
Auxotrophy-Independent Plasmid Shuttle Vectors for Applications in Diverse Yeasts

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Strains, Media, and Culture Conditions
2.2. Optical Density to Cell Count Conversion
2.3. Plasmid Construction
2.4. Evaluation of Transformation Efficiency
2.5. Evaluation of Plasmid Maintenance in Selective and Non-Selective Media
2.6. mNeonGreen Fluorescence Measurements
3. Results & Discussion
3.1. Design and Construction of 18 Drug-Selectable Yeast Plasmid Vectors
3.2. Transformation Efficiencies Are Highly Variable across the Diverse Yeast Panel
3.3. Yeast Origin of Replication Determines Yeast-Plasmid Compatibility
3.4. Plasmid-Derived Gene Expression Levels Vary across the Diverse Yeast Panel
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for modern biology. Science 1988, 240, 1439–1443. [Google Scholar] [CrossRef]
- Botstein, D.; Chervitz, S.A.; Cherry, J.M. Yeast as a model organism. Science 1997, 277, 1259–1260. [Google Scholar] [CrossRef]
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding yeast for budding geneticists: A primer on the Saccharomyces cerevisiae model system. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef]
- Scannell, D.R.; Zill, O.A.; Rokas, A.; Payen, C.; Dunham, M.J.; Eisen, M.B.; Rine, J.; Johnston, M.; Hittinger, C.T. The Awesome Power of Yeast Evolutionary Genetics: New Genome Sequences and Strain Resources for the Saccharomyces sensu stricto Genus. G3 2011, 1, 11–25. [Google Scholar] [CrossRef]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef]
- Hoffman, C.S.; Wood, V.; Fantes, P.A. An Ancient Yeast for Young Geneticists: A Primer on the Schizosaccharomyces pombe Model System. Genetics 2015, 201, 403–423. [Google Scholar] [CrossRef]
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia pastoris: A highly successful expression system for optimal synthesis of heterologous proteins. J. Cell. Physiol. 2020, 235, 5867–5881. [Google Scholar] [CrossRef]
- Averianova, L.A.; Balabanova, L.A.; Son, O.M.; Podvolotskaya, A.B.; Tekutyeva, L.A. Production of Vitamin B2 (Riboflavin) by Microorganisms: An Overview. Front. Bioeng. Biotechnol. 2020, 8, 570828. [Google Scholar] [CrossRef] [PubMed]
- Gnügge, R.; Rudolf, F. Saccharomyces cerevisiae Shuttle vectors. Yeast 2017, 34, 205–221. [Google Scholar] [CrossRef]
- Ma, H.; Kunes, S.; Schatz, P.J.; Botstein, D. Plasmid construction by homologous recombination in yeast. Gene 1987, 58, 201–216. [Google Scholar] [CrossRef]
- Oldenburg, K.R.; Vo, K.T.; Michaelis, S.; Paddon, C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997, 25, 451–452. [Google Scholar] [CrossRef]
- Nora, L.C.; Westmann, C.A.; Martins-Santana, L.; Alves, L.F.; Monteiro, L.M.O.; Guazzaroni, M.E.; Silva-Rocha, R. The art of vector engineering: Towards the construction of next-generation genetic tools. Microb. Biotechnol. 2019, 12, 125–147. [Google Scholar] [CrossRef]
- Cochrane, R.R.; Shrestha, A.; Severo de Almeida, M.M.; Agyare-Tabbi, M.; Brumwell, S.L.; Hamadache Meaney, J.S.; Nucifora, D.P.; Say, H.H.; Sharma, J.; Soltysiak, M.P.M.; et al. Superior Conjugative Plasmids Delivered by Bacteria to Diverse Fungi. BioDesign Res. 2022, 2022, 9802168. [Google Scholar] [CrossRef]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef]
