


Influence of the Core Branching Density on Drug Release from Arborescent Poly(γ-benzyl L-glutamate) End-Grafted with Poly(ethylene oxide)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
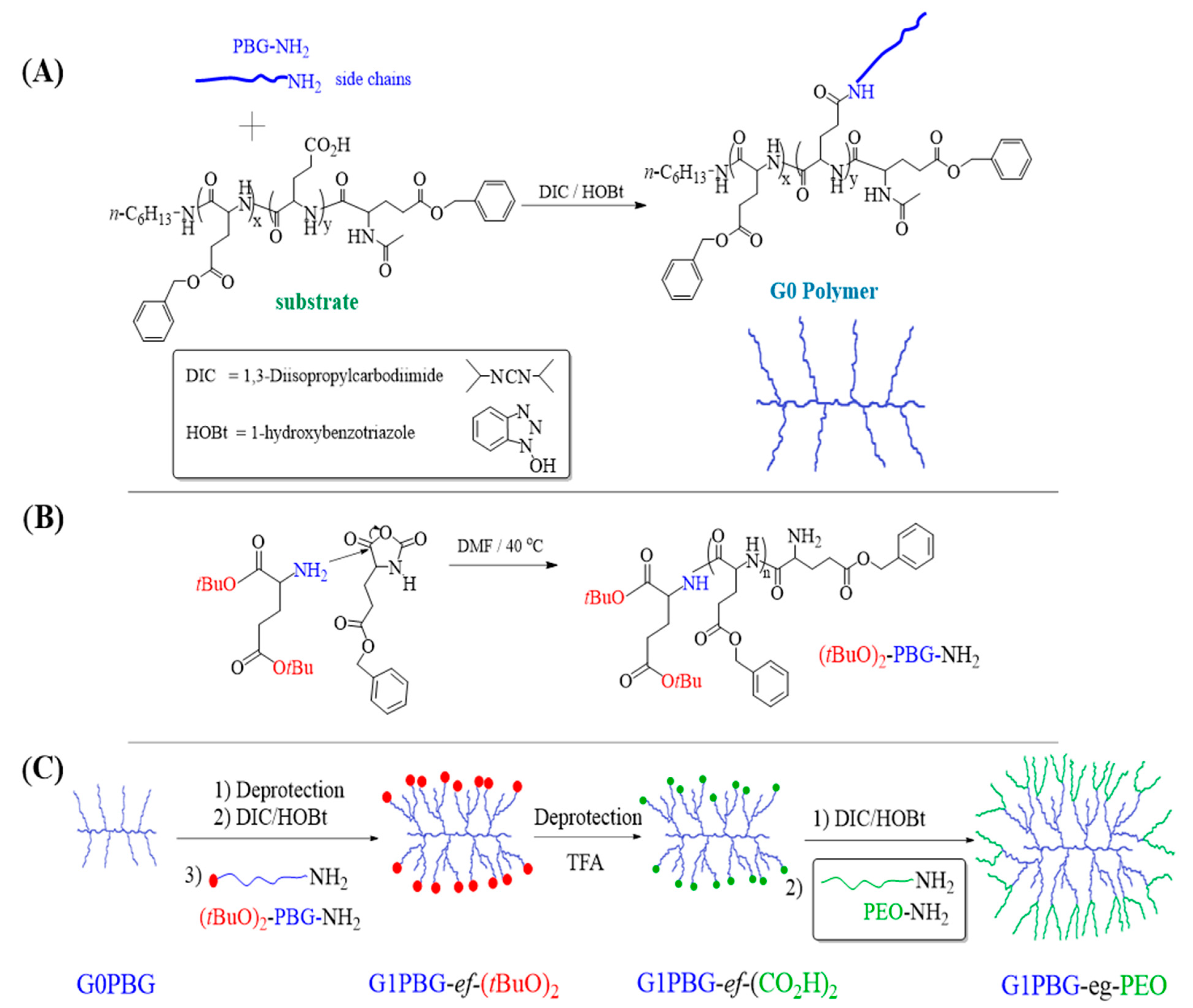
2.3. Synthesis
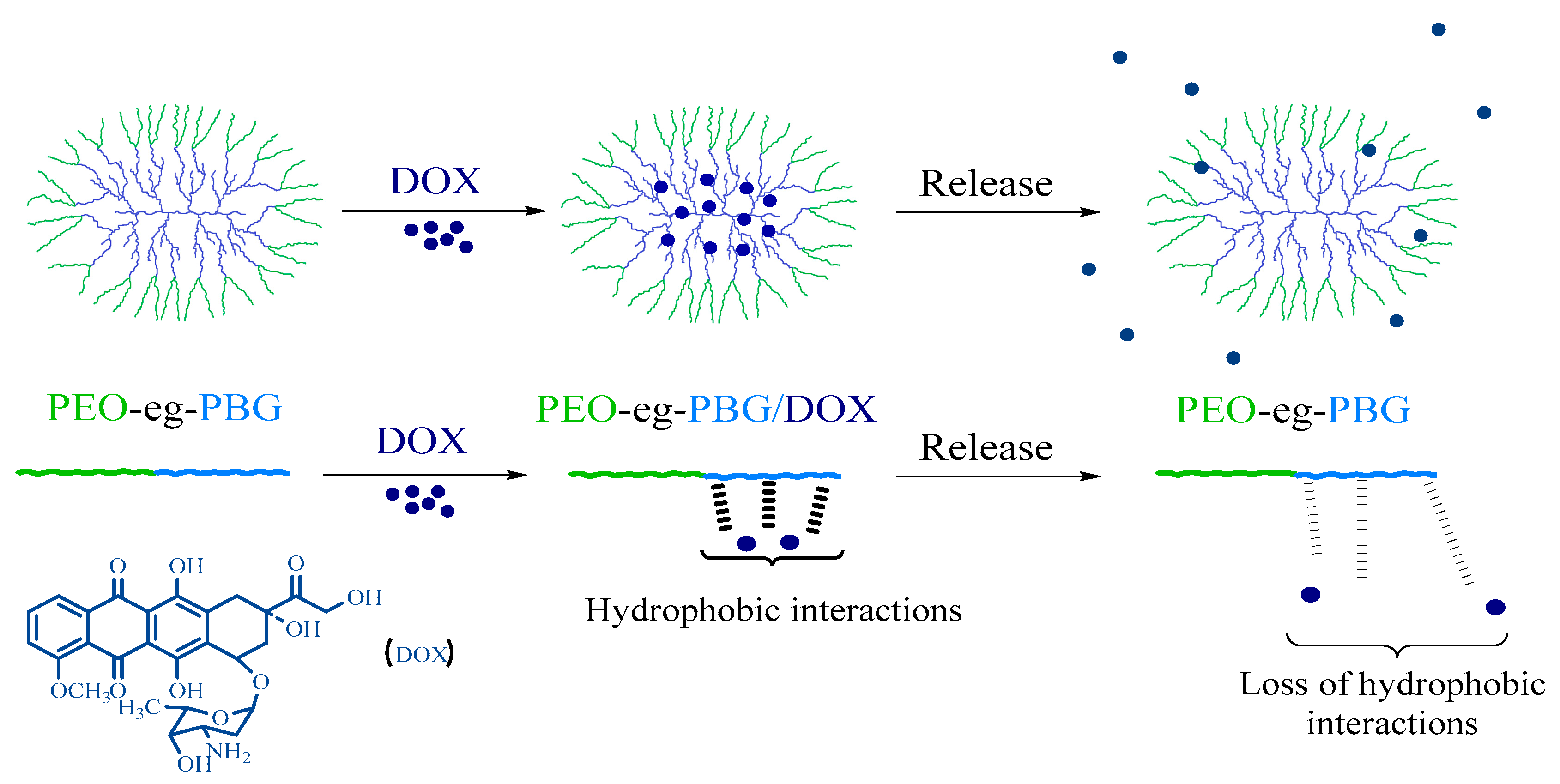
2.4. Preparation of DOX-Loaded Unimolecular Micelles
2.5. In Vitro Release of DOX
3. Results and Discussion
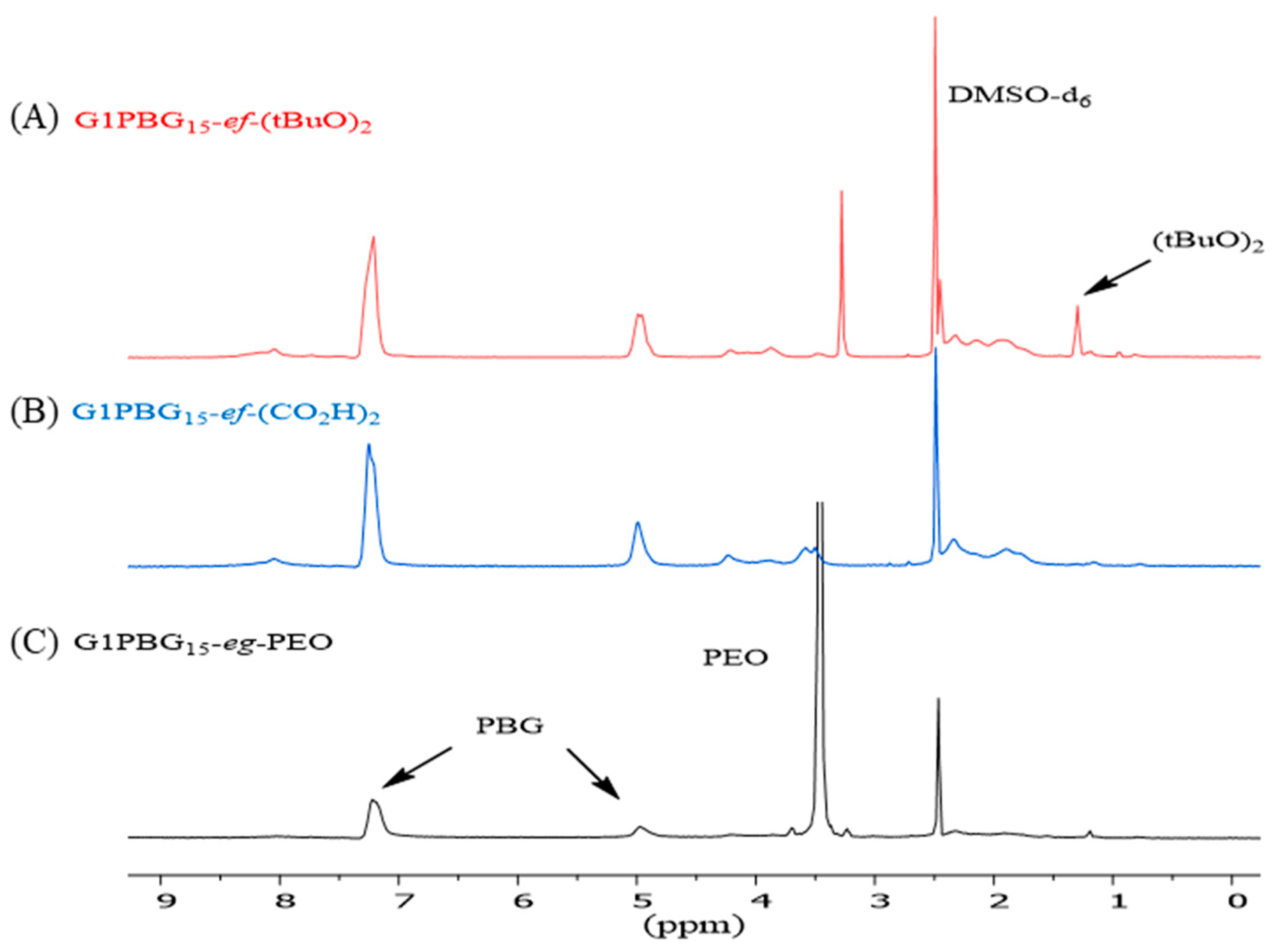
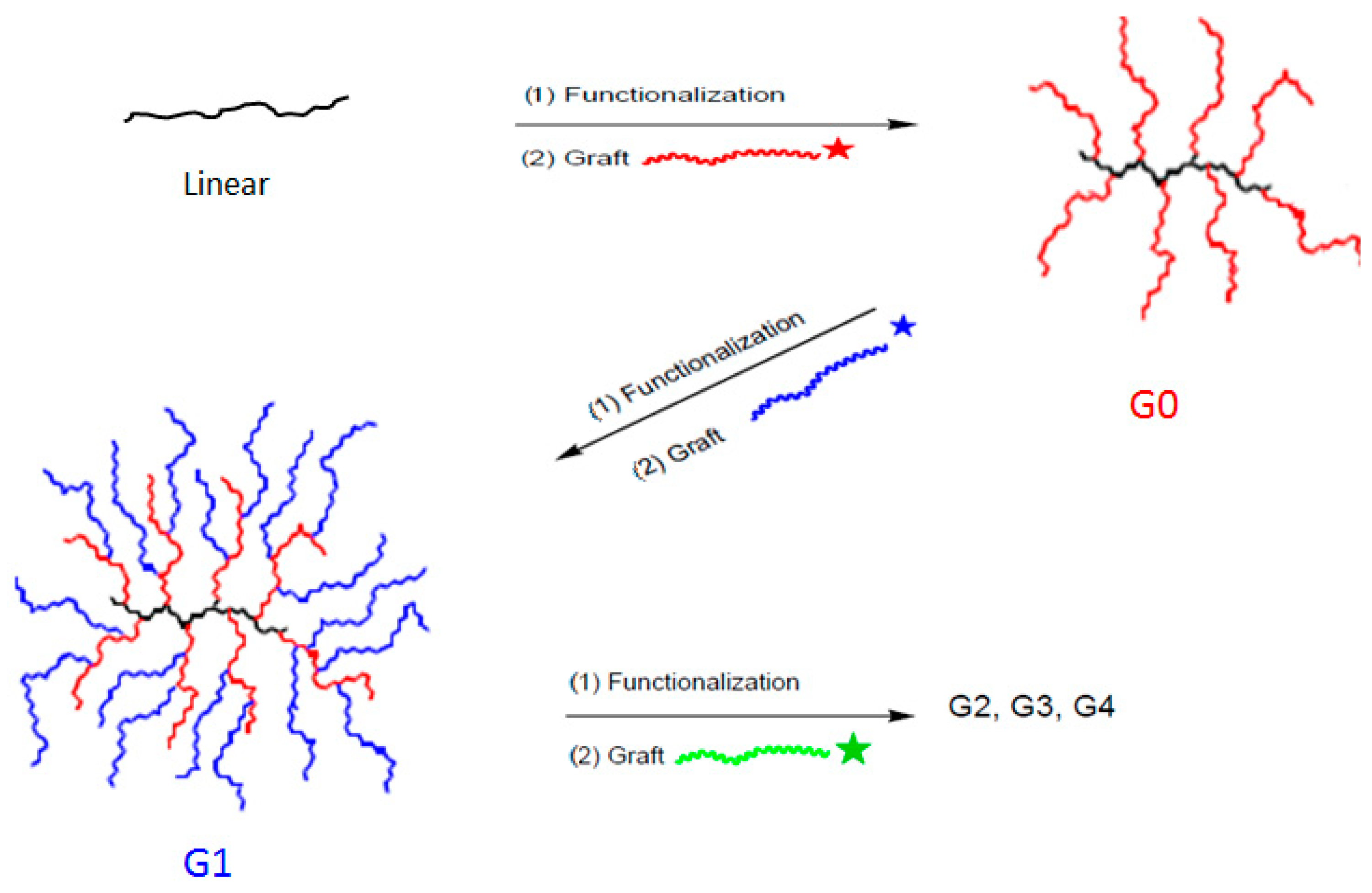
3.1. Synthesis of Linear (tBuO)2-PBG and Arborescent PBG Substrates with Carboxylic Acid Chain Ends
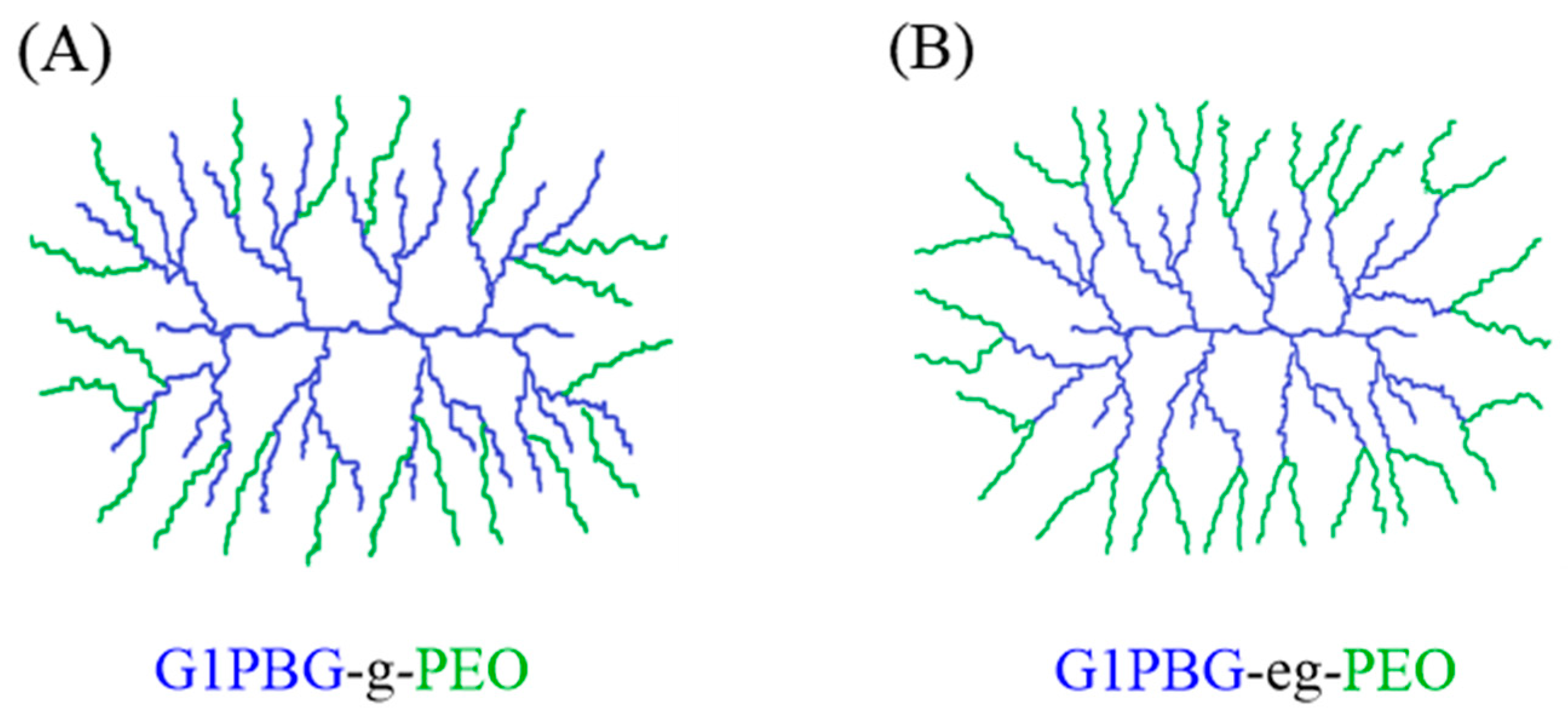
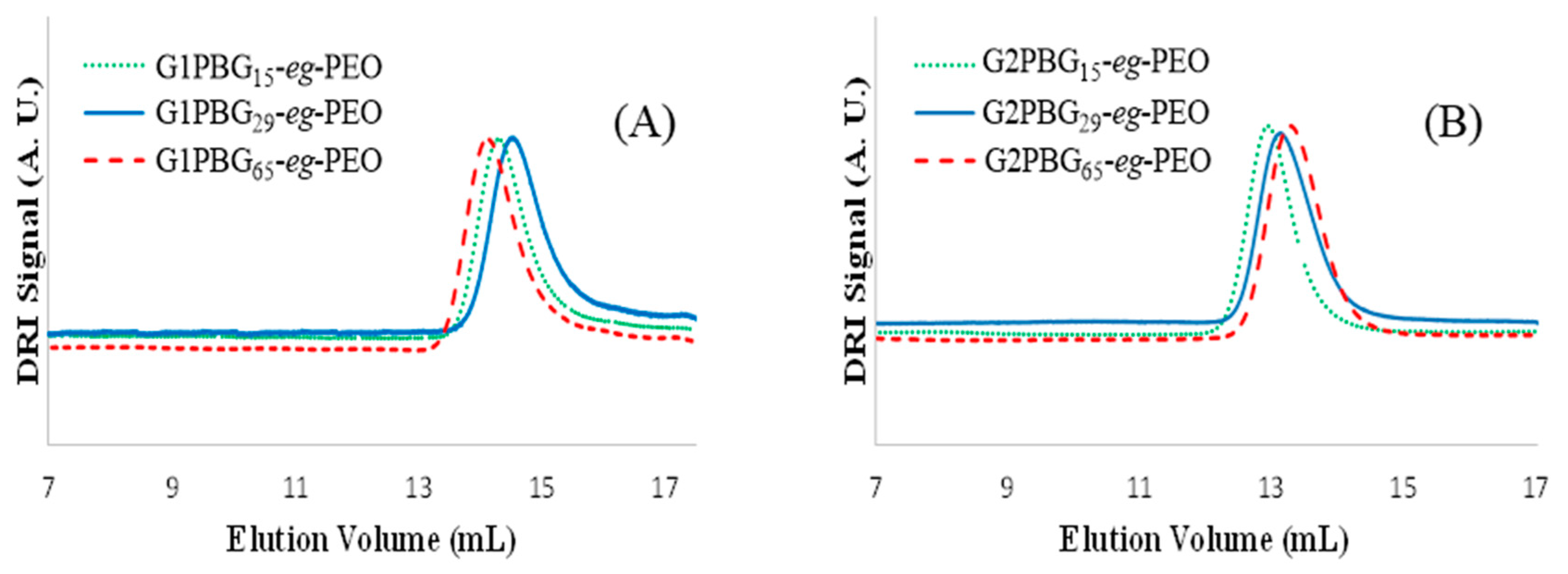
3.2. Synthesis of Chain End-Grafted Arborescent Copolymers
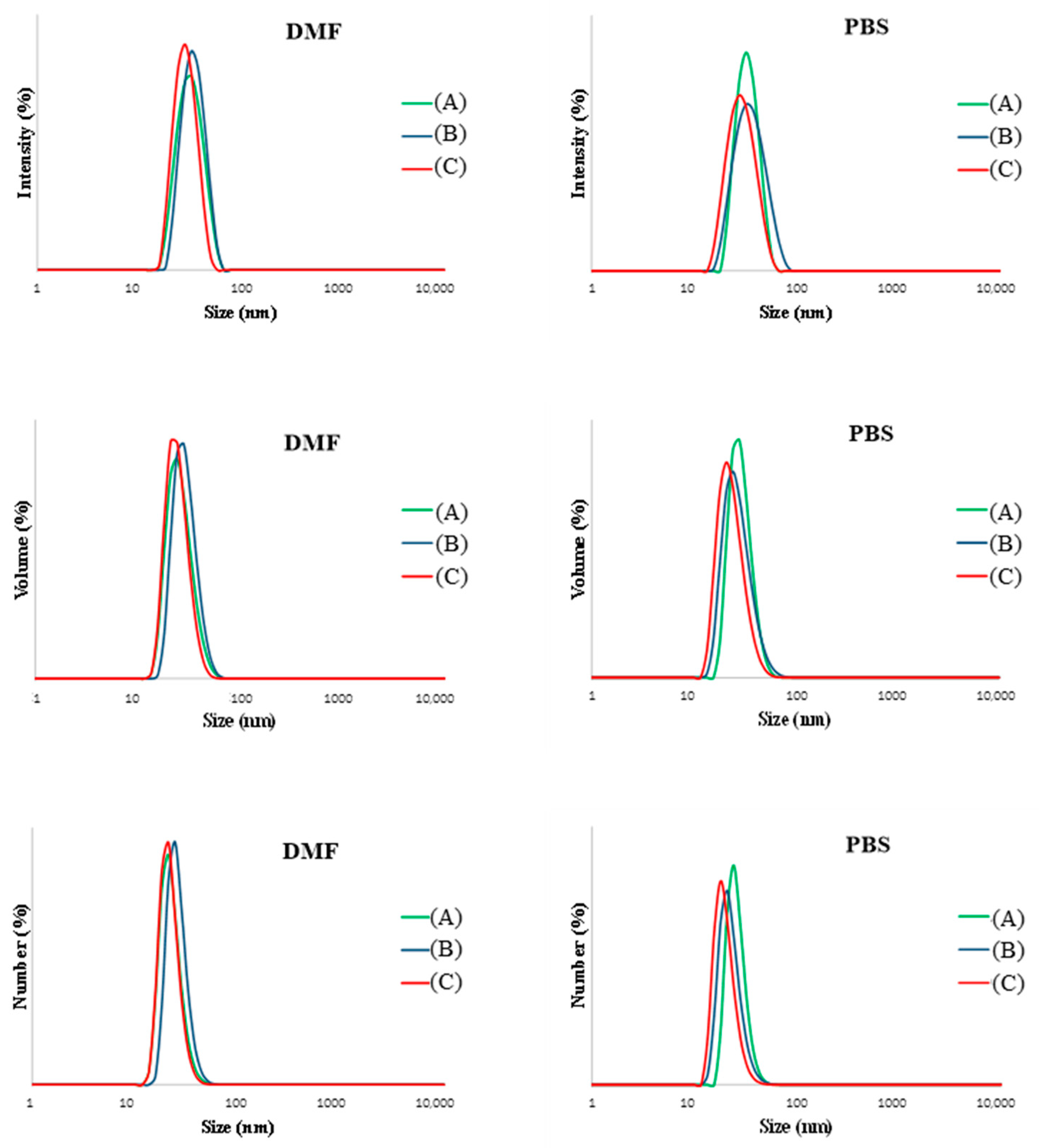
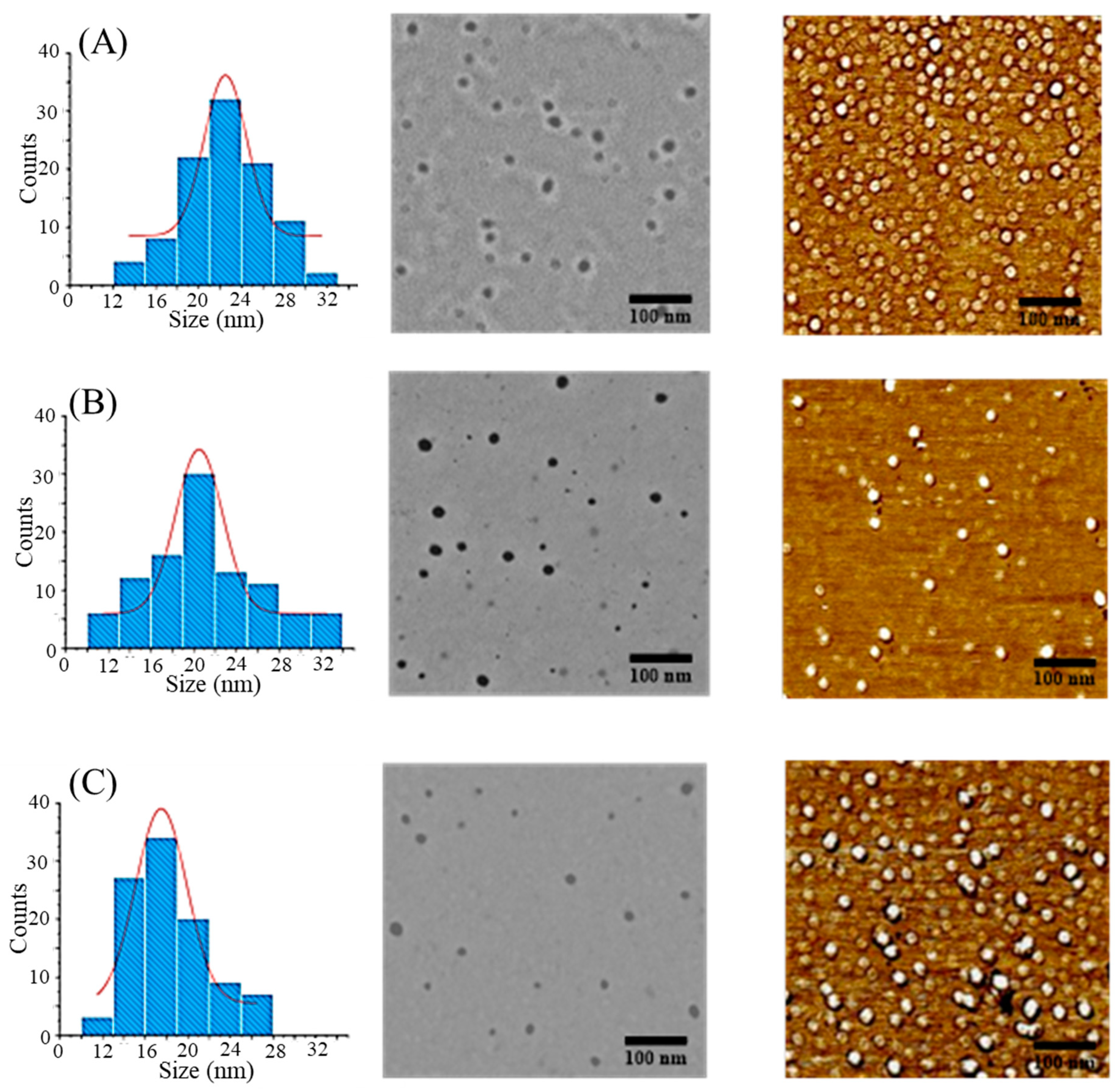
3.3. Properties of Chain End-Grafted Arborescent Copolymer Micelles
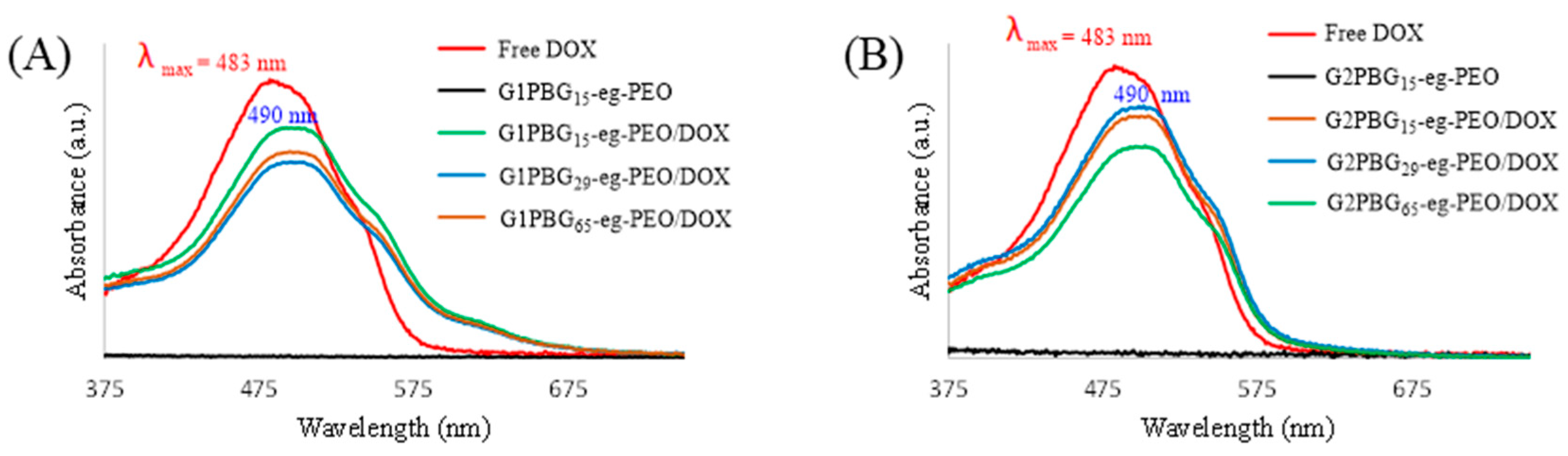
3.4. Drug Loading and Micelle Characterization
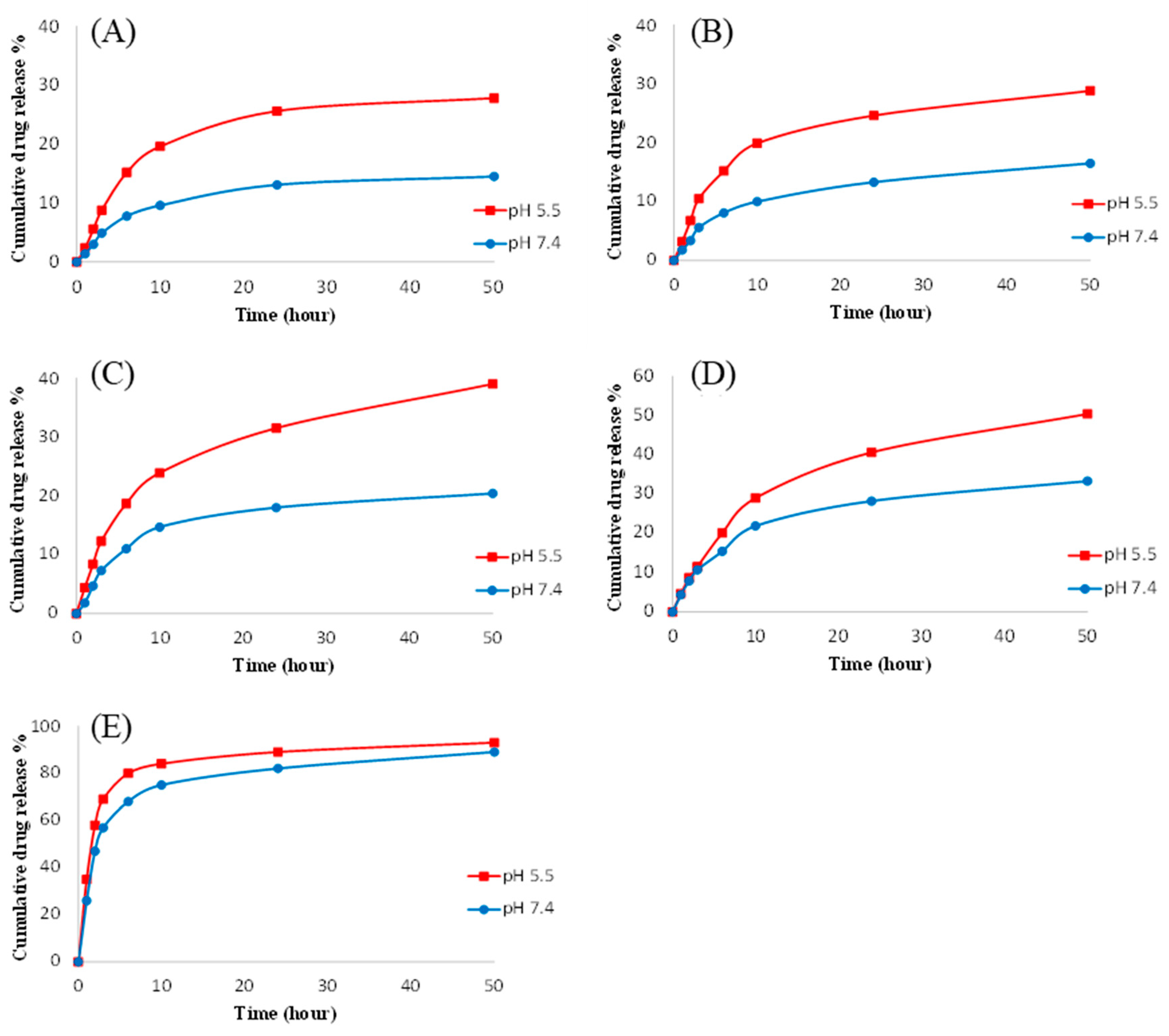
3.5. In Vitro Drug Release Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Souri, M.; Soltani, M.; Moradi Kashkooli, F.; Kiani Shahvandi, M.; Chiani, M.; Shariati, F.S.; Mehrabi, M.R.; Munn, L.L. Towards principled design of cancer nanomedicine to accelerate clinical translation. Mater. Today Bio 2022, 13, 100208. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Jone Kirubavathy, S. Chapter 4—Nanomedicine and nanocarriers for cancer treatment. In Nanotechnology for Drug Delivery and Pharmaceuticals; Pratap Singh, R., Rb Singh, K., Singh, J., Adetunji, C.O., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 71–110. [Google Scholar]
