

Moles of Molecules against Mycobacterium abscessus: A Review of Current Research

Abstract
:1. Introduction
2. Ongoing M. abscessus Therapeutic Regimen
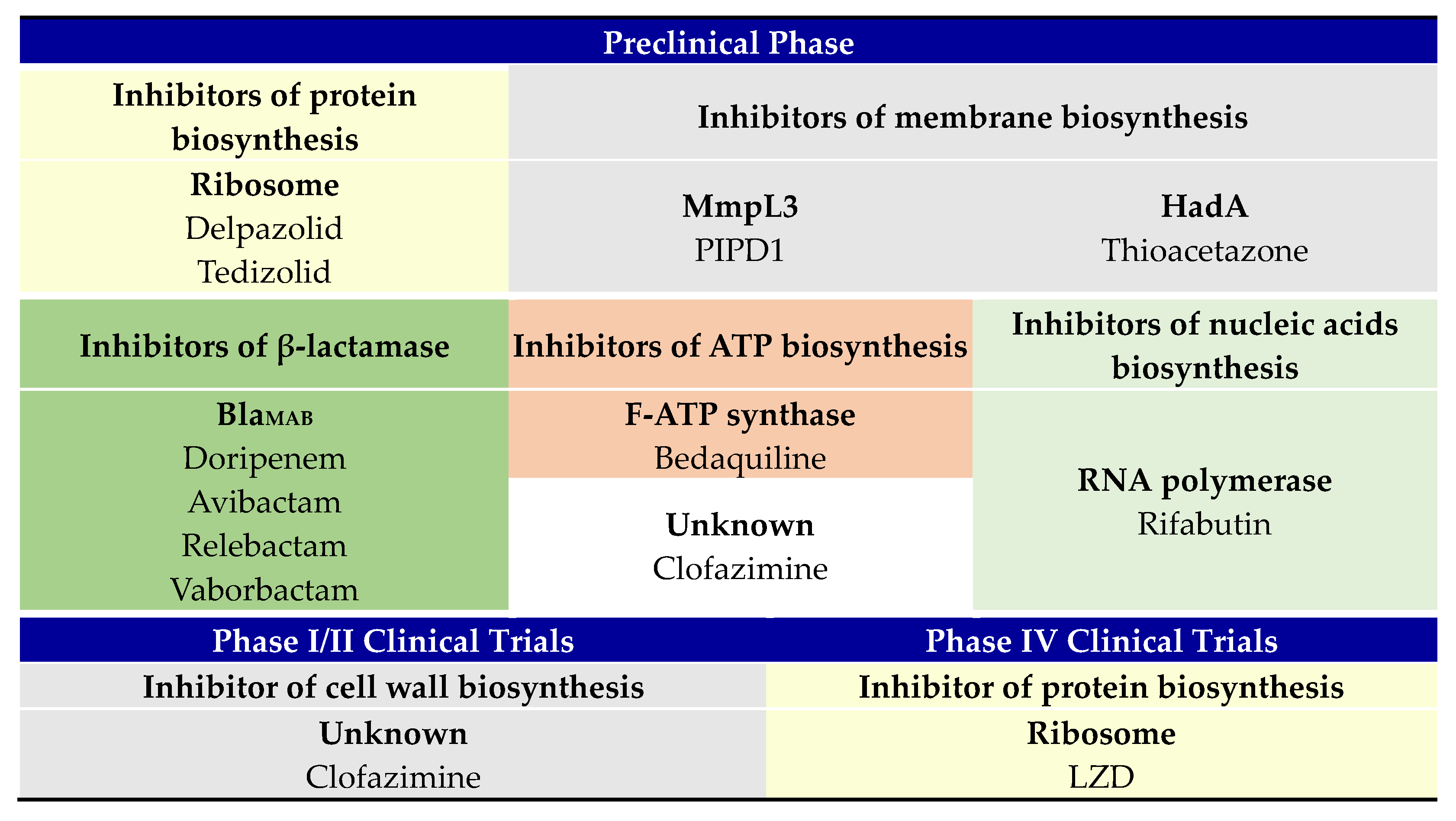
2.1. Preclinical Phase
2.2. Phase II Clinical Trial
2.3. Phase IV Clinical Trial
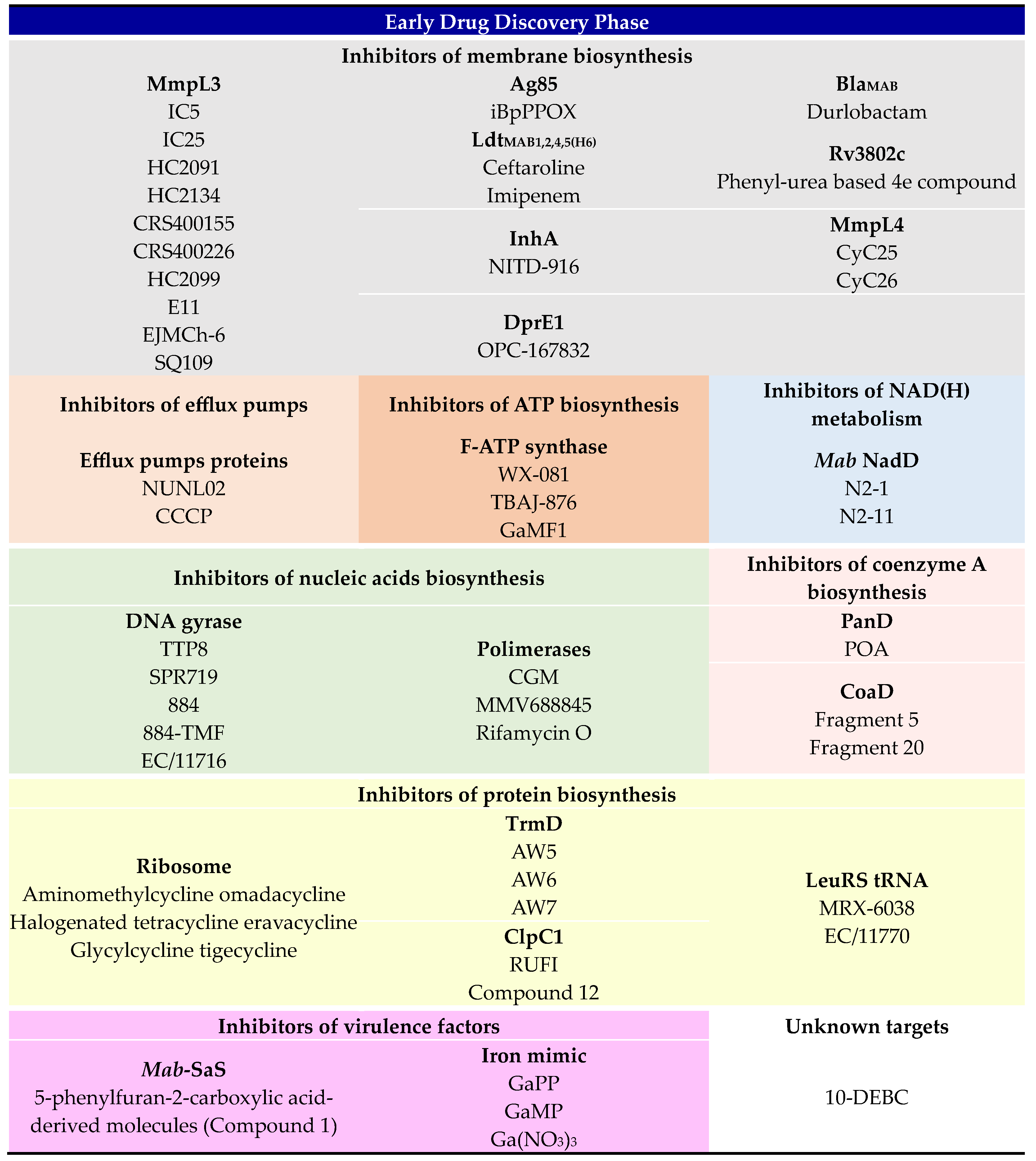
3. Early Drug Discovery Phase
3.1. Inhibitors of Membrane Biosynthesis
3.1.1. Inhibitors of Transporter Proteins
3.1.2. β-Lactamases Inhibitors in Combination with β-Lactams
3.1.3. Other Inhibitors of the Cell Wall Metabolism
3.2. Inhibitors of Efflux Pumps
3.3. Inhibitors of ATP Biosynthesis
3.4. Inhibitors of NAD(H) Metabolism
3.5. Inhibitors of Nucleic Acids Biosynthesis
3.5.1. Inhibitors of DNA Gyrase
3.5.2. Inhibitors of Polymerases
3.6. Inhibitors of Protein Biosynthesis
3.6.1. Molecules That Directly Target the tRNA
3.6.2. Molecules That Target the Enzymes
3.7. Inhibitors of Coenzyme A Biosynthesis
3.8. Targeting Virulence Factors
3.9. Unknown Targets
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Strnad, L.; Winthrop, K.L. Treatment of Mycobacterium abscessus complex. Semin. Respir. Crit. Care Med. 2018, 39, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.; Frerichs, J.B. An unusual acid-fast infection of the knee with subcutaneous, abscess-like lesions of the gluteal region; report of a case with a study of the organism, Mycobacterium abscessus, n. sp. J. Investig. Dermatol. 1953, 20, 133–169. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, F.; Pasek, S.; Schenowitz, C.; Dossat, C.; Barbe, V.; Rottman, M.; Macheras, E.; Heym, B.; Herrmann, J.L.; Daffe, M.; et al. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS ONE 2009, 4, e5660. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Herrmann, J.L.; Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 2020, 18, 392–407. [Google Scholar] [CrossRef]
- Thomson, R.; Tolson, C.; Carter, R.; Coulter, C.; Huygens, F.; Hargreaves, M. Isolation of nontuberculous mycobacteria (NTM) from household water and shower aerosols in patients with pulmonary disease caused by NTM. J. Clin. Microbiol. 2013, 51, 3006–3011. [Google Scholar] [CrossRef]
- Recchia, D.; Stelitano, G.; Stamilla, A.; Gutierrez, D.L.; Degiacomi, G.; Chiarelli, L.R.; Pasca, M.R. Mycobacterium abscessus infections in cystic fibrosis individuals: A review on therapeutic options. Int. J. Mol. Sci. 2023, 24, 4635. [Google Scholar] [CrossRef]
- Pawlik, A.; Garnier, G.; Orgeur, M.; Tong, P.; Lohan, A.; Le Chevalier, F.; Sapriel, G.; Roux, A.L.; Conlon, K.; Honore, N.; et al. Identification and characterization of the genetic changes responsible for the characteristic smooth-to-rough morphotype alterations of clinically persistent Mycobacterium abscessus. Mol. Microbiol. 2013, 90, 612–629. [Google Scholar] [CrossRef]
