2.2. Procedures
Preparation of 3-Iodo-2-(1,1,2,2,2-Pentamethyldisilanyl)thiophene: 2,3-Diiodothiophene (9.646 g, 28.7 mmol) was added to 30 mL of dry THF in a 200-mL three-necked flask fitted with a stirrer, reflux condenser, and dropping funnel. Thereafter, a THF solution comprising 18 mL (36.0 mmol) of 2.0 M ethyl magnesium chloride was added dropwise at room temperature. The mixture was stirred for 1.5 h at room temperature and 5.885 g (35.3 mmol) of chloropentamethyldisilane was added. The resulting mixture was stirred for 6 h and then treated with distilled water. The organic layer was separated, washed with water, and dried over anhydrous magnesium sulfate. The solvent was evaporated, and the residue was chromatographed on a silica gel column and eluted with hexane to obtain 7.268 g (70% yield) of 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene. HR-MS: calcd. for C9H17Si2SI: (M+):340.1806, found: 340.1800. MS m/z 340 (M+); 1H NMR δ(CDCl3) 0.16 (s, 9H, Me3Si), 0.48 (s, 6H, Me2Si), 7.22 (d, 1H, thienyl ring proton, J = 4.4 Hz), 7.38 (d, 1H, thienyl ring proton, J = 4.4 Hz); 13C NMR δ(CDCl3) −2.6 (Me2Si), −1.4 (Me3Si), 86.4, 131.5, 138.1, 139.3 (thienyl ring carbons); 29Si NMR δ(CDCl3) −20.3, −17.9.
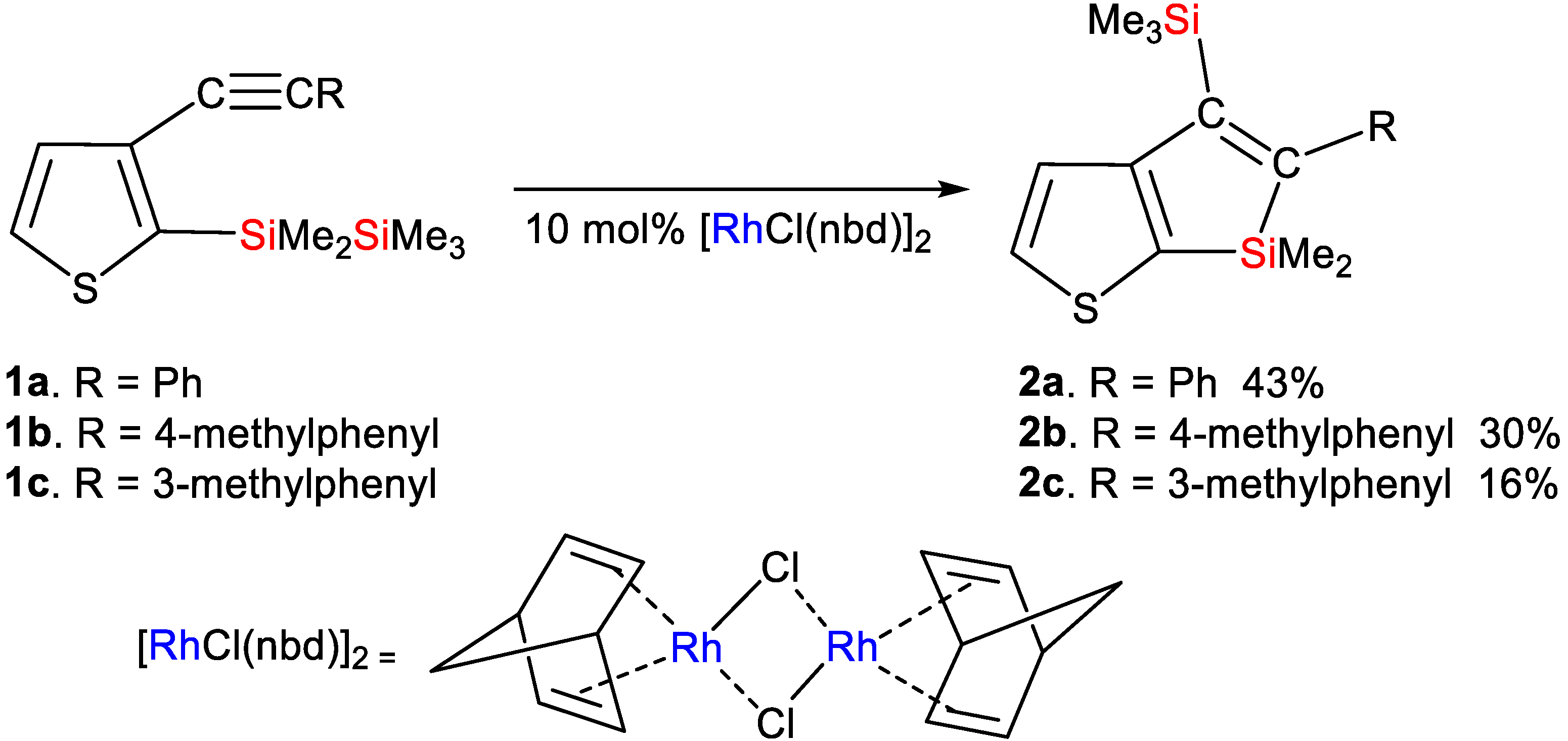
Synthesis of Compound 1a: In a 100-mL three-necked flask equipped with a stirrer, reflux condenser, and dropping funnel, 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene (1.751 g, 5.14 mmol), bis(triphenylphosphine)dichloropalladium (0.181 g, 0.258 mmol), and copper(I) iodide (0.049 g, 0.257 mmol) were combined with 15 mL of dry triethylamine. Following this, ethynylbenzene (1.063 g, 10.4 mmol) was slowly added to the mixture at room temperature via dropwise addition. The resulting mixture was refluxed for 12 h. Afterward, distilled water was added to the mixture, and the organic layer was isolated, washed with water, and dried using anhydrous magnesium sulfate. Evaporation of the solvent followed, and the remaining residue was subjected to chromatography on a silica gel column, eluting