- Negut, I.; Bita, B. Polymeric micellar systems—A special emphasis on “smart” drug delivery. Pharmaceutics 2023, 15, 976. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Shukla, M.K.; Sharma, A.K.; Jayaprakash, G.K.; Tonk, R.K.; Chellappan, D.K.; Singh, S.K.; Dua, K.; Ahmed, F.; Bhattacharyya, S.; et al. Metal-based nanomaterials and nanocomposites as promising frontier in cancer chemotherapy. MedComm 2023, 4, e253. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, M.; Li, Y.; Cong, H.; Yu, B.; Shen, Y. Research status of dendrimer micelles in tumor therapy for drug delivery. Small 2023, e2304006. [Google Scholar] [CrossRef] [PubMed]
- Karaz, S.; Senses, E. Liposomes under shear: Structure, dynamics, and drug delivery applications. Adv. NanoBiomed Res. 2023, 3, 2200101. [Google Scholar] [CrossRef]
- Brindhadevi, K.; Garalleh, H.A.L.; Alalawi, A.; Al-Sarayreh, E.; Pugazhendhi, A. Carbon nanomaterials: Types, synthesis strategies and their application as drug delivery system for cancer therapy. Biochem. Eng. J. 2023, 192, 108828. [Google Scholar] [CrossRef]
- Alsehli, M. Polymeric nanocarriers as stimuli-responsive systems for targeted tumor (cancer) therapy: Recent advances in drug delivery. Saudi Pharm. J. 2020, 28, 255–265. [Google Scholar] [CrossRef]
