunol. 2018, 48, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Poyet, J.-L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar] [PubMed]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 inflammasome and inflammatory diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, H.; Yang, Y.; Tao, J. Therapeutic potential of targeting the NLRP3 inflammasome in rheumatoid arthritis. Inflammation 2023, 46, 835–852. [Google Scholar] [CrossRef]
- Gajofatto, A.; Benedetti, M.D. Treatment strategies for multiple sclerosis: When to start, when to change, when to stop? World J. Clin. Cases 2015, 3, 545. [Google Scholar] [CrossRef]
- Cui, Y.; Yu, H.; Bu, Z.; Wen, L.; Yan, L.; Feng, J. Focus on the role of the NLRP3 inflammasome in multiple sclerosis: Pathogenesis, diagnosis, and therapeutics. Front. Mol. Neurosci. 2022, 15, 894298. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Palomino-Antolin, A.; Narros-Fernández, P.; Farré-Alins, V.; Sevilla-Montero, J.; Decouty-Pérez, C.; Lopez-Rodriguez, A.B.; Fernandez, N.; Monge, L.; Casas, A.I.; Calzada, M.J. Time-dependent dual effect of NLRP3 inflammasome in brain ischaemia. Br. J. Pharmacol. 2022, 179, 1395–1410. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Pinke, K.H.; Zorzella-Pezavento, S.F.G.; de Campos Fraga-Silva, T.F.; Mimura, L.A.N.; De Oliveira, L.R.C.; Ishikawa, L.L.W.; Fernandes, A.A.H.; Lara, V.S.; Sartori, A. Calming down mast cells with ketotifen: A potential strategy for multiple sclerosis therapy? Neurotherapeutics 2020, 17, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Desu, H.L.; Plastini, M.; Illiano, P.; Bramlett, H.M.; Dietrich, W.D.; de Rivero Vaccari, J.P.; Brambilla, R.; Keane, R.W. IC100: A novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J. Neuroinflamm. 2020, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Russo, A.J.; Shivcharan, S.; Rathinam, V.A. AIM2 in health and disease: Inflammasome and beyond. Immunol. Rev. 2020, 297, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, S.; Hu, Y.; Ma, Y.; Wu, Y.; Wu, C.; Liu, X.; Wang, B.; Hu, G.; Zhou, J. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J. Exp. Med. 2021, 218, e20201796. [Google Scholar] [CrossRef]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef]
- Govindarajan, V.; de Rivero Vaccari, J.P.; Keane, R.W. Role of inflammasomes in multiple sclerosis and their potential as therapeutic targets. J. Neuroinflammation 2020, 17, 260. [Google Scholar] [CrossRef]
- Maver, A.; Lavtar, P.; Ristić, S.; Stopinšek, S.; Simčič, S.; Hočevar, K.; Sepčić, J.; Drulović, J.; Pekmezović, T.; Novaković, I. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci. Rep. 2017, 7, 3715. [Google Scholar] [CrossRef]
- Zhang, L.; Jiao, C.; Liu, L.; Wang, A.; Tang, L.; Ren, Y.; Huang, P.; Xu, J.; Mao, D.; Liu, L. NLRC5: A potential target for central nervous system disorders. Front. Immunol. 2021, 12, 704989. [Google Scholar] [CrossRef]
- Beh, S.C.; Greenberg, B.M.; Frohman, T.; Frohman, E.M. Transverse myelitis. Neurol. Clin. 2013, 31, 79–138. [Google Scholar] [CrossRef]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in inflammatory disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [PubMed]
- Kucuksezer, U.C.; Aktas Cetin, E.; Esen, F.; Tahrali, I.; Akdeniz, N.; Gelmez, M.Y.; Deniz, G. The role of natural killer cells in autoimmune diseases. Front. Immunol. 2021, 12, 622306. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, K.A.; Wucherpfennig, K.W. B cells and autoantibodies in the pathogenesis of multiple sclerosis and related inflammatory demyelinating diseases. Adv. Immunol. 2008, 98, 121–149. [Google Scholar] [PubMed]
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Inoue, M.; Shinohara, M.L. Nlrp3 inflammasome and MS/EAE. Autoimmune Dis. 2013, 2013, 859145. [Google Scholar] [CrossRef]
- Barclay, W.E.; Aggarwal, N.; Deerhake, M.E.; Inoue, M.; Nonaka, T.; Nozaki, K.; Luzum, N.A.; Miao, E.A.; Shinohara, M.L. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight 2022, 7, e155563. [Google Scholar] [CrossRef]
