
Precision Medicine in a Community Cancer Center: Pan-Cancer DNA/RNA Sequencing of Tumors Reveals Clinically Relevant Gene Fusions
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Next-Generation Sequencing and Database Development
2.2. Immunohistochemistry
2.3. Data Analysis and Curation
3. Results
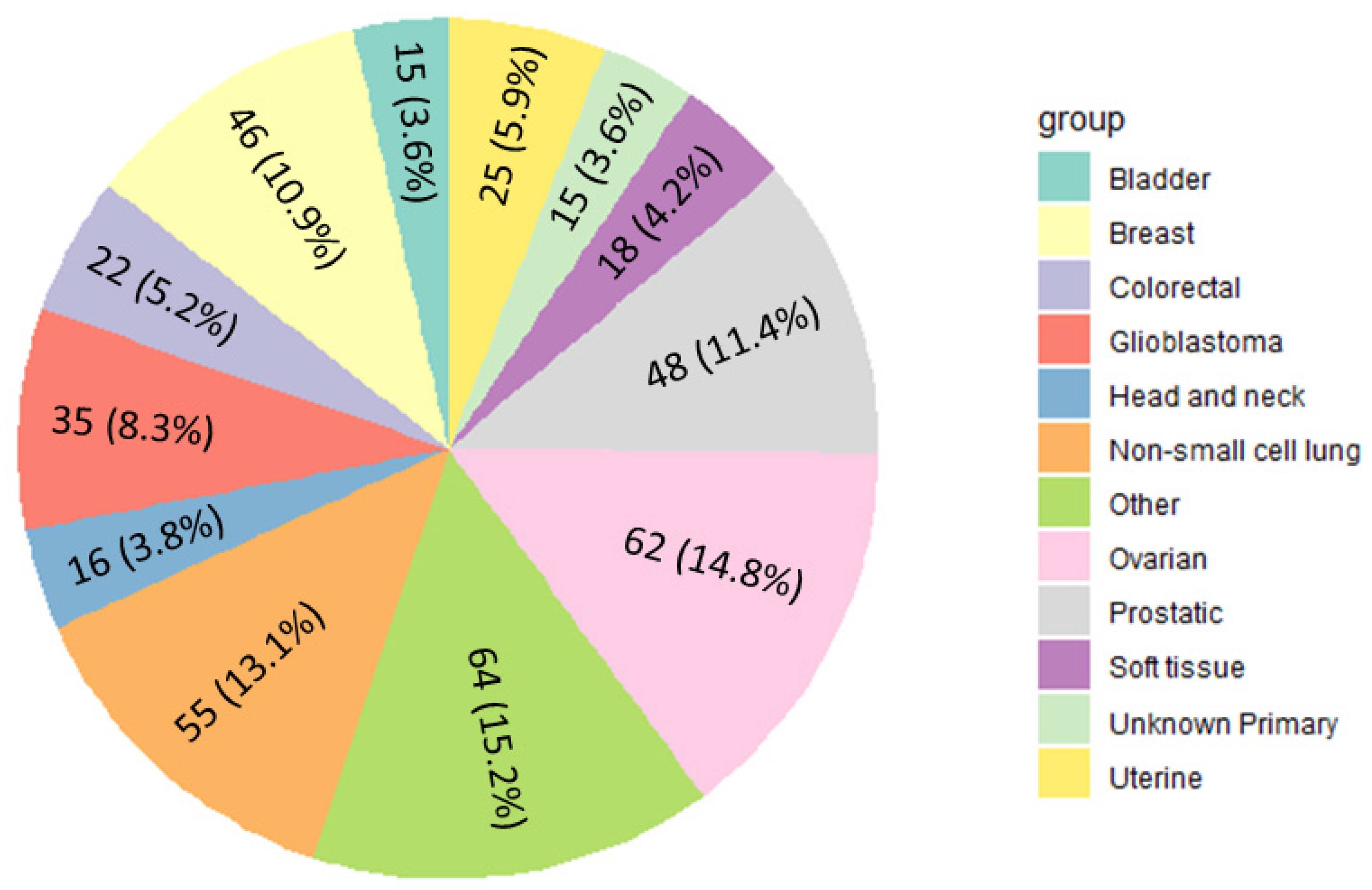
3.1. Identification of Gene Fusion Events across Cancer Types
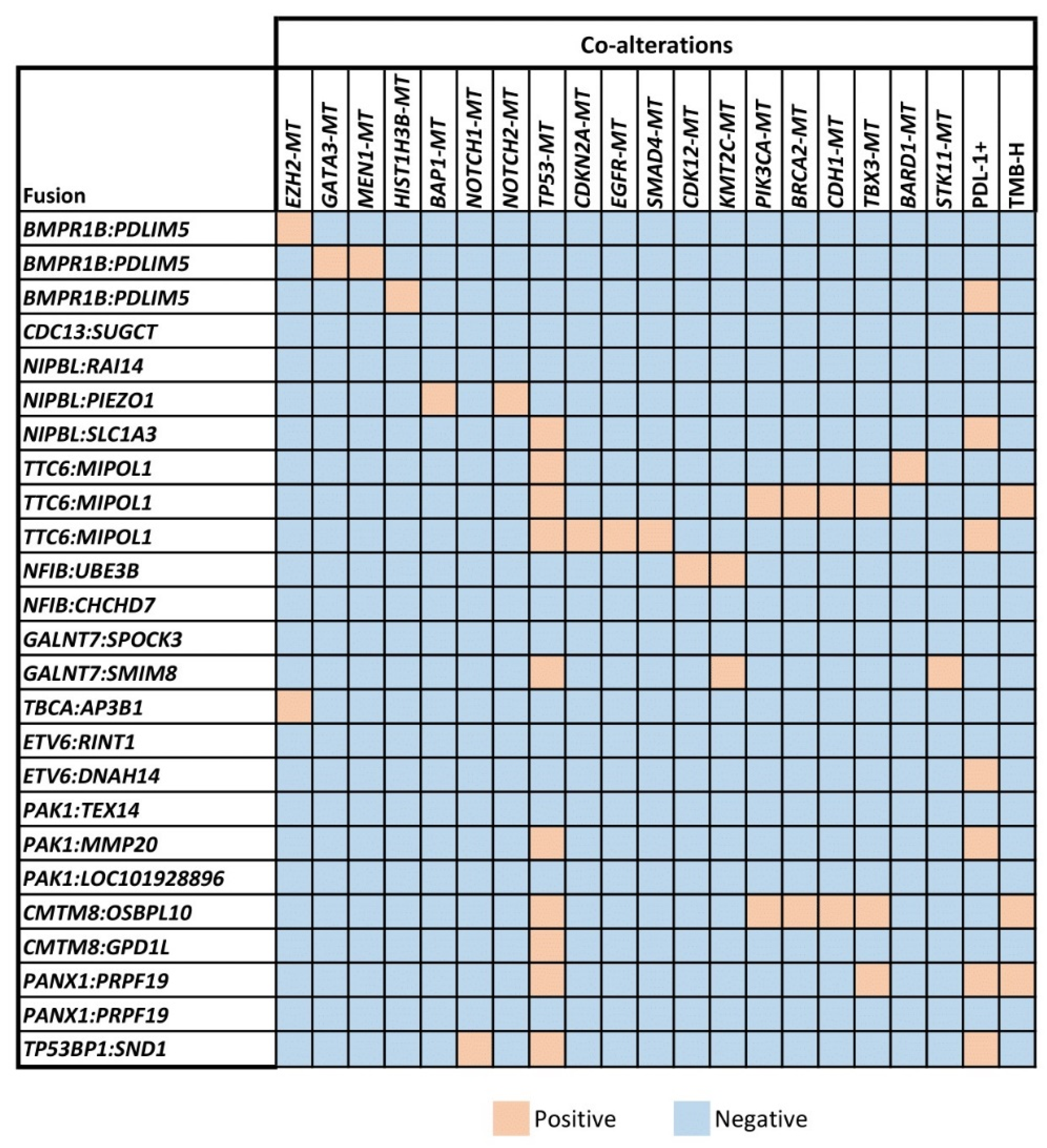
3.2. Detection of Co-Mutations Associated with Unclassified Gene Fusions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quan, V.L.; Panah, E.; Zhang, B.; Shi, K.; Mohan, L.S.; Gerami, P. The role of gene fusions in melanocytic neoplasms. J. Cutan. Pathol. 2019, 46, 878–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobato, M.N.; Metzler, M.; Drynan, L.; Forster, A.; Pannell, R.; Rabbitts, T.H. Modeling chromosomal translocations using conditional alleles to recapitulate initiating events in human leukemias. J. Natl. Cancer Inst. Monogr. 2008, 2008, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, R.; Sugar, L.; Yang, W.; Srivastava, S.; Klotz, L.; Yang, L.; Stanimirovic, A.; Encioiu, E.; Neill, M.; Loblaw, D. Expression of the TMPRSS2: ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br. J. Cancer 2007, 97, 1690–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panicker, S.; Venkatabalasubramanian, S.; Pathak, S.; Ramalingam, S. The impact of fusion genes on cancer stem cells and drug resistance. Mol. Cell. Biochem. 2021, 476, 3771–3783. [Google Scholar] [CrossRef]