- Ali, I.; Althakfi, S.H.; Suhail, M.; Locatelli, M.; Hsieh, M.-F.; Alsehli, M.; Hameed, A.M. Advances in polymeric colloids for cancer treatment. Polymers 2022, 14, 5445. [Google Scholar] [CrossRef]
- Kuperkar, K.; Patel, D.; Atanase, L.I.; Bahadur, P. Amphiphilic block copolymers: Their structures, and self-assembly to polymeric micelles and polymersomes as drug delivery vehicles. Polymers 2022, 14, 4702. [Google Scholar] [CrossRef]
- Perin, F.; Motta, A.; Maniglio, D. Amphiphilic copolymers in biomedical applications: Synthesis routes and property control. Mater. Sci. Eng. C 2021, 123, 111952. [Google Scholar] [CrossRef]
- Kurniasih, I.N.; Keilitz, J.; Haag, R. Dendritic nanocarriers based on hyperbranched polymers. Chem. Soc. Rev. 2015, 44, 4145–4164. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Sahay, G.; Kabanov, A.V.; Bronich, T.K. Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents. Biomacromolecules 2010, 11, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Min, K.H.; Lee, H.J.; Koo, A.N.; Rim, H.P.; Jeon, B.J.; Jeong, S.Y.; Heo, J.S.; Lee, S.C. Ketal cross-linked poly(ethylene glycol)-poly(amino acid)s copolymer micelles for efficient intracellular delivery of doxorubicin. Biomacromolecules 2011, 12, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Grayson, S.M. Approaches for the preparation of non-linear amphiphilic polymers and their applications to drug delivery. Adv. Drug Deliv. Rev. 2012, 64, 852–865. [Google Scholar] [CrossRef]