- Griffith, D.E.; Brown-Elliott, B.A.; Benwill, J.L.; Wallace, R.J., Jr. Mycobacterium abscessus. Pleased to meet you, hope you guess my name. Ann. Am. Thorac. Soc. 2015, 12, 436–439. [Google Scholar] [CrossRef]
- Trias, J.; Jarlier, V.; Benz, R. Porins in the cell wall of mycobacteria. Science 1992, 258, 1479–1481. [Google Scholar] [CrossRef]
- Rominski, A.; Selchow, P.; Becker, K.; Brulle, J.K.; Dal Molin, M.; Sander, P. Elucidation of Mycobacterium abscessus aminoglycoside and capreomycin resistance by targeted deletion of three putative resistance genes. J. Antimicrob. Chemother. 2017, 72, 2191–2200. [Google Scholar] [CrossRef]
- Victoria, L.; Gupta, A.; Gomez, J.L.; Robledo, J. Mycobacterium abscessus complex: A review of recent developments in an emerging pathogen. Front. Cell Infect. Microbiol. 2021, 11, 659997. [Google Scholar] [CrossRef]
- Varley, C.D.; Winthrop, K.L. Nontuberculous mycobacteria: Diagnosis and therapy. Clin. Chest Med. 2022, 43, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Floto, R.A.; Olivier, K.N.; Saiman, L.; Daley, C.L.; Herrmann, J.L.; Nick, J.A.; Noone, P.G.; Bilton, D.; Corris, P.; Gibson, R.L.; et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis: Executive summary. Thorax 2016, 71, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Bentur, L.; Gur, M.; Ashkenazi, M.; Livnat-Levanon, G.; Mizrahi, M.; Tal, A.; Ghaffari, A.; Geffen, Y.; Aviram, M.; Efrati, O. Pilot study to test inhaled nitric oxide in cystic fibrosis patients with refractory Mycobacterium abscessus lung infection. J. Cyst. Fibros. 2020, 19, 225–231. [Google Scholar] [CrossRef]
- Poerio, N.; Riva, C.; Olimpieri, T.; Rossi, M.; Lore, N.I.; De Santis, F.; Henrici De Angelis, L.; Ciciriello, F.; D’Andrea, M.M.; Lucidi, V.; et al. Combined host- and pathogen-directed therapy for the control of Mycobacterium abscessus infection. Microbiol. Spectr. 2022, 10, e0254621. [Google Scholar] [CrossRef]
- Jeong, J.W.; Jung, S.J.; Lee, H.H.; Kim, Y.Z.; Park, T.K.; Cho, Y.L.; Chae, S.E.; Baek, S.Y.; Woo, S.H.; Lee, H.S.; et al. In vitro and in vivo activities of LCB01-0371, a new oxazolidinone. Antimicrob. Agents Chemother. 2010, 54, 5359–5362. [Google Scholar] [CrossRef]
- Study to Evaluate the Efficacy of Delpazolid as Add-on Therapy in Refractory Mycobacterium Abscessus Complex. Available online: https://clinicaltrials.gov/study/NCT06004037?term=delpazolid%20NTM&rank=1 (accessed on 31 August 2023).