with hexane–ethyl acetate (50:1). This process yielded 0.702 g (43% yield) of compound 1a: HR-MS: calcd. for C17H22Si2S (M+): 314.0981, found: 314.0980. MS m/z 314 (M+); 1H NMR δ(CDCl3) 0.11 (s, 9H, Me3Si), 0.51 (s, 6H, Me2Si), 7.29 (d, 1H, thienyl ring proton, J = 4.8 Hz), 7.33–7.35 (m, 3H, phenyl ring protons), 7.48 (d, 1H, thienyl ring proton, J = 4.8 Hz), 7.49–7.51 (m, 2H, phenyl ring protons); 13C NMR δ(CDCl3) −3.1 (Me2Si), −1.9 (Me3Si), 86.9, 90.3 (sp carbons), 123.6, 128.0, 128.4, 128.6, 129.3, 131.2, 132.5, 142.7 (thienyl and phenyl ring carbons); 29Si NMR δ(CDCl3) −22.7, −17.4.
Synthesis of Compound 1b: A total of 1.028 g (3.02 mmol) of 3-Iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, 0.106 g (0.151 mmol) of bis(triphenylphosphine)dichloropalladium, and 0.029 g (0.152 mmol) of copper(I) iodide were combined with 10 mL of dry triethylamine in a 100-mL three-necked flask equipped with a stirrer, reflux condenser, and dropping funnel. While at room temperature, 0.701 g (6.04 mmol) of 4-ethynyltoluene was slowly added dropwise to this mixture. The resulting mixture was then heated under reflux for 12 h. Afterward, distilled water was added to the mixture, and the organic layer was isolated, washed with water, and dried using anhydrous magnesium sulfate. Subsequently, the solvent was evaporated, and the remaining residue was subjected to chromatography on a silica gel column using a hexane–ethyl acetate (50:1) elution system, yielding 0.324 g (33% yield) of compound 1b: HR-MS: calcd. for C18H24Si2S (M+): 328.1137, found: 328.1135. MS m/z 328 (M+); 1H NMR δ(CDCl3) 0.11 (s, 9H, Me3Si), 0.50 (s, 6H, Me2Si), 2.37 (s, 3H, CH3), 7.15 (d, 2H, phenylene ring protons, J = 8.0 Hz), 7.28 (d, 1H, thienyl ring proton, J = 4.8 Hz), 7.39 (d, 2H, phenylene ring protons, J = 8.0 Hz), 7.47 (d, 1H, thienyl ring proton, J = 4.8 Hz); 13C NMR δ(CDCl3) −3.2 (Me2Si), −2.0 (Me3Si), 21.5 (CH3), 86.2, 90.4 (sp carbons), 120.5, 128.8, 129.1, 129.3, 131.1, 132.4, 138.1, 142.3 (thienyl and phenylene ring carbons); 29Si NMR δ(CDCl3) −22.7, −17.5.