- Kikuchi, A.; Nose, T. Unimolecular micelle formation of poly(methyl methacrylate)-graft-polystyrene in mixed selective solvents of acetonitrile/acetoacetic acid ethyl ether. Macromolecules 1996, 29, 6770–6777. [Google Scholar] [CrossRef]
- Alsehli, M.; Gauthier, M. Dendritic polymer micelles for drug delivery. In Bioinspired Materials Science and Engineering; Wiley: Hoboken, NJ, USA, 2018; pp. 311–335. [Google Scholar]
- Vögtle, F.; Richardt, G.; Werner, N. Introduction. In Dendrimer Chemistry; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2009; pp. 1–24. [Google Scholar]
- Tomalia, D.A.; Christensen, J.B.; Boas, U. Dendrimers, Dendrons, and Dendritic Polymers; Cambridge University Press: Cambridge, UK, 2012. [Google Scholar]
- Lukowiak, M.C.; Thota, B.N.S.; Haag, R. Dendritic core–shell systems as soft drug delivery nanocarriers. Biotechnol. Adv. 2015, 33, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.; Möller, M. Uniform highly branched polymers by anionic grafting: Arborescent graft polymers. Macromolecules 1991, 24, 4548–4553. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Hedstrand, D.M.; Ferritto, M.S. Comb-burst dendrimer topology: New macromolecular architecture derived from dendritic grafting. Macromolecules 1991, 24, 1435–1438. [Google Scholar] [CrossRef]