- Darabi, S.; Elliott, A.; Braxton, D.R.; Zeng, J.; Hodges, K.; Poorman, K.; Swensen, J.; Shanthappa, B.U.; Hinton, J.P.; Gibney, G.T. Transcriptional Profiling of Malignant Melanoma Reveals Novel and Potentially Targetable Gene Fusions. Cancers 2022, 14, 1505. [Google Scholar] [CrossRef]
- Nikanjam, M.; Okamura, R.; Barkauskas, D.A.; Kurzrock, R. Targeting fusions for improved outcomes in oncology treatment. Cancer 2020, 126, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of gene fusions in epithelial cancers: Seq and ye shall find. Genome Med. 2015, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Manochakian, R.; James, L.; Azzouqa, A.-G.; Shi, H.; Zhang, Y.; Zhao, Y.; Zhou, K.; Lou, Y. Emerging therapeutic agents for advanced non-small cell lung cancer. J. Hematol. Oncol. 2020, 13, 58. [Google Scholar] [CrossRef]
- Tuna, M.; Amos, C.I.; Mills, G.B. Molecular mechanisms and pathobiology of oncogenic fusion transcripts in epithelial tumors. Oncotarget 2019, 10, 2095. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Chen, Y.; Chi, P. Basket trial of TRK inhibitors demonstrates efficacy in TRK fusion-positive cancers. J. Hematol. Oncol. 2018, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Goto, K.; Oxnard, G.R.; Tan, D.S.-W.; Loong, H.H.; Bauer, T.M.; Gainor, J.F.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C. Selpercatinib (LOXO-292) in patients with RET-fusion+ non-small cell lung cancer. Am. Soc. Clin. Oncol. 2020, 38, 3584. [Google Scholar] [CrossRef]
- Shah, M.H.; Sherman, E.J.; Robinson, B.; Solomon, B.J.; Kang, H.; Lorch, J.H.; Worden, F.P.; Brose, M.S.; Leboulleux, S.; Godbert, Y. Selpercatinib (LOXO-292) in patients with RET-mutant medullary thyroid cancer. Am. Soc. Clin. Oncol. 2020, 4, PO.20.00096. [Google Scholar] [CrossRef]