Synthesis of Compound 1c: In a 100 mL three-necked flask equipped with a stirrer, reflux condenser, and dropping funnel, 1.007 g (2.96 mmol) of 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, along with 0.104 g (0.148 mmol) of bis(triphenylphosphine)dichloropalladium and 0.029 g (0.152 mmol) of copper(I) iodide, were combined with 10 mL of dry triethylamine. To this mixture, 0.679 g (5.85 mmol) of 3-ethynyltoluene was slowly added dropwise at room temperature. The resulting mixture was then refluxed for 12 h. Following this, distilled water was added to the mixture, and the separated organic layer was washed with water and dried using anhydrous magnesium sulfate. After evaporating the solvent, the remaining residue was subjected to chromatography on a silica gel column employing a hexane–ethyl acetate (50:1) elution system, yielding 0.501 g (52% yield) of compound 1c: HR-MS: calcd. for C18H24Si2S (M+): 328.1137, found: 328.1130. MS m/z 328 (M+); 1H NMR δ(CDCl3) 0.11 (s, 9H, Me3Si), 0.51 (s, 6H, Me2Si), 2.32 (s, 3H, CH3), 7.14 (d, 1H, phenylene ring proton, J = 7.6 Hz), 7.23 (t, 1H, phenylene ring proton, J = 7.6 Hz), 7.28 (d, 1H, thienyl ring proton, J = 4.4 Hz), 7.31 (d, 1H, phenylene ring proton, J = 7.6 Hz), 7.32 (s, 1H, phenylene ring proton), 7.47 (d, 1H, thienyl ring proton, J = 4.4 Hz); 13C NMR δ(CDCl3) −3.1 (Me2Si), −2.0 (Me3Si), 21.3 (CH3), 86.5, 90.4 (sp carbons), 123.4, 128.2, 128.3, 128.7, 128.9, 129.3, 131.8, 132.5, 138.0, 142.6 (thienyl and phenylene ring carbons); 29Si NMR δ(CDCl3) −22.7, −17.5.
Reaction of Compound 1a in the Presence of [RhCl(CO)2]2 Catalyst: Compound 1a (0.234 g, 0.745 mmol) and 0.029 g (0.0746 mmol) of [RhCl(CO)2]2 were added to dry toluene (1.5 mL) in a 30 mL two-necked flask fitted with a reflux condenser. Subsequently, the mixture underwent reflux for 12 h. Following this, distilled water was added to the mixture, leading to the isolation of the organic layer, which was washed with water and desiccated using anhydrous magnesium sulfate. The solvent was evaporated, and the resulting residue underwent chromatography on a silica gel column, eluting with hexane–ethyl acetate (50:1). Ultimately, the initial compound 1a was retrieved.
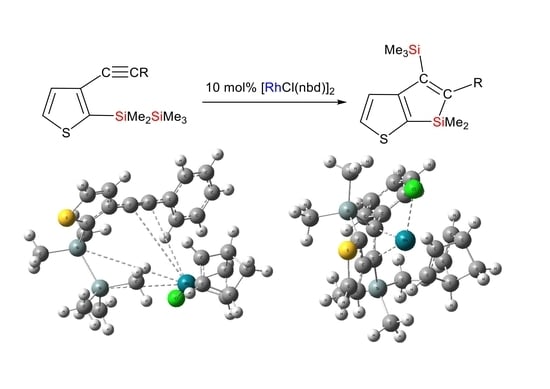
Reaction of Compound 1a in the Presence of [RhCl(nbd)]2 Catalyst: In a 30 mL two-necked flask equipped with a reflux condenser, dry toluene (1.5 mL), compound 1a (0.103 g, 0.327 mmol), and [RhCl(nbd)]2 (0.015 g, 0.0325 mmol) were combined. The mixture was refluxed for 1 h. Subsequently, distilled water was added to the mixture, leading to the isolation of the organic layer, which was washed with water and dried using anhydrous magnesium sulfate. After evaporating the solvent, chromatography on a silica gel column was performed, eluting with hexane, resulting in the yield of 0.044 g (43%) of compound 2a: HR-MS: calcd. for C17H22Si2S (M+): 314.0981, found: 314.0979. MS m/z 314 (M+); 1H NMR δ(CDCl3) 0.01 (s, 9H, Me3Si), 0.31 (s, 6H, Me2Si), 7.04 (dd, 2H, phenyl ring protons, J = 8.2 Hz, 1.6 Hz), 7.22 (tt, 1H, phenyl ring proton, J = 8.2 Hz, 1.6 Hz), 7.30 (t, 2H, phenyl ring protons, J = 8.2 Hz), 7.31 (d, 1H, thienyl ring proton, J = 4.8 Hz), 7.64 (d, 1H, thienyl ring proton, J = 4.8 Hz); 13C NMR δ(CDCl3) −4.1 (Me2Si), 0.8 (Me3Si), 125.2, 125.8, 127.1, 127.9, 132.5, 133.6, 143.0, 150.1, 160.8, 162.4 (thienyl, phenyl ring and olefinic carbons); 29Si NMR δ(CDCl3) −6.8, 0.1.