- Kim, T.S.; Choe, J.H.; Kim, Y.J.; Yang, C.S.; Kwon, H.J.; Jeong, J.; Kim, G.; Park, D.E.; Jo, E.K.; Cho, Y.L.; et al. Activity of LCB01-0371, a novel oxazolidinone, against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Foti, C.; Piperno, A.; Scala, A.; Giuffre, O. Oxazolidinone antibiotics: Chemical, biological and analytical aspects. Molecules 2021, 26, 4280. [Google Scholar] [CrossRef]
- Zong, Z.; Jing, W.; Shi, J.; Wen, S.; Zhang, T.; Huo, F.; Shang, Y.; Liang, Q.; Huang, H.; Pang, Y. Comparison of in vitro activity and MIC distributions between the novel oxazolidinone delpazolid and linezolid against multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis in China. Antimicrob. Agents Chemother. 2018, 62, 10–1128. [Google Scholar] [CrossRef]
- Stec, J.; Onajole, O.K.; Lun, S.; Guo, H.; Merenbloom, B.; Vistoli, G.; Bishai, W.R.; Kozikowski, A.P. Indole-2-carboxamide-based MmpL3 inhibitors show exceptional antitubercular activity in an animal model of tuberculosis infection. J. Med. Chem. 2016, 59, 6232–6247. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Onajole, O.K.; Stec, J.; Dupont, C.; Viljoen, A.; Richard, M.; Chaira, T.; Lun, S.; Bishai, W.; Raj, V.S.; et al. Targeting mycolic acid transport by indole-2-carboxamides for the treatment of Mycobacterium abscessus infections. J. Med. Chem. 2017, 60, 5876–5888. [Google Scholar] [CrossRef] [PubMed]
- Pandya, A.N.; Prathipati, P.K.; Hegde, P.; Li, W.; Graham, K.F.; Mandal, S.; Drescher, K.M.; Destache, C.J.; Ordway, D.; Jackson, M.; et al. Indole-2-carboxamides are active against Mycobacterium abscessus in a mouse model of acute infection. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Viljoen, A.; Dubar, F.; Blaise, M.; Bernut, A.; Pawlik, A.; Bouchier, C.; Brosch, R.; Guerardel, Y.; Lelievre, J.; et al. A new piperidinol derivative targeting mycolic acid transport in Mycobacterium abscessus. Mol. Microbiol. 2016, 101, 515–529. [Google Scholar] [CrossRef]
- Dover, L.G.; Alahari, A.; Gratraud, P.; Gomes, J.M.; Bhowruth, V.; Reynolds, R.C.; Besra, G.S.; Kremer, L. EthA, a common activator of thiocarbamide-containing drugs acting on different mycobacterial targets. Antimicrob. Agents Chemother. 2007, 51, 1055–1063. [Google Scholar] [CrossRef]
- Halloum, I.; Viljoen, A.; Khanna, V.; Craig, D.; Bouchier, C.; Brosch, R.; Coxon, G.; Kremer, L. Resistance to thiacetazone derivatives active against Mycobacterium abscessus involves mutations in the MmpL5 transcriptional repressor MAB_4384. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Calcagno, T.; Hosseini, S.S.; Hematian, A.; Nojookambari, N.Y.; Karimi-Yazdi, M.; Mirsaeidi, M. Role of Clofazimine in treatment of Mycobacterium avium complex. Front. Med. 2021, 8, 638306. [Google Scholar] [CrossRef]
- van Ingen, J.; Totten, S.E.; Helstrom, N.K.; Heifets, L.B.; Boeree, M.J.; Daley, C.L. In vitro synergy between clofazimine and amikacin in treatment of nontuberculous mycobacterial disease. Antimicrob. Agents Chemother. 2012, 56, 6324–6327. [Google Scholar] [CrossRef]
- Dupont, C.; Viljoen, A.; Thomas, S.; Roquet-Baneres, F.; Herrmann, J.L.; Pethe, K.; Kremer, L. Bedaquiline inhibits the ATP synthase in Mycobacterium abscessus and is effective in infected zebrafish. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Obregon-Henao, A.; Arnett, K.A.; Henao-Tamayo, M.; Massoudi, L.; Creissen, E.; Andries, K.; Lenaerts, A.J.; Ordway, D.J. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob. Agents Chemother. 2015, 59, 6904–6912. [Google Scholar] [CrossRef]
- Rominski, A.; Roditscheff, A.; Selchow, P.; Bottger, E.C.; Sander, P. Intrinsic rifamycin resistance of Mycobacterium abscessus is mediated by ADP-ribosyltransferase MAB_0591. J. Antimicrob. Chemother. 2017, 72, 376–384. [Google Scholar] [CrossRef]
- Johansen, M.D.; Daher, W.; Roquet-Baneres, F.; Raynaud, C.; Alcaraz, M.; Maurer, F.P.; Kremer, L. Rifabutin is bactericidal against intracellular and extracellular forms of Mycobacterium abscessus. Antimicrob. Agents Chemother. 2020, 64, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Story-Roller, E.; Galanis, C.; Lamichhane, G. Beta-lactam combinations that exhibit synergy against Mycobacteroides abscessus clinical isolates. Antimicrob. Agents Chemother. 2021, 65, 10–1128. [Google Scholar] [CrossRef]
- Story-Roller, E.; Maggioncalda, E.C.; Lamichhane, G. Synergistic efficacy of beta-lactam combinations against Mycobacterium abscessus pulmonary infection in mice. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Soroka, D.; Dubee, V.; Soulier-Escrihuela, O.; Cuinet, G.; Hugonnet, J.E.; Gutmann, L.; Mainardi, J.L.; Arthur, M. Characterization of broad-spectrum Mycobacterium abscessus class A beta-lactamase. J. Antimicrob. Chemother. 2014, 69, 691–696. [Google Scholar] [CrossRef]
- Shirley, M. Ceftazidime-avibactam: A review in the treatment of serious Gram-negative bacterial infections. Drugs 2018, 78, 675–692. [Google Scholar] [CrossRef]
- Dubee, V.; Bernut, A.; Cortes, M.; Lesne, T.; Dorchene, D.; Lefebvre, A.L.; Hugonnet, J.E.; Gutmann, L.; Mainardi, J.L.; Herrmann, J.L.; et al. Beta-lactamase inhibition by avibactam in Mycobacterium abscessus. J. Antimicrob. Chemother. 2015, 70, 1051–1058. [Google Scholar] [CrossRef]
- Meir, M.; Bifani, P.; Barkan, D. The addition of avibactam renders piperacillin an effective treatment for Mycobacterium abscessus infection in an in vivo model. Antimicrob. Resist. Infect. Control 2018, 7, 151. [Google Scholar] [CrossRef]