- Madsen, P.M.; Desu, H.L.; de Rivero Vaccari, J.P.; Florimon, Y.; Ellman, D.G.; Keane, R.W.; Clausen, B.H.; Lambertsen, K.L.; Brambilla, R. Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav. Immun. 2020, 84, 132–146. [Google Scholar] [CrossRef]
- Jha, S.; Srivastava, S.Y.; Brickey, W.J.; Iocca, H.; Toews, A.; Morrison, J.P.; Chen, V.S.; Gris, D.; Matsushima, G.K.; Ting, J.P.-Y. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J. Neurosci. 2010, 30, 15811–15820. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Rui, W.; Xiao, H.; Fan, Y.; Ma, Z.; Xiao, M.; Li, S.; Shi, J. Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer’s disease. J. Neuroinflammation 2021, 18, 280. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The role of NLRP3 inflammasome in Alzheimer’s disease and potential therapeutic targets. Front. Pharmacol. 2022, 13, 845185. [Google Scholar] [CrossRef] [PubMed]
- Van Zeller, M.; Dias, D.; Sebastião, A.M.; Valente, C.A. NLRP3 inflammasome: A starring role in amyloid-β-and tau-driven pathological events in Alzheimer’s disease. J. Alzheimer’s Dis. 2021, 83, 939–961. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Cornell, J.; Salinas, S.; Huang, H.-Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705. [Google Scholar]
- Sutinen, E.M.; Pirttilä, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J. Neuroinflammation 2012, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Sita, G.; Graziosi, A.; Hrelia, P.; Morroni, F. NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 6984. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, L.; Yang, X. Interaction between autophagy and the NLRP3 inflammasome in Alzheimer’s and Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 1018848. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chan, C. The role of inflammasome in Alzheimer’s disease. Ageing Res. Rev. 2014, 15, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, S.; Qin, Y.; Liang, Z.; Han, S.-N.; Boodapati, S.T.; Li, J.; Lu, Q.; Flavell, R.A.; Mehal, W.Z.; Ouyang, X. GSK3β mediates the spatiotemporal dynamics of NLRP3 inflammasome activation. Cell Death Differ. 2022, 29, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-W.; Kim, S.J.; Kim, M.-S. Oxidative stress with tau hyperphosphorylation in memory impaired 1, 2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The ambiguous relationship of oxidative stress, tau hyperphosphorylation, and autophagy dysfunction in Alzheimer’s disease. Oxidative Med. Cell. Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef]
- Ren, P.; Chen, J.; Li, B.; Zhang, M.; Yang, B.; Guo, X.; Chen, Z.; Cheng, H.; Wang, P.; Wang, S. Nrf2 ablation promotes Alzheimer’s disease-like pathology in APP/PS1 transgenic mice: The role of neuroinflammation and oxidative stress. Oxidative Med. Cell. Longev. 2020, 2020, 3050971. [Google Scholar] [CrossRef]
- Dempsey, C.; Araiz, A.R.; Bryson, K.; Finucane, O.; Larkin, C.; Mills, E.; Robertson, A.; Cooper, M.; O’Neill, L.; Lynch, M. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Beauchet, O.; LeBlanc, A.C. Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging. Nat. Commun. 2020, 11, 4571. [Google Scholar] [CrossRef]
- Shukla, P.K.; Delotterie, D.F.; Xiao, J.; Pierre, J.F.; Rao, R.; McDonald, M.P.; Khan, M.M. Alterations in the gut-microbial-inflammasome-brain axis in a mouse model of Alzheimer’s disease. Cells 2021, 10, 779. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.-L. Gut microbiota and dysbiosis in Alzheimer’s disease: Implications for pathogenesis and treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Ju, D. Inflammasome: A double-edged sword in liver diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef] [PubMed]
- de Brito Toscano, E.C.; Rocha, N.P.; Lopes, B.N.; Suemoto, C.K.; Teixeira, A.L. Neuroinflammation in Alzheimer’s disease: Focus on NLRP1 and NLRP3 inflammasomes. Curr. Protein Pept. Sci. 2021, 22, 584–598. [Google Scholar] [CrossRef]