Reaction of Compound 1a in the Presence of RhCl(PPh3)3 Catalyst: A total of 0.222 g (0.707 mmol) of compound 1a and 0.065 g (0.0703 mmol) of RhCl(PPh3)3 were combined with dry toluene (1.5 mL) in a 30 mL two-necked flask equipped with a reflux condenser. The mixture underwent reflux heating for 12 h. Afterward, distilled water was added to the mixture, followed by separation of the organic layer, washing with water, and drying using anhydrous magnesium sulfate. The solvent was then evaporated, resulting in a residue that underwent chromatography on a silica gel column. Elution was performed using hexane–ethyl acetate (50:1) as the solvent, ultimately leading to the recovery of the initial compound 1a.
Reaction of Compound 1b in the Presence of [RhCl(nbd)]2 Catalyst: In a 30 mL two-necked flask equipped with a reflux condenser, dry toluene (1.5 mL) was combined with compound 1b (0.108 g, 0.329 mmol) and [RhCl(nbd)]2 (0.015 g, 0.0325 mmol). The resulting mixture was refluxed for 1 h. Post-reflux, distilled water was added to the mixture, followed by separation of the organic layer, which was then washed with water and dried using anhydrous magnesium sulfate. The solvent was evaporated, and the remaining residue was subjected to chromatography on a silica gel column, eluting with hexane, yielding 0.032 g (30% yield) of compound 2b: HR-MS: calcd. for C18H24Si2S (M+): 328.1137, found: 328.1135. MS m/z 328 (M+); 1H NMR δ(CDCl3) 0.02 (s, 9H, Me3Si), 0.30 (s, 6H, Me2Si), 2.36 (s, 3H, CH3), 6.93 (d, 2H, phenylene ring protons, J = 8.0 Hz), 7.10 (d, 2H, phenylene ring protons, J = 8.0 Hz), 7.30 (d, 1H, thienyl ring proton, J = 4.8 Hz), 7.63 (d, 1H, thienyl ring proton, J = 4.8 Hz); 13C NMR δ(CDCl3) −4.1 (Me2Si), 0.9 (Me3Si), 21.2 (CH3), 125.2, 127.0, 128.6, 132.4, 133.5, 135.3, 139.9, 149.9, 160.9, 162.5 (thienyl, phenylene ring, and olefinic carbons); 29Si NMR δ(CDCl3) −7.0, −0.3.