- Kaushik, A.; Ammerman, N.C.; Lee, J.; Martins, O.; Kreiswirth, B.N.; Lamichhane, G.; Parrish, N.M.; Nuermberger, E.L. In vitro activity of the new beta-lactamase inhibitors relebactam and vaborbactam in combination with beta-lactams against Mycobacterium abscessus complex clinical isolates. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Tang, Y.W.; Cheng, B.; Yeoh, S.F.; Lin, R.T.P.; Teo, J.W.P. Tedizolid activity against clinical Mycobacterium abscessus complex isolates—An in vitro characterization study. Front. Microbiol. 2018, 9, 2095. [Google Scholar] [CrossRef]
- Ruth, M.M.; Koeken, V.; Pennings, L.J.; Svensson, E.M.; Wertheim, H.F.L.; Hoefsloot, W.; van Ingen, J. Is there a role for tedizolid in the treatment of non-tuberculous mycobacterial disease? J. Antimicrob. Chemother. 2020, 75, 609–617. [Google Scholar] [CrossRef]
- Poon, Y.K.; Monogue, M.; Sanders, J.; Hoz, R.M.L. 1083. Clinical efficacy of tedizolid for the treatment of Mycobacterium abscessus complex infections in solid organ transplant recipients. Open Forum Infect. Dis. 2020, 7 (Suppl. S1), S570–S571. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, H.Y.; Kim, B.J.; Kim, H.; Seok, S.H.; Kim, B.J.; Kook, Y.H. Effect of amikacin on cell wall glycopeptidolipid synthesis in Mycobacterium abscessus. J. Microbiol. 2017, 55, 640–647. [Google Scholar] [CrossRef]
- Rose, S.J.; Neville, M.E.; Gupta, R.; Bermudez, L.E. Delivery of aerosolized liposomal amikacin as a novel approach for the treatment of nontuberculous mycobacteria in an experimental model of pulmonary infection. PLoS ONE 2014, 9, e108703. [Google Scholar] [CrossRef] [PubMed]
- Olivier, K.N.; Griffith, D.E.; Eagle, G.; McGinnis, J.P., 2nd; Micioni, L.; Liu, K.; Daley, C.L.; Winthrop, K.L.; Ruoss, S.; Addrizzo-Harris, D.J.; et al. Randomized trial of liposomal amikacin for inhalation in nontuberculous mycobacterial lung disease. Am. J. Respir. Crit. Care Med. 2017, 195, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsunoda, A.; Nishimoto, E.; Nishida, K.; Komatsubara, Y.; Onoe, R.; Saji, J.; Mineshita, M. Successful use of linezolid for refractory Mycobacterium abcessus infection: A case report. Respir. Med. Case Rep. 2018, 23, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.L.; Jang, J. Development of delpazolid for the treatment of tuberculosis. Appl. Sci. 2020, 10, 2211. [Google Scholar] [CrossRef]
- Bolla, J.R. Targeting MmpL3 for anti-tuberculosis drug development. Biochem. Soc. Trans. 2020, 48, 1463–1472. [Google Scholar] [CrossRef]
- Franz, N.D.; Belardinelli, J.M.; Kaminski, M.A.; Dunn, L.C.; Calado Nogueira de Moura, V.; Blaha, M.A.; Truong, D.D.; Li, W.; Jackson, M.; North, E.J. Design, synthesis and evaluation of indole-2-carboxamides with pan anti-mycobacterial activity. Bioorg Med. Chem. 2017, 25, 3746–3755. [Google Scholar] [CrossRef]
- Ray, R.; Das, S.; Lobo, M.; Birangal, S.R.; Shenoy, G.G. A holistic molecular modelling approach to design novel indole-2-carboxamide derivatives as potential inhibitors of MmpL3. SAR QSAR Environ. Res. 2022, 33, 551–581. [Google Scholar] [CrossRef]
- Williams, J.T.; Haiderer, E.R.; Coulson, G.B.; Conner, K.N.; Ellsworth, E.; Chen, C.; Alvarez-Cabrera, N.; Li, W.; Jackson, M.; Dick, T.; et al. Identification of new MmpL3 inhibitors by untargeted and targeted mutant screens defines MmpL3 domains with differential resistance. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Ganapathy, U.S.; Dick, T. Why matter matters: Fast-tracking Mycobacterium abscessus drug discovery. Molecules 2022, 27, 6948. [Google Scholar] [CrossRef] [PubMed]
- De Groote, M.A.; Jarvis, T.C.; Wong, C.; Graham, J.; Hoang, T.; Young, C.L.; Ribble, W.; Day, J.; Li, W.; Jackson, M.; et al. Optimization and lead selection of benzothiazole amide analogs toward a novel antimycobacterial agent. Front. Microbiol. 2018, 9, 2231. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Xu, Z.; Lakshmanan, U.; Hill, J.; Choong, M.L.; Chng, S.S.; Yamada, Y.; Poulsen, A.; Dick, T.; Gengenbacher, M. Novel acetamide indirectly targets mycobacterial transporter MmpL3 by proton motive force disruption. Front. Microbiol. 2018, 9, 2960. [Google Scholar] [CrossRef]
- Raynaud, C.; Daher, W.; Johansen, M.D.; Roquet-Baneres, F.; Blaise, M.; Onajole, O.K.; Kozikowski, A.P.; Herrmann, J.L.; Dziadek, J.; Gobis, K.; et al. Active benzimidazole derivatives targeting the MmpL3 transporter in Mycobacterium abscessus. ACS Infect. Dis. 2020, 6, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Stampolaki, M.; Malwal, S.R.; Alvarez-Cabrera, N.; Gao, Z.; Moniruzzaman, M.; Babii, S.O.; Naziris, N.; Rey-Cibati, A.; Valladares-Delgado, M.; Turcu, A.L.; et al. Synthesis and testing of analogs of the tuberculosis drug candidate SQ109 against bacteria and protozoa: Identification of lead compounds against Mycobacterium abscessus and malaria parasites. ACS Infect. Dis. 2023, 9, 342–364. [Google Scholar] [CrossRef] [PubMed]
- Madani, A.; Mallick, I.; Guy, A.; Crauste, C.; Durand, T.; Fourquet, P.; Audebert, S.; Camoin, L.; Canaan, S.; Cavalier, J.F. Dissecting the antibacterial activity of oxadiazolone-core derivatives against Mycobacterium abscessus. PLoS ONE 2020, 15, e0238178. [Google Scholar] [CrossRef]
- Story-Roller, E.; Maggioncalda, E.C.; Cohen, K.A.; Lamichhane, G. Mycobacterium abscessus and beta-lactams: Emerging insights and potential opportunities. Front. Microbiol. 2018, 9, 2273. [Google Scholar] [CrossRef]
- Sayed, A.R.M.; Shah, N.R.; Basso, K.B.; Kamat, M.; Jiao, Y.; Moya, B.; Sutaria, D.S.; Lang, Y.; Tao, X.; Liu, W.; et al. First penicillin-binding protein occupancy patterns for 15 β-lactams and β-lactamase inhibitors in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2020, 65, 10–1128. [Google Scholar] [CrossRef]