- Wu, P.-J.; Hung, Y.-F.; Liu, H.-Y.; Hsueh, Y.-P. Deletion of the inflammasome sensor Aim2 mitigates Aβ deposition and microglial activation but increases inflammatory cytokine expression in an Alzheimer disease mouse model. Neuroimmunomodulation 2017, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Saadi, M.; Karkhah, A.; Pourabdolhossein, F.; Ataie, A.; Monif, M.; Nouri, H.R. Involvement of NLRC4 inflammasome through caspase-1 and IL-1β augments neuroinflammation and contributes to memory impairment in an experimental model of Alzheimer’s like disease. Brain Res. Bull. 2020, 154, 81–90. [Google Scholar] [CrossRef] [PubMed]
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 1: Disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. Pharm. Ther. 2015, 40, 504. [Google Scholar]
- Przedborski, S. Inflammation and Parkinson’s disease pathogenesis. Mov. Disord. 2010, 25, S55–S57. [Google Scholar] [CrossRef]
- Nguyen, L.T.N.; Nguyen, H.D.; Kim, Y.J.; Nguyen, T.T.; Lai, T.T.; Lee, Y.K.; Ma, H.-i.; Kim, Y.E. Role of NLRP3 inflammasome in Parkinson’s disease and therapeutic considerations. J. Park. Dis. 2022, 12, 2117–2133. [Google Scholar] [CrossRef]
- Murta, V.; Ferrari, C. Peripheral inflammation and demyelinating diseases. Adv. Exp. Med. Biol. 2016, 949, 263–285. [Google Scholar]
- Jewell, S.; Herath, A.M.; Gordon, R. Inflammasome activation in Parkinson’s disease. J. Park. Dis. 2022, 12, S113–S128. [Google Scholar] [CrossRef]
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β modulate blood-brain barrier permeability and decrease amyloid-β peptide efflux in a human blood-brain barrier model. Int. J. Mol. Sci. 2022, 23, 10235. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Bono, F.; Valerio, A.; Pizzi, M.; Spano, P.; Bellucci, A. Mitochondria and α-synuclein: Friends or foes in the pathogenesis of Parkinson’s disease? Genes 2017, 8, 377. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J. Targeting microglial α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson’s disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef]
- Su, Q.; Ng, W.L.; Goh, S.Y.; Gulam, M.Y.; Wang, L.-F.; Tan, E.-K.; Ahn, M.; Chao, Y.-X. Targeting the inflammasome in Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 957705. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.; LeBlanc, A. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambarè, D.; Bellini, E.; Ordazzo, G. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am. J. Prev. Card. 2020, 4, 100130. [Google Scholar] [CrossRef]
- Packard, R.R.; Lichtman, A.H.; Libby, P. Innate and adaptive immunity in atherosclerosis. Semin. Immunopathol. 2009, 31, 5–22. [Google Scholar] [CrossRef]
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345. [Google Scholar] [CrossRef] [PubMed]
- Piscopiello, M.; Sessa, M.; Anzalone, N.; Castellano, R.; Maisano, F.; Ferrero, E.; Chiesa, R.; Alfieri, O.; Comi, G.; Ferrero, M.E. P2X7 receptor is expressed in human vessels and might play a role in atherosclerosis. Int. J. Cardiol. 2013, 168, 2863–2866. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Liu, L.; Wei, D.; Lv, Y.; Wang, G.; Xiong, W.; Wang, X.; Altaf, A.; Wang, L.; He, D. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int. J. Mol. Med. 2015, 35, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Henkels, K.M.; Wrenshall, L.E.; Kanaho, Y.; Di Paolo, G.; Frohman, M.A.; Gomez-Cambronero, J. Oxidized LDL phagocytosis during foam cell formation in atherosclerotic plaques relies on a PLD2–CD36 functional interdependence. J. Leukoc. Biol. 2018, 103, 867–883. [Google Scholar] [CrossRef]
- Wang, R.; Wu, W.; Li, W.; Huang, S.; Li, Z.; Liu, R.; Shan, Z.; Zhang, C.; Li, W.; Wang, S. Activation of NLRP3 inflammasome promotes foam cell formation in vascular smooth muscle cells and atherogenesis via HMGB1. J. Am. Heart Assoc. 2018, 7, e008596. [Google Scholar] [CrossRef]
