
Hydrofluoric Acid-Free Digestion of Organosilicon Nanoparticles for Bioanalysis by ICP-OES

Abstract
:1. Introduction
2. Materials and Methods
2.1. Standards and Reagents
2.2. ICP-OES Equipment and Instrument Parameters
Other Equipment
2.3. Sample Preparation
2.3.1. Decontamination Procedures
2.3.2. Sample Collection
2.3.3. Digestion for Total Si Determination
2.3.4. Attempted Digestion without H2O2
2.3.5. Standards in 15.2% HNO3/5.3% H2O2
2.3.6. NPs to Spike Tissue
3. Results
3.1. Method Validation and Analytical Quality Control
3.1.1. Limits of Detection and Quantification
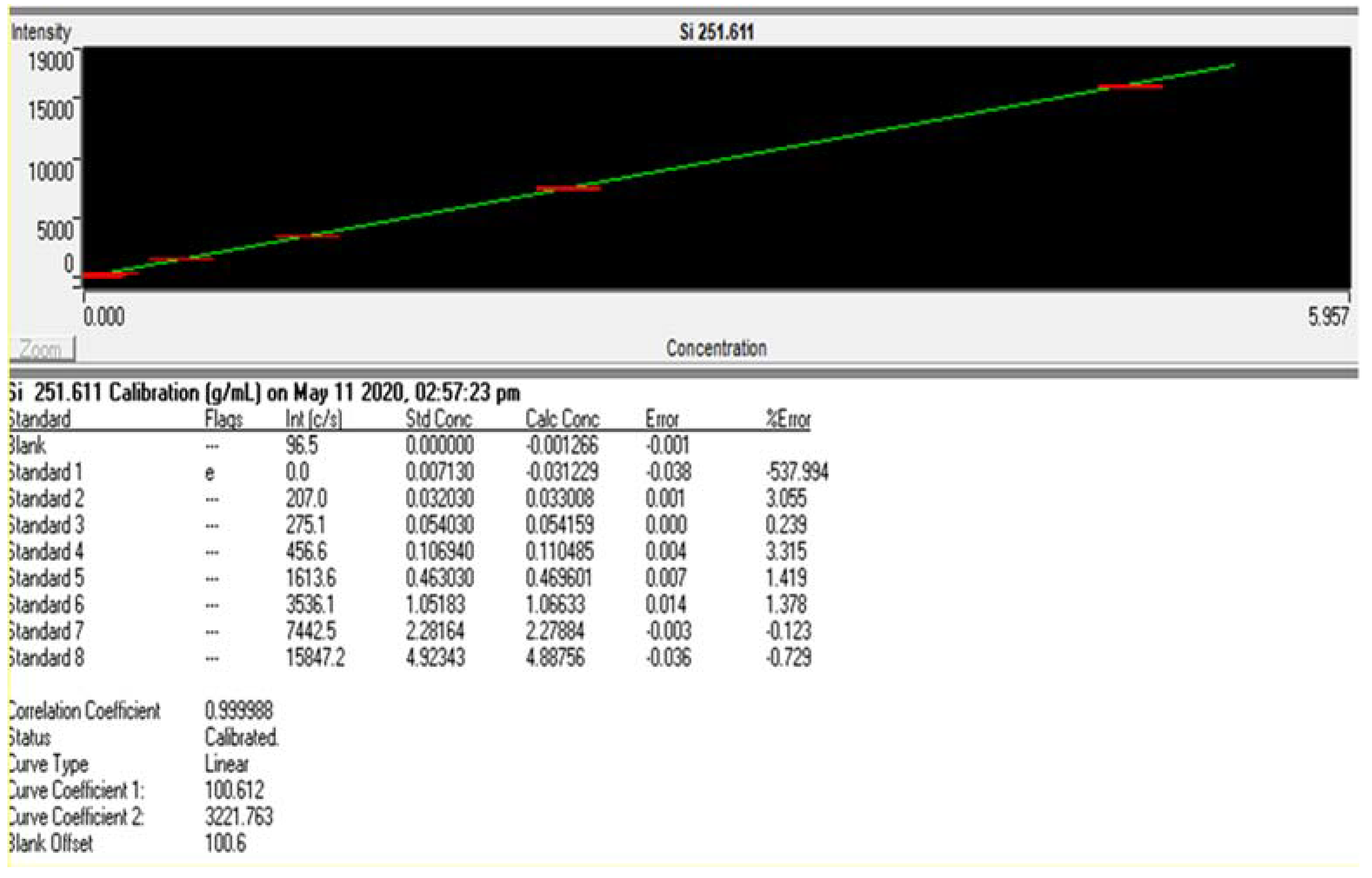
3.1.2. Linearity
3.1.3. Limit of Quantification in Tissue
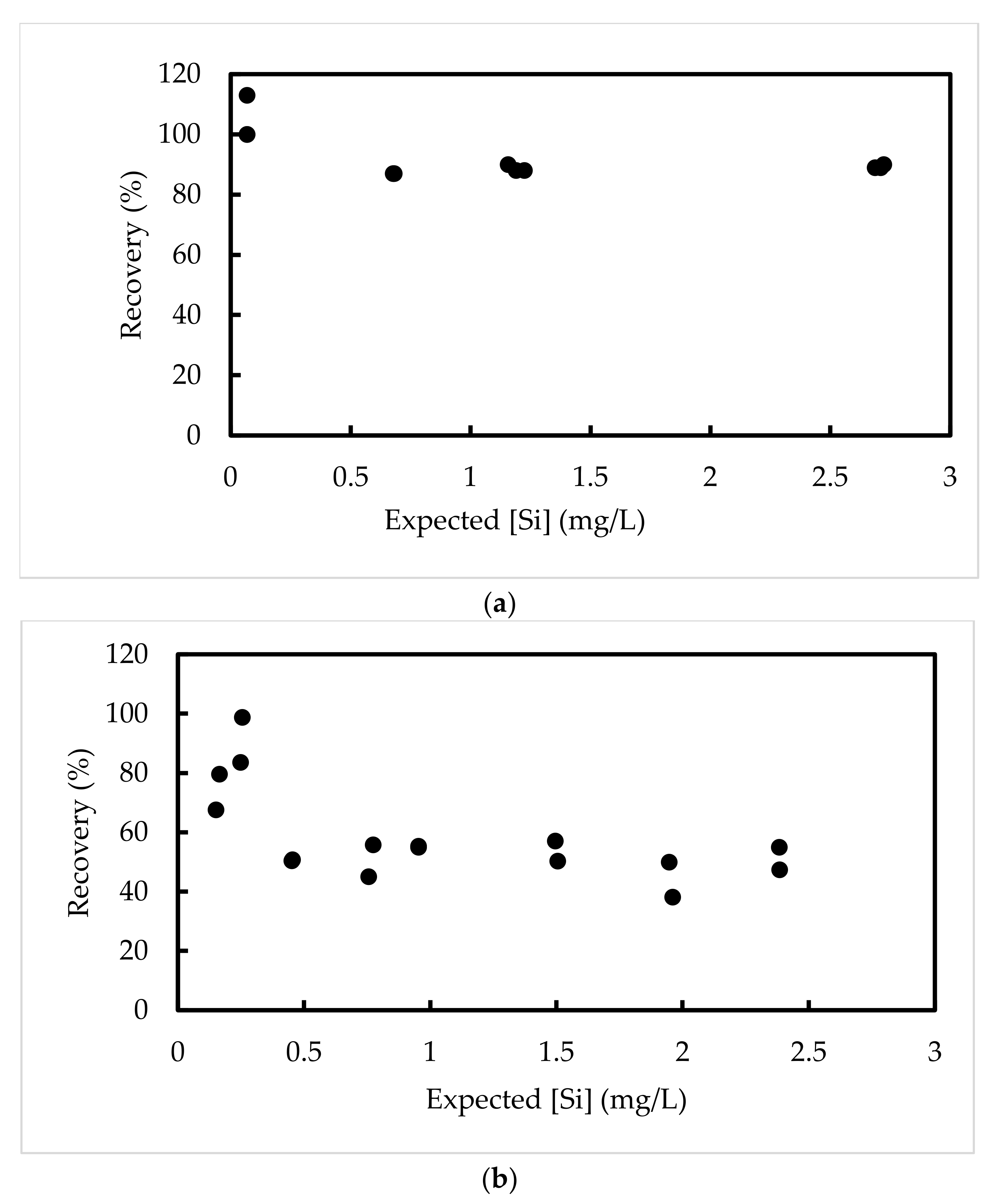
3.1.4. Recovery of Si in Spiked Tissue Using HNO3/H2O2
3.1.5. Attempted Digestion of Colloidal Silica (SiO2)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eriksson, P.-O.; Aaltonen, E.; Petoral, R.; Lauritzson, P.; Miyazaki, H.; Pietras, K.; Månsson, S.; Hansson, L.; Leander, P.; Axelsson, O. Novel Nano-Sized MR Contrast Agent Mediates Strong Tumor Contrast Enhancement in an Oncogene-Driven Breast Cancer Model. PLoS ONE 2014, 9, e107762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.L.; Fassett, J.D.; Lindstrom, A.P. Determination of Si in Standard Reference Material SRM 295× Silica-on-Filter. J. Anal. At. Spectrom. 2003, 18, 738–741. [Google Scholar] [CrossRef]
- Hauptkorn, S.; Pavel, J.; Seltner, H. Determination of Silicon in Biological Samples by ICP-OES after Non-Oxidative Decomposition under Alkaline Conditions. Fresenius J. Anal. Chem. 2001, 370, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Bossert, D.; Urban, D.A.; Maceroni, M.; Ackermann-Hirschi, L.; Haeni, L.; Yajan, P.; Spuch-Calvar, M.; Rothen-Rutishauser, B.; Rodriguez-Lorenzo, L.; Petri-Fink, A.; et al. A Hydrofluoric Acid-Free Method to Dissolve and Quantify Silica