- Dousa, K.M.; Nguyen, D.C.; Kurz, S.G.; Taracila, M.A.; Bethel, C.R.; Schinabeck, W.; Kreiswirth, B.N.; Brown, S.T.; Boom, W.H.; Hotchkiss, R.S.; et al. Inhibiting Mycobacterium abscessus cell wall synthesis: Using a novel diazabicyclooctane beta-lactamase inhibitor to augment beta-lactam action. mBio 2022, 13, e0352921. [Google Scholar] [CrossRef]
- Dousa, K.M.; Kurz, S.G.; Taracila, M.A.; Bonfield, T.; Bethel, C.R.; Barnes, M.D.; Selvaraju, S.; Abdelhamed, A.M.; Kreiswirth, B.N.; Boom, W.H.; et al. Insights into the l,d-transpeptidases and d,d-carboxypeptidase of Mycobacterium abscessus: Ceftaroline, imipenem, and novel diazabicyclooctane inhibitors. Antimicrob. Agents Chemother. 2020, 64, 10–1128. [Google Scholar] [CrossRef]
- Vartak, A.; Goins, C.; de Moura, V.C.N.; Schreidah, C.M.; Landgraf, A.D.; Lin, B.; Du, J.; Jackson, M.; Ronning, D.R.; Sucheck, S.J. Biochemical and microbiological evaluation of N-aryl urea derivatives against mycobacteria and mycobacterial hydrolases. Medchemcomm 2019, 10, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- la Buscato, E.; Blocher, R.; Lamers, C.; Klingler, F.M.; Hahn, S.; Steinhilber, D.; Schubert-Zsilavecz, M.; Proschak, E. Design and synthesis of dual modulators of soluble epoxide hydrolase and peroxisome proliferator-activated receptors. J. Med. Chem. 2012, 55, 10771–10775. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz, M.; Roquet-Baneres, F.; Leon-Icaza, S.A.; Abendroth, J.; Boudehen, Y.M.; Cougoule, C.; Edwards, T.E.; Kremer, L. Efficacy and mode of action of a direct inhibitor of Mycobacterium abscessus InhA. ACS Infect. Dis. 2022, 8, 2171–2186. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, G.; Pasca, M.R.; Chiarelli, L.R.; Manina, G.; Mattevi, A.; Binda, C. The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. [Google Scholar] [CrossRef]
- Mikusova, K.; Makarov, V.; Neres, J. DprE1—From the discovery to the promising tuberculosis drug target. Curr. Pharm. Des. 2014, 20, 4379–4403. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Zimmerman, M.D.; Gengenbacher, M.; Dartois, V.; Dick, T. Mycobacterium tuberculosis DprE1 inhibitor OPC-167832 is active against Mycobacterium abscessus in vitro. Antimicrob. Agents Chemother. 2022, 66, e0123722. [Google Scholar] [CrossRef]
- Nguyen, P.C.; Madani, A.; Santucci, P.; Martin, B.P.; Paudel, R.R.; Delattre, S.; Herrmann, J.L.; Spilling, C.D.; Kremer, L.; Canaan, S.; et al. Cyclophostin and Cyclipostins analogues, new promising molecules to treat mycobacterial-related diseases. Int. J. Antimicrob. Agents 2018, 51, 651–654. [Google Scholar] [CrossRef]
- Madani, A.; Ridenour, J.N.; Martin, B.P.; Paudel, R.R.; Abdul Basir, A.; Le Moigne, V.; Herrmann, J.L.; Audebert, S.; Camoin, L.; Kremer, L.; et al. Cyclipostins and Cyclophostin analogues as multitarget Inhibitors that impair growth of Mycobacterium abscessus. ACS Infect. Dis. 2019, 5, 1597–1608. [Google Scholar] [CrossRef]
- Rindi, L. Efflux pump inhibitors against nontuberculous mycobacteria. Int. J. Mol. Sci. 2020, 21, 4191. [Google Scholar] [CrossRef]
- Silva, L., Jr.; Carrion, L.L.; von Groll, A.; Costa, S.S.; Junqueira, E.; Ramos, D.F.; Cantos, J.; Seus, V.R.; Couto, I.; Fernandes, L.D.; et al. In vitro and in silico analysis of the efficiency of tetrahydropyridines as drug efflux inhibitors in Escherichia coli. Int. J. Antimicrob. Agents 2017, 49, 308–314. [Google Scholar] [CrossRef]
- Vianna, J.S.; Ramis, I.B.; Bierhals, D.; von Groll, A.; Ramos, D.F.; Zanatta, N.; Lourenco, M.C.; Viveiros, M.; Almeida da Silva, P.E. Tetrahydropyridine derivative as efflux inhibitor in Mycobacterium abscessus. J. Glob. Antimicrob. Resist. 2019, 17, 296–299. [Google Scholar] [CrossRef]
- Guo, Q.; Chen, J.; Zhang, S.; Zou, Y.; Zhang, Y.; Huang, D.; Zhang, Z.; Li, B.; Chu, H. Efflux pumps contribute to intrinsic clarithromycin resistance in clinical Mycobacterium abscessus isolates. Infect. Drug Resist. 2020, 13, 447–454. [Google Scholar] [CrossRef]
- Chen, S.; Teng, T.; Zhang, Z.; Shang, Y.; Xiao, H.; Jiang, G.; Wang, F.; Jia, J.; Dong, L.; Zhao, L.; et al. Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) exhibits direct antibacterial activity against Mycobacterium abscessus. Infect. Drug Resist. 2021, 14, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Kamariah, N.; Ragunathan, P.; Shin, J.; Saw, W.G.; Wong, C.F.; Dick, T.; Gruber, G. Unique structural and mechanistic properties of mycobacterial F-ATP synthases: Implications for drug design. Prog. Biophys. Mol. Biol. 2020, 152, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Kamariah, N.; Huber, R.G.; Nartey, W.; Bhushan, S.; Bond, P.J.; Gruber, G. Structure and subunit arrangement of mycobacterial F(1)F(O) ATP synthase and novel features of the unique mycobacterial subunit delta. J. Struct. Biol. 2019, 207, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hotra, A.; Suter, M.; Biukovic, G.; Ragunathan, P.; Kundu, S.; Dick, T.; Gruber, G. Deletion of a unique loop in the mycobacterial F-ATP synthase gamma subunit sheds light on its inhibitory role in ATP hydrolysis-driven H(+) pumping. FEBS J. 2016, 283, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Luo, W.; Xu, D.; Guo, F.; Yang, M.; Zhu, Y.; Shen, L.; Chen, S.; Tang, D.; Li, L.; et al. Discovery and preclinical profile of sudapyridine (WX-081), a novel anti-tuberculosis agent. Bioorg Med. Chem. Lett. 2022, 71, 128824. [Google Scholar] [CrossRef]
- Zhu, R.; Shang, Y.; Chen, S.; Xiao, H.; Ren, R.; Wang, F.; Xue, Y.; Li, L.; Li, Y.; Chu, N.; et al. In vitro activity of the sudapyridine (WX-081) against non-tuberculous mycobacteria isolated in Beijing, China. Microbiol. Spectr. 2022, 10, e0137222. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorganic Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Ganapathy, U.S.; Zimmerman, M.D.; Dartois, V.; Gengenbacher, M.; Dick, T. TBAJ-876, a 3,5-dialkoxypyridine analogue of bedaquiline, is active against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2020, 64, 10–1128. [Google Scholar] [CrossRef]