Reaction of Compound 1c in the Presence of [RhCl(nbd)]2 Catalyst: In a 30 mL two-necked flask with a reflux condenser, compound 1c (0.305 g, 0.928 mmol) and [RhCl(nbd)]2 (0.043 g, 0.0932 mmol) dissolved in 1.5 mL were introduced into dry toluene. The resulting mixture underwent reflux for 1 h. Afterward, distilled water was added to the mixture, leading to the separation of the organic layer, which was subsequently washed with water and dried using anhydrous magnesium sulfate. Evaporation of the solvent followed, and the residue underwent chromatography on a silica gel column, being eluted with hexane, resulting in 0.050 g (16% yield) of compound 2c: HR-MS: calcd. for C18H24Si2S (M+): 328.1137, found: 328.1133. MS m/z 328 (M+); 1H NMR δ(CDCl3) 0.01 (s, 9H, Me3Si), 0.31 (s, 6H, Me2Si), 2.39 (s, 3H, CH3), 6.84 (d, 1H, phenylene ring protons, J = 7.6 Hz), 6.85 (s, 1H, phenylene ring proton), 7.02 (d, 1H, phenylene ring proton, J = 7.6 Hz), 7.18 (t, 1H, phenylene ring proton, J = 7.6 Hz), 7.31 (d, 1H, thienyl ring proton, J = 4.8 Hz), 7.63 (d, 1H, thienyl ring proton, J = 4.8 Hz); 13C NMR δ(CDCl3) −4.0 (Me2Si), 0.9 (Me3Si), 21.5 (CH3), 124.2, 125.2, 126.5, 127.8, 127.9, 132.5, 133.5, 137.3, 142.9, 149.9, 160.9, 162.5 (thienyl, phenylene ring and olefinic carbons); 29Si NMR δ(CDCl3) −7.0, −0.1.
Synthesis of Compound 3: 3-Iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene (0.479 g, 1.41 mmol), bis(triphenylphosphine)dichloropalladium (0.049 g, 0.070 mmol), and copper(I) iodide (0.013 g, 0.070 mmol) were added to 10 mL of dry triethylamine in a 100-mL three-necked flask fitted with a stirrer, reflux condenser, and dropping funnel. 3,3-Dimethyl-1-butyne (0.372 g, 4.53 mmol) was then added dropwise to this mixture at room temperature. The mixture was then heated at reflux for 12 h. Distilled water was added to the mixture and the organic layer was separated, washed with water, and dried over anhydrous magnesium sulfate. The solvent was then evaporated, and the residue was chromatographed on a silica gel column and eluted with hexane–ethyl acetate (50:1) to obtain 0.065 g (16% yield) of compound 3: HR-MS, calculated for C15H26Si2S (M+): 294.1294, found: 294.1290. MS m/z 294 (M+); 1H NMR δ(CDCl3) 0.10 (s, 9H, Me3Si), 0.46 (s, 6H, Me2Si), 1.31 (s, 9H, t-Bu), 7.15 (d, 1H, thienyl ring proton, J = 3.6 Hz), 7.40 (d, 1H, thienyl ring proton, J = 3.6 Hz); 13C NMR δ(CDCl3) −3.2 (Me2Si), −1.9 (Me3Si), 28.0 (CMe3), 31.0 (Me3C), 76.3, 98.9 (sp carbons), 128.9, 129.6, 132.9, 140.3 (thienyl ring carbons); 29Si NMR δ(CDCl3) −23.0, −17.4.
Reaction of Compound 3 in the Presence of [RhCl(nbd)2]2 Catalyst: Dry toluene (1.5 mL), along with compound 3 (0.065 g, 0.221 mmol) and [RhCl(nbd)2]2 (0.010 g, 0.0217 mmol), were combined in a 30 mL two-necked flask equipped with a reflux condenser. The mixture was refluxed for 12 h. Following this, the addition of distilled water to the mixture led to the isolation of the organic layer, which was washed with water and dried using anhydrous magnesium sulfate. Evaporation of the solvent was performed, and the remaining residue underwent chromatography on a silica gel column, eluting with hexane. As a result, compound 3 was obtained once again.
Synthesis of Compound 4: In a 100-mL three-necked flask equipped with a stirrer, reflux condenser, and dropping funnel, 1.019 g (2.99 mmol) of 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, 0.105 g (0.150 mmol) of bis(triphenylphosphine)dichloropalladium, and 0.029 g (0.152 mmol) of copper(I) iodide were combined with 10 mL of dry triethylamine. Subsequently, ethynyltrimethylsilane (0.648 g, 6.60 mmol) was added dropwise at room temperature. The resulting mixture was heated at reflux for 12 h. Following this, distilled water was added to the mixture, and the organic layer was separated, washed with water, and dried using anhydrous magnesium sulfate. Evaporation of the solvent followed, and the remaining residue was subjected to chromatography on a silica gel column, eluting with hexane–ethyl acetate (50:1), yielding 0.213 g (23% yield) of compound 4: HR-MS: calcd. for C14H26Si3S (M+): 310.1063, found: 310.1066. MS m/z 310 (M+); 1H NMR δ(CDCl3) 0.10 (s, 9H, Me3Si), 0.23 (s, 9H, Me3Si), 0.46 (s, 6H, Me2Si), 7.21 (d, 1H, thienyl ring proton, J = 4.4 Hz), 7.40 (d, 1H, thienyl ring proton, J = 4.4 Hz); 13C NMR δ(CDCl3) −3.3 (Me2Si), −1.9, −0.1 (Me3Si), 95.2, 102.3 (sp carbons), 128.7, 129.1, 132.7, 143.7 (thienyl ring carbons); 29Si NMR δ(CDCl3) −22.6, −17.8, −17.1.