Nanoparticles in Aqueous and Solid Matrices. Sci. Rep. 2019, 9, 7938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aureli, F.; Ciprotti, M.; D’Amato, M.; do Nascimento da Silva, E.; Nisi, S.; Passeri, D.; Sorbo, A.; Raggi, A.; Rossi, M.; Cubadda, F. Determination of Total Silicon and SiO2 Particles Using an ICP-MS Based Analytical Platform for Toxicokinetic Studies of Synthetic Amorphous Silica. Nanomaterials 2020, 10, 888. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.J.B.; Oomen, A.G.; van Bemmel, G.; van Vliet, L.; Undas, K.; Munniks, S. Silicon Dioxide and Titanium Dioxide Particles Found in Human Tissues. Nanotoxicology 2020, 14, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xing, B.; Dudas, M.J. Simple Microwave Digestion Technique for Elemental Analysis of Mineral Soil Samples. Can. J. Soil Sci. 1990, 70, 617–620. [Google Scholar] [CrossRef]
- Taber, H.G.; Shogren, D.; Lu, G. Extraction of Silicon from Plant Tissue with Dilute HCl and HF and Measurement by Modified Inductive Coupled Argon Plasma Procedures. Commun. Soil Sci. Plant Anal. 2002, 33, 1661–1670. [Google Scholar] [CrossRef]
- Traoré, S.; Traoré, D.L.; Magassouba, S. Preparation of Silicon from Rice Husk as Renewable Energy Resource by the Use of Microwave Ashing and Acid Digestion. Int. J. Energy Eng. 2018, 8, 25–29. [Google Scholar] [CrossRef]
- Danadurai, K.S.K.; Chellam, S.; Lee, C.-T.; Fraser, M.P. Trace Elemental Analysis of Airborne Particulate Matter Using Dynamic Reaction Cell Inductively Coupled Plasma—Mass Spectrometry: Application to Monitoring Episodic Industrial Emission Events. Anal. Chim. Acta 2011, 686, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.M.; Sakamoto, H. Comparison of the Acid Combinations in Microwave-Assisted Digestion of Marine Sediments for Heavy Metal Analyses. Anal. Sci. 2005, 21, 1181–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viveros, R.D.; Liberman, A.; Trogler, W.C.; Kummel, A.C. Alkaline and Ultrasonic Dissolution of Biological Materials for Trace Silicon Determination. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 2015, 33, 031803. [Google Scholar] [CrossRef] [Green Version]