- Hotra, A.; Ragunathan, P.; Ng, P.S.; Seankongsuk, P.; Harikishore, A.; Sarathy, J.P.; Saw, W.G.; Lakshmanan, U.; Sae-Lao, P.; Kalia, N.P.; et al. Discovery of a novel mycobacterial F-ATP synthase inhibitor and its potency in combination with diarylquinolines. Angew. Chem. Int. Ed. Engl. 2020, 59, 13295–13304. [Google Scholar] [CrossRef] [PubMed]
- Ragunathan, P.; Dick, T.; Gruber, G. Anti-Mycobacterium abscessus activity of tuberculosis F-ATP synthase inhibitor GaMF1. Antimicrob. Agents Chemother. 2022, 66, e0001822. [Google Scholar] [CrossRef] [PubMed]
- Rodionova, I.A.; Schuster, B.M.; Guinn, K.M.; Sorci, L.; Scott, D.A.; Li, X.; Kheterpal, I.; Shoen, C.; Cynamon, M.; Locher, C.; et al. Metabolic and bactericidal effects of targeted suppression of NadD and NadE enzymes in mycobacteria. mBio 2014, 5, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; O’Brien, K.M.; Sharma, R.; Boshoff, H.I.; Rehren, G.; Chakraborty, S.; Wallach, J.B.; Monteleone, M.; Wilson, D.J.; Aldrich, C.C.; et al. A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. USA 2013, 110, 19095–19100. [Google Scholar] [CrossRef] [PubMed]
- Sorci, L.; Blaby, I.; De Ingeniis, J.; Gerdes, S.; Raffaelli, N.; de Crecy Lagard, V.; Osterman, A. Genomics-driven reconstruction of acinetobacter NAD metabolism: Insights for antibacterial target selection. J. Biol. Chem. 2010, 285, 39490–39499. [Google Scholar] [CrossRef]
- Osterman, A.L.; Rodionova, I.; Li, X.; Sergienko, E.; Ma, C.T.; Catanzaro, A.; Pettigrove, M.E.; Reed, R.W.; Gupta, R.; Rohde, K.H.; et al. Novel antimycobacterial compounds suppress NAD biogenesis by targeting a unique pocket of NaMN adenylyltransferase. ACS Chem. Biol. 2019, 14, 949–958. [Google Scholar] [CrossRef]
- Cambau, E.; Guillard, T. Antimicrobials that affect the synthesis and conformation of nucleic acids. Rev. Sci. Tech. 2012, 31, 65–87. [Google Scholar] [CrossRef]
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA topoisomerases: Advances in understanding of cellular roles and multi-protein complexes via structure-function analysis. Bioessays 2021, 43, e2000286. [Google Scholar] [CrossRef]
- Henderson, S.R.; Stevenson, C.E.M.; Malone, B.; Zholnerovych, Y.; Mitchenall, L.A.; Pichowicz, M.; McGarry, D.H.; Cooper, I.R.; Charrier, C.; Salisbury, A.M.; et al. Structural and mechanistic analysis of ATPase inhibitors targeting mycobacterial DNA gyrase. J. Antimicrob. Chemother. 2020, 75, 2835–2842. [Google Scholar] [CrossRef]
- Madani, A.; Negatu, D.A.; El Marrouni, A.; Miller, R.R.; Boyce, C.W.; Murgolo, N.; Bungard, C.J.; Zimmerman, M.D.; Dartois, V.; Gengenbacher, M.; et al. Activity of tricyclic pyrrolopyrimidine Gyrase B inhibitor against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2022, 66, e0066922. [Google Scholar] [CrossRef]
- Aragaw, W.W.; Cotroneo, N.; Stokes, S.; Pucci, M.; Critchley, I.; Gengenbacher, M.; Dick, T. In vitro resistance against DNA gyrase inhibitor SPR719 in Mycobacterium avium and Mycobacterium abscessus. Microbiol. Spectr. 2022, 10, e0132121. [Google Scholar] [CrossRef] [PubMed]
- Stokes, S.S.; Vemula, R.; Pucci, M.J. Advancement of GyrB inhibitors for treatment of infections caused by Mycobacterium tuberculosis and non-tuberculous mycobacteria. ACS Infect. Dis. 2020, 6, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Negatu, D.A.; Beuchel, A.; Madani, A.; Alvarez, N.; Chen, C.; Aragaw, W.W.; Zimmerman, M.D.; Laleu, B.; Gengenbacher, M.; Dartois, V.; et al. Piperidine-4-carboxamides target DNA gyrase in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2021, 65, e0067621. [Google Scholar] [CrossRef]
- Gibson, E.G.; Bax, B.; Chan, P.F.; Osheroff, N. Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect. Dis. 2019, 5, 570–581. [Google Scholar] [CrossRef]
- Ganapathy, U.S.; Del Rio, R.G.; Cacho-Izquierdo, M.; Ortega, F.; Lelievre, J.; Barros-Aguirre, D.; Aragaw, W.W.; Zimmerman, M.D.; Lindman, M.; Dartois, V.; et al. A Mycobacterium tuberculosis NBTI DNA gyrase inhibitor is active against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2021, 65, e0151421. [Google Scholar] [CrossRef] [PubMed]
- Ditse, Z.; Lamers, M.H.; Warner, D.F. DNA replication in Mycobacterium tuberculosis. Microbiol. Spectr. 2017, 5, 10–1128. [Google Scholar] [CrossRef]
- Cossar, P.J.; Lewis, P.J.; McCluskey, A. Protein-protein interactions as antibiotic targets: A medicinal chemistry perspective. Med. Res. Rev. 2020, 40, 469–494. [Google Scholar] [CrossRef]
- Terlain, B.; Thomas, J.P. Structure of griselimycin, polypeptide antibiotic extracted from streptomyces cultures. II. Structure of griselimycin. Bull. Soc. Chim. Fr. 1971, 6, 2357–2362. [Google Scholar]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; Konig, C.; et al. Antibiotics targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef]
- Aragaw, W.W.; Roubert, C.; Fontaine, E.; Lagrange, S.; Zimmerman, M.D.; Dartois, V.; Gengenbacher, M.; Dick, T. Cyclohexyl-griselimycin is active against Mycobacterium abscessus in mice. Antimicrob. Agents Chemother. 2022, 66, e0140021. [Google Scholar] [CrossRef]
- Mann, L.; Ganapathy, U.S.; Abdelaziz, R.; Lang, M.; Zimmerman, M.D.; Dartois, V.; Dick, T.; Richter, A. In vitro profiling of the synthetic RNA polymerase inhibitor MMV688845 against Mycobacterium abscessus. Microbiol. Spectr. 2022, 10, e0276022. [Google Scholar] [CrossRef]