- Varghese, J.F.; Patel, R.; Yadav, U.C. Sterol regulatory element binding protein (SREBP)-1 mediates oxidized low-density lipoprotein (oxLDL) induced macrophage foam cell formation through NLRP3 inflammasome activation. Cell. Signal. 2019, 53, 316–326. [Google Scholar] [CrossRef]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calò, L.A. The role of oxidized low-density lipoproteins in atherosclerosis: The myths and the facts. Mediat. Inflamm. 2013, 2013, 714653. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 inflammasomes in atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef]
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef]
- Rezaieyazdi, Z.; AkbariRad, M.; Saadati, N.; Salari, M.; Orang, R.; Sedighi, S.; Esmaily, H.; Azarpazhooh, M.R.; Firoozi, A.; Akbarpour, E. Serum interleukin-18 and its relationship with subclinical atherosclerosis in systemic lupus erythematosus. ARYA Atheroscler. 2021, 17, 1–6. [Google Scholar] [PubMed]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef]
- Vargas, A.M.; Rivera-Rodriguez, D.E.; Martinez, L.R. Methamphetamine alters the TLR4 signaling pathway, NF-κB activation, and pro-inflammatory cytokine production in LPS-challenged NR-9460 microglia-like cells. Mol. Immunol. 2020, 121, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Čokić, V.P.; Mitrović-Ajtić, O.; Beleslin-Čokić, B.B.; Marković, D.; Buač, M.; Diklić, M.; Kraguljac-Kurtović, N.; Damjanović, S.; Milenković, P.; Gotić, M. Proinflammatory cytokine IL-6 and JAK-STAT signaling pathway in myeloproliferative neoplasms. Mediat. Inflamm. 2015, 2015, 453020. [Google Scholar] [CrossRef]
- Xu, J.; Lu, X.; Shi, G.-P. Vasa vasorum in atherosclerosis and clinical significance. Int. J. Mol. Sci. 2015, 16, 11574–11608. [Google Scholar] [CrossRef]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015, 6, 598. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Sjöholm, Å.; Nyström, T. Inflammation and the etiology of type 2 diabetes. Diabetes. Metab. Res. Rev. 2006, 22, 4–10. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, D.; Li, Y.; Wang, W.; Bei, W.; Guo, J. NLRP3 inflammasome and IL-1β pathway in type 2 diabetes and atherosclerosis: Friend or foe? Pharmacol. Res. 2021, 173, 105885. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wang, W.; Okla, M.; Kang, I.; Moreau, R.; Chung, S. Suppression of NLRP3 inflammasome by γ-tocotrienol ameliorates type 2 diabetes. J. Lipid Res. 2016, 57, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef]
- Sokolova, M.; Sahraoui, A.; Høyem, M.; Øgaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. NLRP3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E912–E923. [Google Scholar] [CrossRef]
- Marchetti, P.; Bugliani, M.; De Tata, V.; Suleiman, M.; Marselli, L. Pancreatic beta cell identity in humans and the role of type 2 diabetes. Front. Cell Dev. Biol. 2017, 5, 55. [Google Scholar] [CrossRef]
- Eguchi, N.; Vaziri, N.D.; Dafoe, D.C.; Ichii, H. The role of oxidative stress in pancreatic β cell dysfunction in diabetes. Int. J. Mol. Sci. 2021, 22, 1509. [Google Scholar] [CrossRef]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The role of inflammation in diabetes: Current concepts and future perspectives. Eur. Cardiol. Rev. 2019, 14, 50. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Giraud, J.; Davis, R.J.; White, M.F. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J. Biol. Chem. 2003, 278, 2896–2902. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, B.; Wang, L.; Yang, M.; Xia, Z.; Wei, W.; Zhang, F.; Yuan, X. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLoS ONE 2017, 12, e0181248. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Guo, R.; Mehmood, A.; Zhang, L.; Yin, B.; Yuan, C.; Zhang, H.; Guo, L.; Li, B. Liraglutide attenuate central nervous inflammation and demyelination through AMPK and pyroptosis-related NLRP3 pathway. CNS Neurosci. Ther. 2022, 28, 422–434. [Google Scholar] [CrossRef]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: Causes and therapeutic strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef]