- Coradin, T.; Eglin, D.; Livage, J. The Silicomolybdic Acid Spectrophotometric Method and Its Application to Silicate/Biopolymer Interaction Studies. Spectroscopy 2004, 18, 567–576. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Instrument Parameter | Value |
|---|---|
| RF power | 1.3 kW |
| Plasma gas flow | 15 L min−1 |
| Auxiliary gas flow | 1.5 L min−1 |
| Replicate read time | 5 s |
| Sample uptake delay time | 35 s |
| Instr. stabilization delay | 20 s |
| No. of replicates | 3 |
| Nebulizer flow | 0.75 L min−1 |
| Peristaltic pump flow rate | 15 rpm |
| Blank samples between samples | 1% HNO3 + 5% HCl with detergent was included between samples. |
| Wash between samples | Rinse for 20 s with 1% HNO3. |
| Calibration curve | Weighted linear and maximal 5% error allowed. Recalibration was made every 30 samples. |
| AVR Intensity of Blank (int) | STD Deviation (int) | Slope (int/ppm) | LOD (mg/L) | LOQ (mg/L) |
|---|---|---|---|---|
| 106.5 | 10.8 | 2977.66 | 0.012 | 0.036 |
| Statistical Analysis | Observation |
|---|---|
| Slope | 3221.76 |
| Y-Intercept | 100.6 |
| Correlation Coefficient | 0.9999 |
| Samples | Expected ppm | Raw Data ppm | Ratio |
|---|---|---|---|
| LLOQ Si | 0.0684 | 0.0689 | 1.00 |
| LLOQ Si | 0.0682 | 0.0685 | 1.00 |
| LLOQ Si | 0.0684 | 0.0775 | 1.13 |
| Low-level Si | 0.6782 | 0.5880 | 0.87 |
| Low-level Si | 0.6820 | 0.5882 | 0.87 |
| Medium level Si | 1.2254 | 1.080 | 0.88 |
| Medium level Si | 1.1904 | 1.0521 | 0.88 |
| Medium level Si | 1.1566 | 1.0411 | 0.90 |
| High level Si | 2.7097 | 2.4116 | 0.89 |
| High level Si | 2.6867 | 2.3979 | 0.89 |