- Lin, W.; Mandal, S.; Degen, D.; Liu, Y.; Ebright, Y.W.; Li, S.; Feng, Y.; Zhang, Y.; Mandal, S.; Jiang, Y.; et al. Structural basis of Mycobacterium tuberculosis transcription and transcription inhibition. Mol. Cell 2017, 66, 169–179. [Google Scholar] [CrossRef]
- Hanh, B.T.B.; Park, J.W.; Kim, T.H.; Kim, J.S.; Yang, C.S.; Jang, K.; Cui, J.; Oh, D.C.; Jang, J. Rifamycin O, an alternative anti-Mycobacterium abscessus agent. Molecules 2020, 25, 1597. [Google Scholar] [CrossRef]
- Bacchi, A.; Pelizzi, G.; Nebuloni, M.; Ferrari, P. Comprehensive study on structure-activity relationships of rifamycins: Discussion of molecular and crystal structure and spectroscopic and thermochemical properties of rifamycin O. J. Med. Chem. 1998, 41, 2319–2332. [Google Scholar] [CrossRef]
- Ramakrishnan, V. Ribosome structure and the mechanism of translation. Cell 2002, 108, 557–572. [Google Scholar] [CrossRef]
- Brandi, L.; Fabbretti, A.; Pon, C.L.; Dahlberg, A.E.; Gualerzi, C.O. Initiation of protein synthesis: A target for antimicrobials. Expert. Opin. Ther. Targets 2008, 12, 519–534. [Google Scholar] [CrossRef]
- Jayasekera, M.M.; Onheiber, K.; Keith, J.; Venkatesan, H.; Santillan, A.; Stocking, E.M.; Tang, L.; Miller, J.; Gomez, L.; Rhead, B.; et al. Identification of novel inhibitors of bacterial translation elongation factors. Antimicrob. Agents Chemother. 2005, 49, 131–136. [Google Scholar] [CrossRef]
- Song, S.; Gao, P.; Sun, L.; Kang, D.; Kongsted, J.; Poongavanam, V.; Zhan, P.; Liu, X. Recent developments in the medicinal chemistry of single boron atom-containing compounds. Acta Pharm. Sin. B 2021, 11, 3035–3059. [Google Scholar] [CrossRef]
- Wu, W.; He, S.; Li, A.; Guo, Q.; Tan, Z.; Liu, S.; Wang, X.; Zhang, Z.; Li, B.; Chu, H. A novel leucyl-tRNA synthetase inhibitor, MRX-6038, expresses anti-Mycobacterium abscessus activity in vitro and in vivo. Antimicrob. Agents Chemother. 2022, 66, e0060122. [Google Scholar] [CrossRef]
- Ganapathy, U.S.; Del Rio, R.G.; Cacho-Izquierdo, M.; Ortega, F.; Lelievre, J.; Barros-Aguirre, D.; Lindman, M.; Dartois, V.; Gengenbacher, M.; Dick, T. A leucyl-tRNA synthetase inhibitor with broad-spectrum anti-mycobacterial activity. Antimicrob. Agents Chemother. 2021, 65, 10–1128. [Google Scholar] [CrossRef]
- Ganapathy, U.S.; Gengenbacher, M.; Dick, T. Epetraborole is active against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2021, 65, e0115621. [Google Scholar] [CrossRef]
- Whitehouse, A.J.; Thomas, S.E.; Brown, K.P.; Fanourakis, A.; Chan, D.S.; Libardo, M.D.J.; Mendes, V.; Boshoff, H.I.M.; Floto, R.A.; Abell, C.; et al. Development of inhibitors against Mycobacterium abscessus tRNA (m(1)G37) methyltransferase (TrmD) using fragment-based approaches. J. Med. Chem. 2019, 62, 7210–7232. [Google Scholar] [CrossRef]
- Thomas, S.E.; Whitehouse, A.J.; Brown, K.; Burbaud, S.; Belardinelli, J.M.; Sangen, J.; Lahiri, R.; Libardo, M.D.J.; Gupta, P.; Malhotra, S.; et al. Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification. Nucleic Acids Res. 2020, 48, 8099–8112. [Google Scholar] [CrossRef]
- Day, L.E. Tetracycline inhibition of cell-free protein synthesis I binding of tetracycline to components of the system. J. Bacteriol. 1966, 91, 1917–1923. [Google Scholar] [CrossRef]
- Honeyman, L.; Ismail, M.; Nelson, M.L.; Bhatia, B.; Bowser, T.E.; Chen, J.; Mechiche, R.; Ohemeng, K.; Verma, A.K.; Cannon, E.P.; et al. Structure-activity relationship of the aminomethylcyclines and the discovery of omadacycline. Antimicrob. Agents Chemother. 2015, 59, 7044–7053. [Google Scholar] [CrossRef]
- Ronn, M.; Zhu, Z.; Hogan, P.C.; Zhang, W.-Y.; Niu, J.; Katz, C.E.; Dunwoody, N.; Gilicky, O.; Deng, Y.; Hunt, D.K.; et al. Process R&D of eravacycline: The first fully synthetic fluorocycline in clinical development. Org. Process Res. Dev. 2013, 17, 838–845. [Google Scholar] [CrossRef]
- Greer, N.D. Tigecycline (Tygacil): The first in the glycylcycline class of antibiotics. Bayl. Univ. Med. Cent. Proc. 2006, 19, 155–161. [Google Scholar] [CrossRef]
- Kaushik, A.; Ammerman, N.C.; Martins, O.; Parrish, N.M.; Nuermberger, E.L. In vitro activity of new tetracycline analogs omadacycline and eravacycline against drug-resistant clinical isolates of Mycobacterium abscessus. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Bax, H.I.; de Vogel, C.P.; Mouton, J.W.; de Steenwinkel, J.E.M. Omadacycline as a promising new agent for the treatment of infections with Mycobacterium abscessus. J. Antimicrob. Chemother. 2019, 74, 2930–2933. [Google Scholar] [CrossRef]
- Brown-Elliott, B.A.; Wallace, R.J., Jr. In vitro susceptibility testing of Omadacycline against nontuberculous mycobacteria. Antimicrob. Agents Chemother. 2021, 65, 10–1128. [Google Scholar] [CrossRef]
- Choules, M.P.; Wolf, N.M.; Lee, H.; Anderson, J.R.; Grzelak, E.M.; Wang, Y.; Ma, R.; Gao, W.; McAlpine, J.B.; Jin, Y.Y.; et al. Rufomycin targets ClpC1 proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Shetye, G.; Yu, Y.; Santarsiero, B.D.; Klein, L.L.; Abad-Zapatero, C.; Wolf, N.M.; Cheng, J.; Jin, Y.; Lee, H.; et al. Antimycobacterial rufomycin analogues from Streptomyces atratus strain MJM3502. J. Nat. Prod. 2020, 83, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Tuck, K.L.; Saldanha, S.A.; Birch, L.M.; Smith, A.G.; Abell, C. The design and synthesis of inhibitors of pantothenate synthetase. Org. Biomol. Chem. 2006, 4, 3598–3610. [Google Scholar] [CrossRef] [PubMed]
- Saw, W.G.; Leow, C.Y.; Harikishore, A.; Shin, J.; Cole, M.S.; Aragaw, W.W.; Ragunathan, P.; Hegde, P.; Aldrich, C.C.; Dick, T.; et al. Structural and mechanistic insights into Mycobacterium abscessus aspartate decarboxylase PanD and a pyrazinoic acid-derived inhibitor. ACS Infect. Dis. 2022, 8, 1324–1335. [Google Scholar] [CrossRef]