Reaction of Compound 4 in the Presence of [RhCl(nbd)2]2 Catalyst: In a 30 mL two-necked flask equipped with a reflux condenser, compound 4 (0.101 g, 0.325 mmol) and 0.015 g (0.0325 mmol) of [RhCl(nbd)2]2 were introduced into dry toluene (1.5 mL). The resulting mixture was then subjected to reflux for 12 h. Afterward, distilled water was added to the mixture, leading to the separation of the organic layer, which was subsequently washed with water and dried using anhydrous magnesium sulfate. Evaporation of the solvent followed, and the residue underwent chromatography on a silica gel column, being eluted with hexane. Ultimately, the initial compound 4 was recovered.
Synthesis of Compound 5: In a 100-mL three-necked flask equipped with a stirrer, reflux condenser, and dropping funnel, 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene (1.007 g, 2.96 mmol), bis(triphenylphosphine)dichloropalladium (0.104 g, 0.148 mmol), and copper(I) iodide (0.029 g, 0.152 mmol) were combined with 10 mL of dry triethylamine. Gradually, 3-phenyl-1-propyne (0.722 g, 6.21 mmol) was added dropwise to this mixture at room temperature. The resulting solution was refluxed for 12 h. Post-reflux, distilled water was added to the mixture, leading to the separation of the organic layer, which was subsequently washed with water and desiccated using anhydrous magnesium sulfate. Evaporation of the solvent ensued, and the remaining residue underwent chromatography on a silica gel column, eluting with hexane–ethyl acetate (50:1). The resulting yield was 0.070 g (7%) of compound 5: HR-MS, calculated for C18H24Si2S (M+): 328.1137, found: 328.1130. MS m/z 328 (M+); 1H NMR δ(CDCl3) 0.07 (s, 9H, Me3Si), 0.41 (s, 6H, Me2Si), 3.81 (s, 2H, CH2), 7.20 (d, 1H, thienyl ring proton, J = 4.4 Hz), 7.24 (t, 1H, phenyl ring proton, J = 8.0 Hz), 7.33 (t, 2H, phenyl ring protons, J = 8.0 Hz), 7.39 (d, 2H, phenyl ring protons, J = 8.0 Hz), 7.42 (d, 1H, thienyl ring proton, J = 4.4 Hz); 13C NMR δ(CDCl3) −3.2 (Me2Si), −2.0 (Me3Si), 26.0 (CH2), 79.8, 88.4 (sp carbons), 126.6, 128.1, 128.5, 129.0, 129.1, 132.6, 136.6, 141.8 (phenyl and thienyl ring carbons); 29Si NMR δ(CDCl3) −23.1, −17.6.
Reaction of Compound 5 in the Presence of [RhCl(nbd)2]2 Catalyst: In a 30 mL two-necked flask equipped with a reflux condenser, dry toluene (1.5 mL), compound 5 (0.101 g, 0.307 mmol), and [RhCl(nbd)2]2 (0.015 g, 0.0325 mmol) were combined and refluxed for 1 h. Afterward, distilled water was added to the mixture, leading to the isolation of the organic layer, which was washed with water and dried using anhydrous magnesium sulfate. Evaporation of the solvent followed by chromatography on a silica gel column, eluting with hexane, revealed consumption of the initial compound 5. However, upon analysis using gas–liquid chromatography (GLC) and gel permeation chromatography (GPC), numerous products were detected in the reaction mixture. No thiophene-fused silole derivatives were found in the reaction mixture.