- Christianson, T.W.; Sikorski, R.S.; Dante, M.; Shero, J.H.; Hieter, P. Multifunctional yeast high-copy-number shuttle vectors. Gene 1992, 110, 119–122. [Google Scholar] [CrossRef]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]
- Chee, M.K.; Haase, S.B. New and Redesigned pRS Plasmid Shuttle Vectors for Genetic Manipulation of Saccharomyces cerevisiae. G3 2012, 2, 515–526. [Google Scholar] [CrossRef]
- Siewers, V. An overview on selection marker genes for transformation of Saccharomyces cerevisiae. Methods Mol. Biol. 2014, 1152, 3–15. [Google Scholar] [CrossRef]
- Pronk, J.T. Auxotrophic yeast strains in fundamental and applied research. Appl. Environ. Microbiol. 2002, 68, 2095–2100. [Google Scholar] [CrossRef]
- Lewis, A.; Caldwell, R.; Rogers, J.; Ingaramo, M.; Wang, R.; Soifer, I.; Hendrickson, D.; McIsaac, S.R.; Botstein, D.; Gibney, P.A. Loss of major nutrient sensing and signaling pathways suppresses starvation lethality in electron transport chain mutants. Mol. Biol. Cell 2021, 32, ar39. [Google Scholar] [CrossRef]
- Haupt, I.; Hubener, R.; Thrum, H. Streptothricin F, an inhibitor of protein synthesis with miscoding activity. J. Antibiot. 1978, 31, 1137–1142. [Google Scholar] [CrossRef]
- Eustice, D.C.; Wilhelm, J.M. Mechanisms of action of aminoglycoside antibiotics in eucaryotic protein synthesis. Antimicrob. Agents Chemother. 1984, 26, 53–60. [Google Scholar] [CrossRef]
- Steiner, S.; Philippsen, P. Sequence and promoter analysis of the highly expressed TEF gene of the filamentous fungus Ashbya gossypii. Mol. Gen. Genet. 1994, 242, 263–271. [Google Scholar] [CrossRef]
- Jimenez, A.; Davies, J. Expression of a transposable antibiotic resistance element in Saccharomyces. Nature 1980, 287, 869–871. [Google Scholar] [CrossRef]
- Wach, A.; Brachat, A.; Pöhlmann, R.; Philippsen, P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 1994, 10, 1793–1808. [Google Scholar] [CrossRef]
- Gritz, L.; Davies, J. Plasmid-encoded hygromycin B resistance: The sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene 1983, 25, 179–188. [Google Scholar] [CrossRef]
- Goldstein, A.L.; McCusker, J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 1999, 15, 1541–1553. [Google Scholar] [CrossRef]
- Taxis, C.; Knop, M. System of centromeric, episomal, and integrative vectors based on drug resistance markers for Saccharomyces cerevisiae. Biotechniques 2006, 40, 73–78. [Google Scholar] [CrossRef]
- Cottarel, G.; Shero, J.H.; Hieter, P.; Hegemann, J.H. A 125-base-pair CEN6 DNA fragment is sufficient for complete meiotic and mitotic centromere functions in Saccharomyces cerevisiae. Mol. Cell. Biol. 1989, 9, 3342–3349. [Google Scholar] [CrossRef]
- Liachko, I.; Dunham, M.J. An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res. 2014, 14, 364–367. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Hajra, S.; Paek, A.; Jayaram, M. Mechanisms for chromosome and plasmid segregation. Annu. Rev. Biochem. 2006, 75, 211–241. [Google Scholar] [CrossRef]
- Nieduszynski, C.A.; Knox, Y.; Donaldson, A.D. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 2006, 20, 1874–1879. [Google Scholar] [CrossRef]
- Müller, C.A.; Nieduszynski, C.A. Conservation of replication timing reveals global and local regulation of replication origin activity. Genome Res. 2012, 22, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Talarek, N.; Louis, E.J.; Cullin, C.; Aigle, M. Developing methods and strains for genetic studies in the Saccharomyces bayanus var. uvarum species. Yeast 2004, 21, 1195–1203. [Google Scholar] [CrossRef]