- Thomas, S.E.; McCarthy, W.J.; El Bakali, J.; Brown, K.P.; Kim, S.Y.; Blaszczyk, M.; Mendes, V.; Abell, C.; Floto, R.A.; Coyne, A.G.; et al. Structural characterization of Mycobacterium abscessus phosphopantetheine adenylyl transferase ligand interactions: Implications for fragment-based drug design. Front. Mol. Biosci. 2022, 9, 880432. [Google Scholar] [CrossRef]
- Stelitano, G.; Cocorullo, M.; Mori, M.; Villa, S.; Meneghetti, F.; Chiarelli, L.R. Iron acquisition and metabolism as a promising target for antimicrobials (bottlenecks and opportunities): Where do we stand? Int. J. Mol. Sci. 2023, 24, 6181. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Cazzaniga, G.; Gelain, A.; Tresoldi, A.; Cocorullo, M.; Roversi, M.; Chiarelli, L.R.; Tomaiuolo, M.; Del Re, P.; et al. Targeting siderophore-mediated iron uptake in M. abscessus: A new strategy to limit the virulence of non-tuberculous mycobacteria. Pharmaceutics 2023, 15, 502. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Griego, A.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Pini, E.; Camera, M.; Canzano, P.; Fumagalli, A.; et al. Synthesis and assessment of the in vitro and ex vivo activity of salicylate synthase (Mbti) inhibitors as new candidates for the treatment of mycobacterial infections. Pharmaceuticals 2022, 15, 992. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Gelain, A.; Pini, E.; Chiarelli, L.R.; Sammartino, J.C.; Poli, G.; Tuccinardi, T.; Beretta, G.; Porta, A.; et al. Shedding X-ray light on the role of magnesium in the activity of Mycobacterium tuberculosis salicylate synthase (MbtI) for drug design. J. Med. Chem. 2020, 63, 7066–7080. [Google Scholar] [CrossRef]
- Shyam, M.; Shilkar, D.; Verma, H.; Dev, A.; Sinha, B.N.; Brucoli, F.; Bhakta, S.; Jayaprakash, V. The mycobactin biosynthesis pathway: A prospective therapeutic target in the battle against tuberculosis. J. Med. Chem. 2021, 64, 71–100. [Google Scholar] [CrossRef]
- Scaccaglia, M.; Rega, M.; Vescovi, M.; Pinelli, S.; Tegoni, M.; Bacci, C.; Pelosi, G.; Bisceglie, F. Gallium(III)-pyridoxal thiosemicarbazone derivatives as nontoxic agents against Gram-negative bacteria. Metallomics 2022, 14, mfac070. [Google Scholar] [CrossRef] [PubMed]
- Chitambar, C.R. Gallium and its competing roles with iron in biological systems. Biochim. Biophys. Acta 2016, 1863, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Kelson, A.B.; Carnevali, M.; Truong-Le, V. Gallium-based anti-infectives: Targeting microbial iron-uptake mechanisms. Curr. Opin. Pharmacol. 2013, 13, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.R.; Switzer, B.; Britigan, B.E.; Narayanasamy, P. Gallium porphyrin and gallium nitrate synergistically inhibit mycobacterial species by targeting different aspects of iron/heme metabolism. ACS Infect. Dis. 2020, 6, 2582–2591. [Google Scholar] [CrossRef]
- Hanh, B.T.B.; Kim, T.H.; Park, J.W.; Lee, D.G.; Kim, J.S.; Du, Y.E.; Yang, C.S.; Oh, D.C.; Jang, J. Etamycin as a novel Mycobacterium abscessus inhibitor. Int. J. Mol. Sci. 2020, 21, 6908. [Google Scholar] [CrossRef]
- Han, H.W.; Seo, H.H.; Jo, H.Y.; Han, H.J.; Falcao, V.C.A.; Delorme, V.; Heo, J.; Shum, D.; Choi, J.H.; Lee, J.M.; et al. Drug discovery platform targeting M. tuberculosis with human embryonic stem cell-derived macrophages. Stem Cell Rep. 2019, 13, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Thimmaiah, K.N.; Easton, J.B.; Germain, G.S.; Morton, C.L.; Kamath, S.; Buolamwini, J.K.; Houghton, P.J. Identification of N10-substituted phenoxazines as potent and specific inhibitors of Akt signaling. J. Biol. Chem. 2005, 280, 31924–31935. [Google Scholar] [CrossRef]
- Lee, D.-G.; Kim, H.-J.; Lee, Y.; Kim, J.-H.; Hwang, Y.; Ha, J.; Ryoo, S. 10-DEBC hydrochloride as a promising new agent against infection of Mycobacterium abscessus. Int. J. Mol. Sci. 2022, 23, 591. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Code | Structure | Code | Structure |
|---|---|---|---|
| IC5 |  | IC25 |  |
| HC2091 |  | CRS400226 |  |
| E11 |  | EJMCh-6 |  |
| SQ109 |  | iBpPPOX |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| 4e |  | NITD-916 |  |
| OPC-167832 |  | CysC26 |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| NUNL02 |  | CCCP |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| WX-081 |  | GaMF1 |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| N2-1 |  | N2-11 |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| TPP8 |  | SPR719 |  |
| 844-TMF |  | EC/11716 |  |
| MMV688845 |  | Ryfamicin O |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| EC/11770 |  | AW6 |  |
| RUFI |  | Compound 12 |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| Fragment 5 |  | Fragment 20 |  |
| Code | Structure | Code | Structure |
|---|---|---|---|
| Compound 1 |  | GaPP |  |
| Code | Structure |
|---|---|
| 10-DEBC |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cocorullo, M.; Bettoni, C.; Foiadelli, S.; Stelitano, G. Moles of Molecules against Mycobacterium abscessus: A Review of Current Research. Future Pharmacol. 2023, 3, 637-663. https://doi.org/10.3390/futurepharmacol3030041
Cocorullo M, Bettoni C, Foiadelli S, Stelitano G. Moles of Molecules against Mycobacterium abscessus: A Review of Current Research. Future Pharmacology. 2023; 3(3):637-663. https://doi.org/10.3390/futurepharmacol3030041
Chicago/Turabian StyleCocorullo, Mario, Christian Bettoni, Sara Foiadelli, and Giovanni Stelitano. 2023. "Moles of Molecules against Mycobacterium abscessus: A Review of Current Research" Future Pharmacology 3, no. 3: 637-663. https://doi.org/10.3390/futurepharmacol3030041