- Welsh, P.; Polisecki, E.; Robertson, M.; Jahn, S.; Buckley, B.M.; de Craen, A.J.; Ford, I.; Jukema, J.W.; Macfarlane, P.W.; Packard, C.J. Unraveling the directional link between adiposity and inflammation: A bidirectional Mendelian randomization approach. J. Clin. Endocrinol. Metab. 2010, 95, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011, 152, 4039–4045. [Google Scholar] [CrossRef]
- Sokolova, M.; Yang, K.; Hansen, S.H.; Louwe, M.C.; Kummen, M.; Hov, J.E.; Sjaastad, I.; Berge, R.K.; Halvorsen, B.; Aukrust, P. NLRP3 inflammasome deficiency attenuates metabolic disturbances involving alterations in the gut microbial profile in mice exposed to high fat diet. Sci. Rep. 2020, 10, 21006. [Google Scholar] [CrossRef] [PubMed]
- Javaid, H.M.A.; Sahar, N.E.; ZhuGe, D.-L.; Huh, J.Y. Exercise inhibits NLRP3 inflammasome activation in obese mice via the anti-inflammatory effect of meteorin-like. Cells 2021, 10, 3480. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, P.; Brüning, J.C. Contribution of specific ceramides to obesity-associated metabolic diseases. Cell. Mol. Life Sci. 2022, 79, 395. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; Van De Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef]
- Nagar, A.; Rahman, T.; Harton, J.A. The ASC speck and NLRP3 inflammasome function are spatially and temporally distinct. Front. Immunol. 2021, 12, 752482. [Google Scholar] [CrossRef]
- Beckley, A.J.; Lan, L.-Q.; Aono, S.; Wang, L.; Shi, J.N. Caspase-1 activation and mature interleukin-1β release are uncoupled events in monocytes. World J. Biol. Chem. 2013, 4, 30–34. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Picó, C.; Palou, M.; Pomar, C.A.; Rodríguez, A.M.; Palou, A. Leptin as a key regulator of the adipose organ. Rev. Endocr. Metab. Disord. 2022, 23, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.-G. Role of insulin in health and disease: An update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Baral, A.; Park, P.-H. Leptin Induces Apoptotic and Pyroptotic Cell Death via NLRP3 Inflammasome Activation in Rat Hepatocytes. Int. J. Mol. Sci. 2021, 22, 12589. [Google Scholar] [CrossRef] [PubMed]
- Iikuni, N.; Kwan Lam, Q.L.; Lu, L.; Matarese, G.; Cava, A.L. Leptin and inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory mechanisms of the NLRP3 inflammasomes in diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-W.; Hung, L.-C.; Chen, Y.-C.; Wang, W.-H.; Lin, C.-Y.; Tzeng, H.-H.; Suen, J.-L.; Chen, Y.-H. Insulin reduces inflammation by regulating the activation of the NLRP3 inflammasome. Front. Immunol. 2021, 11, 587229. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Fu, R.; Wang, S.; Huang, Y.; Li, X.; Zhou, M.; Zhao, J.; Yang, N. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 2018, 194, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Spel, L.; Martinon, F. Inflammasomes contributing to inflammation in arthritis. Immunol. Rev. 2020, 294, 48–62. [Google Scholar] [CrossRef]
- Tourkochristou, E.; Aggeletopoulou, I.; Konstantakis, C.; Triantos, C. Role of NLRP3 inflammasome in inflammatory bowel diseases. World J. Gastroenterol. 2019, 25, 4796. [Google Scholar] [CrossRef]
- Kingsbury, S.R.; Conaghan, P.G.; McDermott, M.F. The role of the NLRP3 inflammasome in gout. J. Inflamm. Res. 2011, 4, 39–49. [Google Scholar]
- Ciążyńska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. The role of NLRP1, NLRP3, and AIM2 inflammasomes in psoriasis. Int. J. Mol. Sci. 2021, 22, 5898. [Google Scholar] [CrossRef]
- Forouzandeh, M.; Besen, J.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome signaling proteins ASC and IL-18 as biomarkers of psoriasis. Front. Pharmacol. 2020, 11, 1238. [Google Scholar] [CrossRef]
- Verma, D.; Fekri, S.Z.; Sigurdardottir, G.; Eding, C.B.; Sandin, C.; Enerbäck, C. Enhanced inflammasome activity in patients with psoriasis promotes systemic inflammation. J. Invest. Dermatol. 2021, 141, 586–595.e5. [Google Scholar] [CrossRef] [PubMed]