Synthesis of Compound 6: In a 100 mL two-necked flask equipped with a reflux condenser, 1.007 g (2.96 mmol) of 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, 0.104 g (0.148 mmol) of bis(triphenylphosphine)dichloropalladium, and 0.029 g (0.152 mmol) of copper(I) iodide were combined with 10 mL of dry triethylamine. To this mixture, 1-ethynylcyclohexene (0.628 g, 5.92 mmol) was slowly added dropwise at room temperature, followed by heating at reflux for 12 h. After the reflux, distilled water was added to the mixture, and the resulting organic layer was separated, washed with water, and dried over anhydrous magnesium sulfate. Evaporation of the solvent ensued, and the remaining residue was subjected to chromatography on a silica gel column, eluting with hexane–ethyl acetate (50:1), yielding 0.598 g (63% yield) of compound 6: HR-MS, calculated for C17H26Si2S (M+): 318.1294, found: 328.1291. MS m/z 318 (M+); 1H NMR δ(CDCl3) 0.10 (s, 9H, Me3Si), 0.45 (s, 6H, Me2Si), 1.58–1.70 (m, 4H, CH2), 2.11–2.22 (m, 4H, CH2), 6.13–6.16 (m, 1H, olefinic proton), 7.18 (d, 1H, thienyl ring proton, J = 4.4 Hz), 7.41 (d, 1H, thienyl ring proton, J = 4.4 Hz); 13C NMR δ(CDCl3) −3.2 (Me2Si), −2.0 (Me3Si), 21.5, 22.3, 25.7, 29.1 (CH2), 84.1, 92.2 (sp carbons), 120.9, 129.1, 129.2, 132.4, 134.3, 141.4 (thienyl ring and olefinic carbons); 29Si NMR δ(CDCl3) −23.0, −17.6.
Reaction of Compound 6 in the Presence of [RhCl(nbd)2]2 Catalyst: In a 30 mL two-necked flask equipped with a reflux condenser, compound 6 (0.110 g, 0.345 mmol) and 0.016 g (0.0347 mmol) of [RhCl(nbd)2]2 were introduced into dry toluene (1.5 mL). The resulting mixture was heated under reflux for 1 h. Post-reflux, distilled water was added to the mixture, leading to the separation of the organic layer, which was subsequently washed with water and dried using anhydrous magnesium sulfate. Evaporation of the solvent followed, and the remaining residue underwent chromatography on a silica gel column, being eluted with hexane. Compound 6 was consumed in the reaction; however, multiple products were detected in the reaction mixture via gas–liquid chromatography (GLC) and gel permeation chromatography (GPC). No derivatives of thiophene-fused silole were identified in the reaction mixture.
Reaction of 3-Iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene with 1-Hexyne in the Presence of Palladium and Copper Catalysts: In a 100 mL two-necked flask equipped with a reflux condenser, 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene (1.003 g, 2.95 mmol), bis(triphenylphosphine)dichloropalladium (0.104 g, 0.148 mmol), and copper(I) iodide (0.028 g, 0.147 mmol) were combined with 10 mL of dry triethylamine. Gradual addition of 1-hexyne (0.487 g, 5.93 mmol) to this mixture at room temperature was followed by refluxing for 12 h. Analysis via GLC and GPC revealed the presence of numerous products in the reaction mixture. Subsequently, the addition of distilled water to the mixture led to the separation of the organic layer, which underwent washing with water and drying using anhydrous magnesium sulfate. Evaporation of the solvent and chromatography on a silica gel column, eluting with hexane–ethyl acetate (10:1), were performed. Surprisingly, 3-(hex-1-yn-1-yl)-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, an expected product from the Sonogashira coupling reaction between 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene and 1-hexyne akin to compounds 1a–1c, was not detected in the mixture. All efforts to isolate this compound were unsuccessful.
Reaction of 3-Iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene with 1-Octyne in the Presence of Palladium and Copper Catalysts: In a 100 mL two-necked flask equipped with a reflux condenser, 1.005 g (2.95 mmol) of 3-iodo-3-(1,1,2,2,2-pentamethyldisilanyl)thiophene, 0.104 g (0.148 mmol) of bis(triphenylphosphine)dichloropalladium, and 0.028 g (0.147 mmol) of copper(I) iodide were combined with 10 mL of dry triethylamine. Gradually, 1-octyne (0.651 g, 5.91 mmol) was added dropwise to this mixture at room temperature, and the resulting mixture was heated at reflux for 12 h. Multiple products were identified in the reaction mixture via GLC and GPC. Afterward, distilled water was added to the mixture, and the organic layer was separated, washed with water, and dried over anhydrous magnesium sulfate. Evaporation of the solvent followed, and the remaining residue was subjected to chromatography on a silica gel column, eluting with hexane–ethyl acetate (10:1). Interestingly, the expected 3-(oct-1-yn-1-yl)-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, anticipated from the Sonogashira coupling reaction of 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene and 1-octyne, similar to compounds 1a–1c, was not detected.