| High level Si | 2.7236 | 2.4425 | 0.90 |
| Samples | Expected Si mg/L | Raw Data ICP Si mg/L | Ratio |
|---|---|---|---|
| Ctrl-chicken liver | 0.052 ± 0.01 | ||
| Spike chicken liver #1 | 0.18 | 0.17 | 0.98 |
| Spike chicken liver #2 | 0.20 | 0.19 | 0.94 |
| Spike chicken liver #3 | 0.40 | 0.40 | 0.99 |
| Spike chicken liver #4 | 0.41 | 0.41 | 1.01 |
| Spike chicken liver #5 | 0.80 | 0.70 | 0.87 |
| Spike chicken liver #6 | 0.87 | 0.75 | 0.86 |
| Spike chicken liver #7 | 1.49 | 1.40 | 0.94 |
| Spike chicken liver #8 | 1.52 | 1.35 | 0.89 |
| Spike chicken liver #9 | 2.69 | 2.57 | 0.96 |
| Study #, Sample ID | Expected Si mg/L | Raw Data ICP Si mg/L | Ratio |
|---|---|---|---|
| Ctrl-mouse liver | <LOD | ||
| Low spike mouse liver #1 | 0.45 | 0.42 | 0.92 |
| Medium spike mouse liver #2 | 0.69 | 0.64 | 0.92 |
| High spike mouse liver #3 | 1.01 | 0.93 | 0.92 |
| High spike mouse liver #4 | 2.24 | 2.07 | 0.92 |
| Samples | Expected ppm | Raw Data ppm | Ratio |
|---|---|---|---|
| Ctrl- mouse carcass | 1.64 ± 0.14 | ||
| Spike mouse carcass #1 | 2.66 | 2.60 | 0.98 |
| Spike mouse carcass #2 | 2.57 | 2.28 | 0.89 |
| Spike mouse carcass #3 | 2.61 | 2.40 | 0.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousefpour, N.; Björnberg, O.; Yao Mattisson, I.; Axelsson, O. Hydrofluoric Acid-Free Digestion of Organosilicon Nanoparticles for Bioanalysis by ICP-OES. Micro 2023, 3, 264-273. https://doi.org/10.3390/micro3010019
Yousefpour N, Björnberg O, Yao Mattisson I, Axelsson O. Hydrofluoric Acid-Free Digestion of Organosilicon Nanoparticles for Bioanalysis by ICP-OES. Micro. 2023; 3(1):264-273. https://doi.org/10.3390/micro3010019
Chicago/Turabian StyleYousefpour, Nooshin, Olof Björnberg, Ingrid Yao Mattisson, and Oskar Axelsson. 2023. "Hydrofluoric Acid-Free Digestion of Organosilicon Nanoparticles for Bioanalysis by ICP-OES" Micro 3, no. 1: 264-273. https://doi.org/10.3390/micro3010019