- An, X.; Tiwari, A.K.; Sun, Y.; Ding, P.-R.; Ashby Jr, C.R.; Chen, Z.-S. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: A review. Leuk. Res. 2010, 34, 1255–1268. [Google Scholar] [CrossRef]
- Cuellar, S.; Vozniak, M.; Rhodes, J.; Forcello, N.; Olszta, D. BCR-ABL1 tyrosine kinase inhibitors for the treatment of chronic myeloid leukemia. J. Oncol. Pharm. Pract. 2018, 24, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Torres-Ruiz, R.; Puig-Serra, P.; Moreno-Gaona, P.; Martin, M.; Moya, F.; Quintana-Bustamante, O.; Garcia-Silva, S.; Carcaboso, A.; Petazzi, P. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat. Commun. 2020, 11, 5060. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Consortium, A.P.G.; Consortium, A.P.G.; André, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Pugh, T.J.; Bell, J.L.; Bruce, J.P.; Doherty, G.J.; Galvin, M.; Green, M.F.; Hunter-Zinck, H.; Kumari, P.; Lenoue-Newton, M.L.; Li, M.M. AACR Project GENIE: 100,000 cases and beyond. Cancer Discov. 2022, 12, 2044–2057. [Google Scholar] [CrossRef]
- Melas, M.; Subbiah, S.; Saadat, S.; Rajurkar, S.; McDonnell, K.J. The community oncology and academic medical center alliance in the age of precision medicine: Cancer genetics and genomics considerations. J. Clin. Med. 2020, 9, 2125. [Google Scholar] [CrossRef]
- Darabi, S.; Braxton, D.; Homer, J.; Brodie, T.; Holnagel, D.; Eisenberg, B.; Demeure, M.J. Programmatic Efforts Increase Adoption of Genomic Precision Medicine in Cancer Care in a Community Cancer Center. JCO Precis. Oncol. 2022, 6, e2200090. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2022. Available online: https://mitelmandatabase.isb-cgc.org/ (accessed on 2 February 2023).