- Whitton, G.; Gauthier, M. Arborescent micelles: Dendritic poly(γ-benzyl l-glutamate) cores grafted with hydrophilic chain segments. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 1197–1209. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Alsehli, M.; Gauthier, M. Unimolecular micelles from randomly grafted arborescent copolymers with different core branching densities: Encapsulation of doxorubicin and in vitro release study. Materials 2023, 16, 2461. [Google Scholar] [CrossRef] [PubMed]
- Knobler, Y.; Bittner, S.; Frankel, M. Reaction of N-carboxy-α-amino-acid anhydrides with hydrochlorides of hydroxylamine, O-alkylhydroxylamines, and amines; syntheses of amino-hydroxamic acids, amido-oxy-peptides, and α-amino-acid amides. J. Chem. Soc. 1964, 3941–3951. [Google Scholar] [CrossRef]
- Dimitrov, I.; Schlaad, H. Synthesis of nearly monodisperse polystyrene–polypeptide block copolymers via polymerisation of N-carboxyanhydrides. Chem. Commun. 2003, 2944–2945. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.F.; Schütt, D.; Kubowicz, S. Preparation of well-defined diblock copolymers with short polypeptide segments by polymerization of N-carboxy anhydrides. Macromol. Rapid Commun. 2005, 26, 23–28. [Google Scholar] [CrossRef]
- Guo, J.; Hong, H.; Chen, G.; Shi, S.; Nayak, T.R.; Theuer, C.P.; Barnhart, T.E.; Cai, W.; Gong, S. Theranostic unimolecular micelles based on brush-shaped amphiphilic block copolymers for tumor-targeted drug delivery and positron emission tomography imaging. ACS Appl. Mater. Interfaces 2014, 6, 21769–21779. [Google Scholar] [CrossRef]