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S. NOD-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci. Rep. 2016, 6, 22745. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yu, C.; Yang, Z.; Wei, Q.; Mu, K.; Zhang, Y.; Zhao, W.; Wang, X.; Huai, W.; Han, L. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J. Rheumatol. 2014, 41, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Girardelli, M.; Kamada, A.J.; Pancotto, J.A.; Donadi, E.A.; Crovella, S.; Sandrin-Garcia, P. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity 2012, 45, 271–278. [Google Scholar] [CrossRef] [PubMed]
- da Cruz, H.L.A.; Cavalcanti, C.A.J.; de Azêvedo Silva, J.; de Lima, C.A.D.; Fragoso, T.S.; Barbosa, A.D.; Dantas, A.T.; de Ataíde Mariz, H.; Duarte, A.L.B.P.; Pontillo, A. Differential expression of the inflammasome complex genes in systemic lupus erythematosus. Immunogenetics 2020, 72, 217–224. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Thacker, S.G.; Berthier, C.C.; Cohen, C.D.; Kretzler, M.; Kaplan, M.J. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J. Immunol. 2011, 187, 6143–6156. [Google Scholar] [CrossRef]
- Ma, Z.-Z.; Sun, H.-S.; Lv, J.-C.; Guo, L.; Yang, Q.-R. Expression and clinical significance of the NEK7-NLRP3 inflammasome signaling pathway in patients with systemic lupus erythematosus. J. Inflamm. 2018, 15, 16. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, C.; Jiang, N.; He, D.; Bai, Y.; Xin, Y. Rapamycin combined with MCC950 to treat multiple sclerosis in experimental autoimmune encephalomyelitis. J. Cell. Biochem. 2019, 120, 5160–5168. [Google Scholar] [CrossRef]
- Chen, R.; Yin, C.; Fang, J.; Liu, B. The NLRP3 inflammasome: An emerging therapeutic target for chronic pain. J. Neuroinflammation 2021, 18, 84. [Google Scholar] [CrossRef]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef]
- Zhai, Y.; Meng, X.; Ye, T.; Xie, W.; Sun, G.; Sun, X. Inhibiting the NLRP3 inflammasome activation with MCC950 ameliorates diabetic encephalopathy in db/db mice. Molecules 2018, 23, 522. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, X.; Li, L.; Ma, T.; Shi, M.; Yang, Y.; Fan, Q. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes Metab. Syndr. Obes. 2019, 12, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [PubMed]
- Soma, J.; Sato, K.; Saito, H.; Tsuchiya, Y. Effect of tranilast in early-stage diabetic nephropathy. Nephrol. Dial. Transplant. 2006, 21, 2795–2799. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Peng, Q. NLRP3 inhibitor tranilast attenuates gestational diabetes mellitus in a genetic mouse model. Drugs R D 2022, 22, 105–112. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W. Tranilast directly targets NLRP 3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Pan, Y.; Liu, Y.; Zheng, S.; Ding, K.; Mu, K.; Yuan, Y.; Li, Z.; Song, H. Novel Role for Tranilast in Regulating NLRP 3 Ubiquitination, Vascular Inflammation, and Atherosclerosis. J. Am. Heart Assoc. 2020, 9, e015513. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle-Deschner, J.B.; Haug, I. Canakinumab in patients with cryopyrin-associated periodic syndrome: An update for clinicians. Ther. Adv. Musculoskelet. Dis. 2013, 5, 315–329. [Google Scholar] [CrossRef]
- Davies, K.; Bukhari, M.A. Recent pharmacological advances in the management of gout. Rheumatology 2018, 57, 951–958. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Nasoohi, S.; Ishrat, T. MCC950, the selective inhibitor of nucleotide oligomerization domain-like receptor protein-3 inflammasome, protects mice against traumatic brain injury. J. Neurotrauma 2018, 35, 1294–1303. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Zheng, Y.; Yu, Q.; Zeng, M.; Bai, L.; Yang, L.; Guo, M.; Jiang, X.; Gan, J. Inhibitors of the NLRP3 inflammasome pathway as promising therapeutic candidates for inflammatory diseases. Int. J. Mol. Med. 2023, 51, 35. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.-S.; Ho, Y.-C.; Hsu, C.-L.; Pan, S.-L. Spinal cord injury and Alzheimer’s disease risk: A population-based, retrospective cohort study. Spinal Cord 2018, 56, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Zhao, G.; Wang, Y.; Ren, P.; Wu, M. MCC950, a selective inhibitor of NLRP3 inflammasome, reduces the inflammatory response and improves neurological outcomes in mice model of spinal cord injury. Front. Mol. Biosci. 2020, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E–deficient mice—Brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, H.; Song, L.; Yang, Z.; Qiu, M.; Wang, J.; Shi, S. Anthocyanins: Promising natural products with diverse pharmacological activities. Molecules 2021, 26, 3807. [Google Scholar] [CrossRef]