Reaction of 3-Iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene with ethynylcyclohexane in the presence of palladium and copper catalysts: In a 100 mL two-necked flask equipped with a reflux condenser, 1.001 g (2.94 mmol) of 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, 0.104 g (0.148 mmol) of bis(triphenylphosphine)dichloropalladium, and 0.029 g (0.152 mmol) of copper(I) iodide were combined with 10 mL of dry triethylamine. Following this, ethynylcyclohexane (0.641 g, 5.93 mmol) was added dropwise to this mixture at room temperature, and the resulting mixture was heated at reflux for 12 h. Multiple products were identified in the reaction mixture through GLC and GPC. Post-reaction, distilled water was added to the mixture, leading to the separation of the organic layer, which was subsequently washed with water and dried over anhydrous magnesium sulfate. Evaporation of the solvent ensued, and the remaining residue underwent chromatography on a silica gel column, being eluted with hexane–ethyl acetate (10:1). Surprisingly, the anticipated 3-(cyclohexylethynyl)-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene, expected from the Sonogashira coupling reaction of 3-iodo-2-(1,1,2,2,2-pentamethyldisilanyl)thiophene and ethynylcyclohexane, akin to compounds 1a-1c, was not produced.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
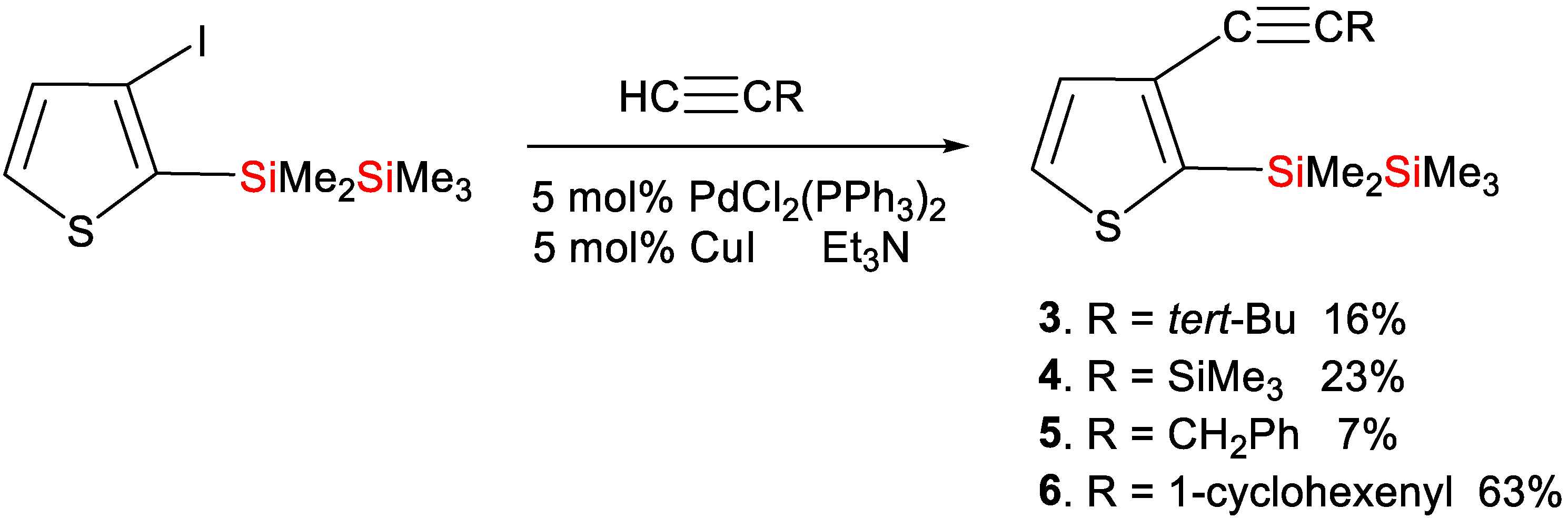{kind=link}
{kind=link}