- Sheiko, S.S.; Borisov, O.V.; Prokhorova, S.A.; Möller, M. Cylindrical molecular brushes under poor solvent conditions: Microscopic observation and scaling analysis. Eur. Phys. J. E 2004, 13, 125–131. [Google Scholar] [CrossRef]
- Han, D.; Tong, X.; Zhao, Y. One-pot synthesis of brush diblock copolymers through simultaneous ATRP and click coupling. Macromolecules 2011, 44, 5531–5536. [Google Scholar] [CrossRef]
- Lv, S.; Li, M.; Tang, Z.; Song, W.; Sun, H.; Liu, H.; Chen, X. Doxorubicin-loaded amphiphilic polypeptide-based nanoparticles as an efficient drug delivery system for cancer therapy. Acta Biomater. 2013, 9, 9330–9342. [Google Scholar] [CrossRef]
- Kwon, G.; Naito, M.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Block copolymer micelles for drug delivery: Loading and release of doxorubicin. J. Control. Release 1997, 48, 195–201. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, Q.; Jin, Y.; Qiu, L. High loading of hydrophilic/hydrophobic doxorubicin into polyphosphazene polymersome for breast cancer therapy. Nanomedicine 2014, 10, 349–358. [Google Scholar] [CrossRef]
- Pouton, C.W.; Wagstaff, K.M.; Roth, D.M.; Moseley, G.W.; Jans, D.A. Targeted delivery to the nucleus. Adv. Drug Deliv. Rev. 2007, 59, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Prabaharan, M.; Grailer, J.J.; Pilla, S.; Steeber, D.A.; Gong, S. Folate-conjugated amphiphilic hyperbranched block copolymers based on Boltorn H40, poly(L-lactide) and poly(ethylene glycol) for tumor-targeted drug delivery. Biomaterials 2009, 30, 3009–3019. [Google Scholar] [CrossRef]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Alsehli, M.; Gauthier, M. Arborescent Polypeptides for Sustained Drug Delivery. MRS Online Proc. Libr. (OPL) 2016, 1819, 70. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | PBG Substrate | Graft Copolymer | |||||
|---|---|---|---|---|---|---|---|
| Mn (g/mol) a | mol % −CO2H b | Gy c | Mn (g/mol) a | Mw/Mn a | fnd | wt% PEO e | |
| G1PBG15-eg-PEO | 220,000 | 9 | 70 | 890,000 | 1.06 | 67 | 75 |
| G1PBG29-eg-PEO | 210,000 | 8 | 68 | 760,000 | 1.09 | 55 | 72 |
| G1PBG65-eg-PEO | 270,000 | 9 | 63 | 1.0 × 106 | 1.05 | 73 | 73 |
| G2PBG15-eg-PEO | 1.2 × 106 | 9 | 24 | 2.4 × 106 | 1.05 | 120 | 50 |
| G2PBG29-eg-PEO | 970,000 | 9 | 31 | 2.3 × 106 | 1.09 | 133 | 57 |
| G2PBG65-eg-PEO | 1.3 × 106 | 7 | 20 | 2.2 × 106 | 1.07 | 90 | 40 |
| Copolymer | DMF | PBS | ||||||
|---|---|---|---|---|---|---|---|---|
| Dh, number | Dh, volume | Dh, intensity | PDI | Dh, number | Dh, volume | Dh, intensity | PDI | |
| G1PBG15-eg-PEO | 11 ± 1 | 15 ± 1 | 17 ± 1 | 0.08 | 13 ± 2 | 17 ± 2 | 26 ± 2 | 0.19 |
| G1PBG29-eg-PEO | 12 ± 1 | 13 ± 1 | 15 ± 1 | 0.10 | 14 ± 2 | 17 ± 2 | 22 ± 2 | 0.31 |
| G1PBG65-eg-PEO | 14 ± 2 | 16 ± 2 | 18 ± 2 | 0.12 | 14± 2 | 16 ± 2 | 19 ± 3 | 0.27 |
| G2PBG15-eg-PEO | 25 ± 2 | 27 ± 2 | 32 ± 2 | 0.04 | 29 ± 1 | 33 ± 1 | 37 ± 1 | 0.19 |
| G2PBG29-eg-PEO | 25 ± 1 | 29 ± 1 | 35 ± 1 | 0.05 | 26 ± 1 | 31 ± 3 | 39 ± 1 | 0.27 |
| G2PBG65-eg-PEO | 21 ± 1 | 23 ± 1 | 29 ± 1 | 0.09 | 22 ± 1 | 27 ± 1 | 31 ± 1 | 0.23 |