- Molagoda, I.M.N.; Lee, K.T.; Choi, Y.H.; Jayasingha, J.A.C.C.; Kim, G.-Y. Anthocyanins from Hibiscus syriacus L. inhibit NLRP3 inflammasome in BV2 microglia cells by alleviating NF-κB-and ER stress-induced Ca2+ accumulation and mitochondrial ROS production. Oxidative Med. Cell. Longev. 2021, 2021, 1246491. [Google Scholar] [CrossRef]
- Zhu, X.; Lin, X.; Zhang, P.; Liu, Y.; Ling, W.; Guo, H. Upregulated NLRP3 inflammasome activation is attenuated by anthocyanins in patients with nonalcoholic fatty liver disease: A case-control and an intervention study. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101843. [Google Scholar] [CrossRef]
- Salehi, B.; Sharifi-Rad, J.; Cappellini, F.; Reiner, Ž.; Zorzan, D.; Imran, M.; Sener, B.; Kilic, M.; El-Shazly, M.; Fahmy, N.M. The therapeutic potential of anthocyanins: Current approaches based on their molecular mechanism of action. Front. Pharmacol. 2020, 11, 1300. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Liu, H.; Quan, H.; Sui, H.; Jia, Y. Development of novel oridonin analogs as specifically targeted NLRP3 inflammasome inhibitors for the treatment of dextran sulfate sodium-induced colitis. Eur. J. Med. Chem. 2023, 245, 114919. [Google Scholar] [CrossRef]
- Domiciano, T.P.; Wakita, D.; Jones, H.D.; Crother, T.R.; Verri Jr, W.A.; Arditi, M.; Shimada, K. Quercetin inhibits inflammasome activation by interfering with ASC oligomerization and prevents interleukin-1 mediated mouse vasculitis. Sci. Rep. 2017, 7, 41539. [Google Scholar] [CrossRef]
- Xue, Y.; Du, M.; Zhu, M.-J. Quercetin suppresses NLRP3 inflammasome activation in epithelial cells triggered by Escherichia coli O157: H7. Free Radic. Biol. Med. 2017, 108, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed]
- So, A.; De Smedt, T.; Revaz, S.; Tschopp, J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 2007, 9, 1–6. [Google Scholar] [CrossRef]
- Ottaviani, S.; Moltó, A.; Ea, H.-K.; Neveu, S.; Gill, G.; Brunier, L.; Palazzo, E.; Meyer, O.; Richette, P.; Bardin, T. Efficacy of anakinra in gouty arthritis: A retrospective study of 40 cases. Arthritis Res. Ther. 2013, 15, R28. [Google Scholar] [CrossRef]
- Bertoni, A.; Penco, F.; Mollica, H.; Bocca, P.; Prigione, I.; Corcione, A.; Cangelosi, D.; Schena, F.; Del Zotto, G.; Amaro, A. Spontaneous NLRP3 inflammasome-driven IL-1-β secretion is induced in severe COVID-19 patients and responds to anakinra treatment. J. Allergy Clin. Immunol. 2022, 150, 796–805. [Google Scholar] [CrossRef]
- Dhimolea, E. MAbs, 2010. In Canakinumab; Taylor & Francis: Abingdon, UK, 2010; pp. 3–13. [Google Scholar]
- Riddle, M.C. sulfonylureas differ in effects on ischemic preconditioning—Is it time to retire glyburide? J. Clin. Endocrinol. Metab. 2003, 88, 528–530. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflammation 2020, 17, 280. [Google Scholar] [CrossRef]
- Han, Y.-M.; Ramprasath, T.; Zou, M.-H. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp. Mol. Med. 2020, 52, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Yamanashi, T.; Iwata, M.; Shibushita, M.; Tsunetomi, K.; Nagata, M.; Kajitani, N.; Miura, A.; Matsuo, R.; Nishiguchi, T.; Kato, T.A. Beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, attenuates anxiety-related behavior in a rodent post-traumatic stress disorder model. Sci. Rep. 2020, 10, 21629. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agents | Diseases | Targeted Inflammasomes | Functions |
|---|---|---|---|
| MCC95 (CP-456773, CRID3) | MS | NLRP3, IL-1β | MCC950 could reduce clinical symptom of MS [208]. |