- Wang, Z.; Wang, Y.; Zhang, J.; Hu, Q.; Zhi, F.; Zhang, S.; Mao, D.; Zhang, Y.; Liang, H. Significance of the TMPRSS2: ERG gene fusion in prostate cancer. Mol. Med. Rep. 2017, 16, 5450–5458. [Google Scholar] [CrossRef] [Green Version]
- Adashek, J.J.; Desai, A.P.; Andreev-Drakhlin, A.Y.; Roszik, J.; Cote, G.J.; Subbiah, V. Hallmarks of RET and Co-occuring Genomic Alterations in RET-aberrant Cancers. Mol. Cancer Ther. 2021, 20, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Reeser, J.W.; Martin, D.; Miya, J.; Kautto, E.A.; Lyon, E.; Zhu, E.; Wing, M.R.; Smith, A.; Reeder, M.; Samorodnitsky, E. Validation of a targeted RNA sequencing assay for kinase fusion detection in solid tumors. J. Mol. Diagn. 2017, 19, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lu, H.; Ng, P.K.-S.; Pantazi, A.; Ip, C.K.M.; Jeong, K.J.; Amador, B.; Tran, R.; Tsang, Y.H.; Yang, L. A functional genomic approach to actionable gene fusions for precision oncology. Sci. Adv. 2022, 8, eabm2382. [Google Scholar] [CrossRef]
- Zhao, X.; Kotch, C.; Fox, E.; Surrey, L.F.; Wertheim, G.B.; Baloch, Z.W.; Lin, F.; Pillai, V.; Luo, M.; Kreiger, P.A. NTRK fusions identified in pediatric tumors: The frequency, fusion partners, and clinical outcome. JCO Precis. Oncol. 2021, 1, 204–214. [Google Scholar] [CrossRef]
- Hamosh, A.; Scott, A.F.; Amberger, J.; Valle, D.; McKusick, V.A. Online Mendelian inheritance in man (OMIM). Hum. Mutat. 2000, 15, 57–61. [Google Scholar] [CrossRef]
- Bach, D.-H.; Park, H.J.; Lee, S.K. The dual role of bone morphogenetic proteins in cancer. Mol. Ther. Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.; Bibeau, F.; Dietel, M.; Hechtman, J.; Troiani, T.; López-Rios, F.; Douillard, J.-Y. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Saliba, J.; Church, A.J.; Rao, S.; Danos, A.; Furtado, L.V.; Laetsch, T.; Zhang, L.; Nardi, V.; Lin, W.-H.; Ritter, D.I. Standardized evidence-based approach for assessment of oncogenic and clinical significance of NTRK fusions. Cancer Genet. 2022, 264, 50–59. [Google Scholar] [CrossRef]
- Gene Fusion Curation. Available online: https://cancervariants.org/projects/fusions/ (accessed on 25 June 2023).
- Liu, J.; Krantz, I. Cornelia de Lange syndrome, cohesin, and beyond. Clin. Genet. 2009, 76, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Argani, P.; Palsgrove, D.N.; Anders, R.A.; Smith, S.C.; Saoud, C.; Kwon, R.; Voltaggio, L.; Assarzadegan, N.; Oshima, K.; Rooper, L. A novel NIPBL-NACC1 gene fusion is characteristic of the cholangioblastic variant of intrahepatic cholangiocarcinoma. Am. J. Surg. Pathol. 2021, 45, 1550–1560. [Google Scholar] [CrossRef]
- Braxton, D.R.; Saxe, D.; Damjanov, N.; Stashek, K.; Shroff, S.; Morrissette, J.D.; Tondon, R.; Furth, E.E. Molecular and cytogenomic profiling of hepatic adenocarcinoma expressing inhibinA, a mimicker of neuroendocrine tumors: Proposal to reclassify as “cholangioblastic variant of intrahepatic cholangiocarcinoma”. Hum. Pathol. 2017, 62, 232–241. [Google Scholar] [CrossRef]
- Huang, G.; Howard, L.N.; Alonsozana, E.; Sill, D.; Bose, D.; Lai, J. Molecular Characteristics and Immunogenomic Profiling of Cholangioblastic Variant of Intrahepatic Cholangiocarcinoma in a 68-year-old Patient. Anticancer Res. 2022, 42, 5475–5478. [Google Scholar] [CrossRef]