| Copolymer | PBG | PBG-eg-PEO | PBG-eg-PEO/DOX | |||||
|---|---|---|---|---|---|---|---|---|
| Dh (nm) | PDI | Dh (nm) | PDI | Dh (nm) | PDI | DLC (wt%) | DLE (%) | |
| G1PBG15-eg-PEO/DOX | - | - | 13 | 0.19 | 14 | 0.41 | 7.9 ± 0.3 | 47 ± 4 |
| G1PBG29-eg-PEO/DOX | - | - | 14 | 0.31 | 14 | 0.32 | 7.5 ± 0.2 | 45 ± 4 |
| G1PBG65-eg-PEO/DOX | - | - | 14 | 0.27 | 15 | 0.35 | 8.1 ± 0.2 | 49 ± 5 |
| G2PBG15-eg-PEO/DOX | 14 | 0.18 | 29 | 0.19 | 31 | 0.28 | 11.2 ± 0.3 | 67 ± 4 |
| G2PBG29-eg-PEO/DOX | 10 | 0.05 | 26 | 0.27 | 27 | 0.27 | 9.7 ± 0.2 | 58 ± 3 |
| G2PBG65-eg-PEO/DOX | 12 | 0.04 | 22 | 0.23 | 24 | 0.29 | 10.4 ± 0.3 | 62 ± 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsehli, M.; Gauthier, M. Influence of the Core Branching Density on Drug Release from Arborescent Poly(γ-benzyl L-glutamate) End-Grafted with Poly(ethylene oxide). Int. J. Transl. Med. 2023, 3, 496-515. https://doi.org/10.3390/ijtm3040035
Alsehli M, Gauthier M. Influence of the Core Branching Density on Drug Release from Arborescent Poly(γ-benzyl L-glutamate) End-Grafted with Poly(ethylene oxide). International Journal of Translational Medicine. 2023; 3(4):496-515. https://doi.org/10.3390/ijtm3040035
Chicago/Turabian StyleAlsehli, Mosa, and Mario Gauthier. 2023. "Influence of the Core Branching Density on Drug Release from Arborescent Poly(γ-benzyl L-glutamate) End-Grafted with Poly(ethylene oxide)" International Journal of Translational Medicine 3, no. 4: 496-515. https://doi.org/10.3390/ijtm3040035