| AD | NLRP3 | MCC950 reduced Aβ pathology and improved cognitive function in AD mice [118]. | |
| Gout | IL-1β | MCC950 significantly reduced the production of IL-1β and neutrophil infiltration in the inflamed joint [209]. | |
| Atherosclerosis | NLRP3, ASC, Caspase-1, GSDMD-N, IL-1β, IL-18 | MCC950 treatment reduced plaque areas and macrophage contents [210]. | |
| Diabetic encephalopathy | NLRP3, ASC, caspase-1, IL-1β | MCC950 treatment improved insulin sensitivity in db/db mice, thereby alleviating diabetic encephalopathy [211]. | |
| Diabetic nephropathy | NLRP3, caspase-1, IL-1β | MCC950 treatment reduced kidney injury in diabetic nephropathy [212]. | |
| Ketotifen (Zaditor®) | MS | NLRP3 | Ketotifen treatment reduced both the prevalence and severity of EAE disease [81]. Ketotifen restored balance of oxidative stress, and reduced infiltration of T cells in the CNS [81]. |
| IC100 (IgG4) | MS | ASC | IC100 decreased the trafficking of CD4+, CD8+ T cells, and CD11b + MHCII+ cells into the CNS [82]. IC100 treatment reduced the number and activation state of CNS resident microglia [82]. |
| Fenamate NSAIDs (flufenamic acid and mefenamic acid) | AD | NLRP3 | Fenamate NSAIDs showed therapeutic benefits in a model of memory loss caused by amyloid beta and in a transgenic mouse model of AD [213]. |
| Tranilast (N-[3′,4′-dimethoxycinnamoyl]-anthranilic acid) | Diabetic nephropathy | NLRP3 | Tranilast effectively decreased urinary albumin excretion, a significant clinical indicator of diabetic nephropathy [214]. |
| Gestational diabetes mellitus | NLRP3, TNF-α, IL-6 | Tranilast exhibited a significant amelioration of GDM symptoms in mice [215]. | |
| T2D | NLRP3 | Tranilast showed therapeutic effects in mouse models of T2D [216]. | |
| Gouty arthritis | NLRP3 | Tranilast showed therapeutic effects in mouse models of gouty arthritis [216]. | |
| Atherosclerosis | NLRP3 | Tranilast showed notable efficacy in ameliorating vascular inflammation and reducing atherosclerosis in both low-density lipoprotein receptor-deficient and apolipoprotein E-deficient mouse models [217]. | |
| Canakinumab (ILARIS®) | Gouty arthritis | NLRP3, IL-1β | Canakinumab is used in the treatment of gouty arthritis [218]. Canakinumab is effective in reducing the number of gout flares in patients with a history of gout [219]. |
| Glyburide | Gouty arthritis, silicosis, and AD | NLRP3 | Glyburide is thought to be effective against conditions like gouty arthritis, silicosis, and AD, where excessive IL-1β production via Cryopyrin-dependent pathways plays a significant role in the pathology [220,221,222]. |
| Pioglitazone | Diabetes mellitus | NLRP3 | Pioglitazone is effective against diabetic renal damage [174]. |
| Liraglutide | EAE | NLRP3 | Liraglutide treatment improves the disease score in EAE mice [175]. |
| Rosuvastatin | Diabetic cardiomyopathy | NLRP3 | Rosuvastatin induced reduction of diabetic cardiomyopathy in a rat model of T2D [165]. |
| γ-Tocotrienol | T2D | NLRP3 | γ-Tocotrienol (γT3) is effective in slowing down the advancement of T2D [165]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultana, S.; Viet, T.D.; Amin, T.; Kazi, E.; Micolucci, L.; Mollah, A.K.M.M.; Akhtar, M.M.; Islam, M.S. Exploring Inflammasome Complex as a Therapeutic Approach in Inflammatory Diseases. Future Pharmacol. 2023, 3, 789-818. https://doi.org/10.3390/futurepharmacol3040048
Sultana S, Viet TD, Amin T, Kazi E, Micolucci L, Mollah AKMM, Akhtar MM, Islam MS. Exploring Inflammasome Complex as a Therapeutic Approach in Inflammatory Diseases. Future Pharmacology. 2023; 3(4):789-818. https://doi.org/10.3390/futurepharmacol3040048
Chicago/Turabian StyleSultana, Sharmim, Thanh Doan Viet, Tasmiha Amin, Esha Kazi, Luigina Micolucci, Abul Kalam Mohammad Moniruzzaman Mollah, Most Mauluda Akhtar, and Md Soriful Islam. 2023. "Exploring Inflammasome Complex as a Therapeutic Approach in Inflammatory Diseases" Future Pharmacology 3, no. 4: 789-818. https://doi.org/10.3390/futurepharmacol3040048