- Xu, W.; Ying, Y.; Shan, L.; Feng, J.; Zhang, S.; Gao, Y.; Xu, X.; Yao, Y.; Zhu, C.; Mao, W. Enhanced expression of cohesin loading factor NIPBL confers poor prognosis and chemotherapy resistance in non-small cell lung cancer. J. Transl. Med. 2015, 13, 153. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Peng, F.; Chang, Y.; Liu, T.; Shen, J.; Chen, Z.; Dong, Q.; Zhou, P.; Jiang, F.; Xiang, H. Cohesin mutation sensitizes cancer cells to anti-PD-1 therapy through endogenous retrovirus-mediated PD-L1 upregulation. bioRxiv 2022. [Google Scholar]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Yu, H.; Ness, S.; Mao, P.; Guo, F.; Tang, J.; Guo, Y. Comprehensive analysis of co-mutations identifies cooperating mechanisms of tumorigenesis. Cancers 2022, 14, 415. [Google Scholar] [CrossRef]
- Yu, H.; Boyle, T.A.; Zhou, C.; Rimm, D.L.; Hirsch, F.R. PD-L1 expression in lung cancer. J. Thorac. Oncol. 2016, 11, 964–975. [Google Scholar] [CrossRef] [Green Version]
- Morris, V.K.; Overman, M.J.; Lam, M.; Parseghian, C.M.; Johnson, B.; Dasari, A.; Raghav, K.; Kee, B.K.; Huey, R.; Wolff, R.A. Bintrafusp Alfa, an Anti-PD-L1: TGFβ Trap Fusion Protein, in Patients with ctDNA-positive, Liver-limited Metastatic Colorectal Cancer. Cancer Res. Commun. 2022, 2, 979–986. [Google Scholar] [CrossRef]
- Heyer, E.E.; Deveson, I.W.; Wooi, D.; Selinger, C.I.; Lyons, R.J.; Hayes, V.M.; O’Toole, S.A.; Ballinger, M.L.; Gill, D.; Thomas, D.M. Diagnosis of fusion genes using targeted RNA sequencing. Nat. Commun. 2019, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Tsang, E.S.; Grisdale, C.J.; Pleasance, E.; Topham, J.T.; Mungall, K.; Reisle, C.; Choo, C.; Carreira, M.; Bowlby, R.; Karasinska, J.M. Uncovering Clinically Relevant Gene Fusions with Integrated Genomic and Transcriptomic Profiling of Metastatic CancersLandscape of Genomic Fusions. Clin. Cancer Res. 2021, 27, 522–531. [Google Scholar] [CrossRef]
- Yi, K.; Ju, Y.S. Patterns and mechanisms of structural variations in human cancer. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Tumor Type (Total # of Samples Profiled) | Fusion-Positive Samples | Unclassified Gene Fusions |
|---|---|---|
| Prostate adenocarcinoma (89) | 8 (32%) | BMPR1B-PDLIM5 (2 patients), CDC13-SUGCT, NIPBL-RAI14, TTC6-MIPOL1, NFIB-UBE3B, GALNT7-SPOCK3, TBCA-AP3B1 |
| Breast cancer (224) | 8 (32%) | NIPBL-PIEZO1, ETV6-RINT1, PAK1-TEX14, PAK1-MMP20, PAK1-LOC101928896, TTC6-MIPOL1, BMPR1B-PDLIM5, CMTM8-OSBPL10 |
| Non-small cell lung cancer (751) | 4 (16%) | PANX1-PRPF19 (2 patients), TTC6-MIPOL1, GALNT7-SMIM8 |
| Head and neck cancer (177) | 2 (8%) | ETV6-DNAH14, TP53BP1-SND1 |
| Salivary gland tumor (22) | 1 (4%) | NFIB-CHCHD7 |
| Pancreatic adenocarcinoma (186) | 1 (4%) | CMTM8-GPD1L |
| Ovarian cancer (362) | 1 (4%) | NIPBL-SLC1A3 |
| Total Number of Fusions | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darabi, S.; Zuazo, C.E.; Braxton, D.R.; Eisenberg, B.L.; Demeure, M.J. Precision Medicine in a Community Cancer Center: Pan-Cancer DNA/RNA Sequencing of Tumors Reveals Clinically Relevant Gene Fusions. Biologics 2023, 3, 198-208. https://doi.org/10.3390/biologics3030011
Darabi S, Zuazo CE, Braxton DR, Eisenberg BL, Demeure MJ. Precision Medicine in a Community Cancer Center: Pan-Cancer DNA/RNA Sequencing of Tumors Reveals Clinically Relevant Gene Fusions. Biologics. 2023; 3(3):198-208. https://doi.org/10.3390/biologics3030011
Chicago/Turabian StyleDarabi, Sourat, Carlos E. Zuazo, David R. Braxton, Burton L. Eisenberg, and Michael J. Demeure. 2023. "Precision Medicine in a Community Cancer Center: Pan-Cancer DNA/RNA Sequencing of Tumors Reveals Clinically Relevant Gene Fusions" Biologics 3, no. 3: 198-208. https://doi.org/10.3390/biologics3030011