- Kitagawa, T.; Tokuhiro, K.; Sugiyama, H.; Kohda, K.; Isono, N.; Hisamatsu, M.; Takahashi, H.; Imaeda, T. Construction of a β-glucosidase expression system using the multistress-tolerant yeast Issatchenkia orientalis. Appl. Microbiol. Biotechnol. 2010, 87, 1841–1853. [Google Scholar] [CrossRef] [PubMed]
- Maria, H.; Kapoor, S.; Liu, T.; Rusche, L.N. Conservation of a DNA replication motif among phylogenetically distant budding yeast species. Genome Biol. Evol. 2021, 13, evab137. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Huang, C.-C.; Hajra, S.; Jayaram, M. Yeast cohesin complex embraces 2 micron plasmid sisters in a tri-linked catenane complex. Nucleic Acids Res. 2010, 38, 570–584. [Google Scholar] [CrossRef]
- Rizvi, S.M.A.; Prajapati, H.K.; Ghosh, S.K. The 2 micron plasmid: A selfish genetic element with an optimized survival strategy within Saccharomyces cerevisiae. Curr. Genet. 2018, 64, 25–42. [Google Scholar] [CrossRef]
- McQuaid, M.E.; Mereshchuk, A.; Dobson, M.J. Insights into the DNA sequence elements required for partitioning and copy number control of the yeast 2-micron plasmid. Curr. Genet. 2019, 65, 887–892. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Hickman, M.J.; Spatt, D.; Winston, F. The Hog1 mitogen-activated protein kinase mediates a hypoxic response in Saccharomyces cerevisiae. Genetics 2011, 188, 325–338. [Google Scholar] [CrossRef]
- Cliften, P.; Sudarsanam, P.; Desikan, A.; Fulton, L.; Fulton, B.; Majors, J.; Waterston, R.; Cohen, B.A.; Johnston, M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 2003, 301, 71–76. [Google Scholar] [CrossRef]
- Gibson, D.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., III; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002, 350, 87–96. [Google Scholar] [CrossRef]
- Kawai, S.; Hashimoto, W.; Murata, K. Transformation of Saccharomyces cerevisiae and other fungi: Methods and possible underlying mechanism. Bioeng. Bugs. 2010, 1, 395–403. [Google Scholar] [CrossRef]
- Andersson Rasmussen, A.; Kandasamy, D.; Beck, H.; Crosby, S.D.; Björnberg, O.; Schnackerz, K.D.; Piškur, J. Global expression analysis of the yeast Lachancea (Saccharomyces) kluyveri reveals new URC genes involved in pyrimidine catabolism. Eukaryot. Cell 2014, 13, 31–42. [Google Scholar] [CrossRef]
- Bleuven, C.; Dubé, A.K.; Nguyen, G.Q.; Gagnon-Arsenault, I.; Martin, H.; Landry, C.R. A collection of barcoded natural isolates of Saccharomyces paradoxus to study microbial evolutionary ecology. MicrobiologyOpen 2019, 8, e773. [Google Scholar] [CrossRef]
- Moreno-Beltrán, M.; Gore-Lloyd, D.; Chuck, C.; Henk, D. Variation among Metschnikowia pulcherrima isolates for genetic modification and homologous recombination. Microorganisms 2021, 9, 290. [Google Scholar] [CrossRef]
- Karim, A.S.; Curran, K.A.; Alper, H.S. Characterization of plasmid burden and copy number in Saccharomyces cerevisiae for optimization of metabolic engineering applications. FEMS Yeast Res. 2013, 13, 107–116. [Google Scholar] [CrossRef]
- Eguchi, Y.; Makanae, K.; Hasunuma, T.; Ishibashi, Y.; Kito, K.; Moriya, H. Estimating the protein burden limit of yeast cells by measuring the expression limits of glycolytic proteins. eLife 2018, 7, e34595. [Google Scholar] [CrossRef]



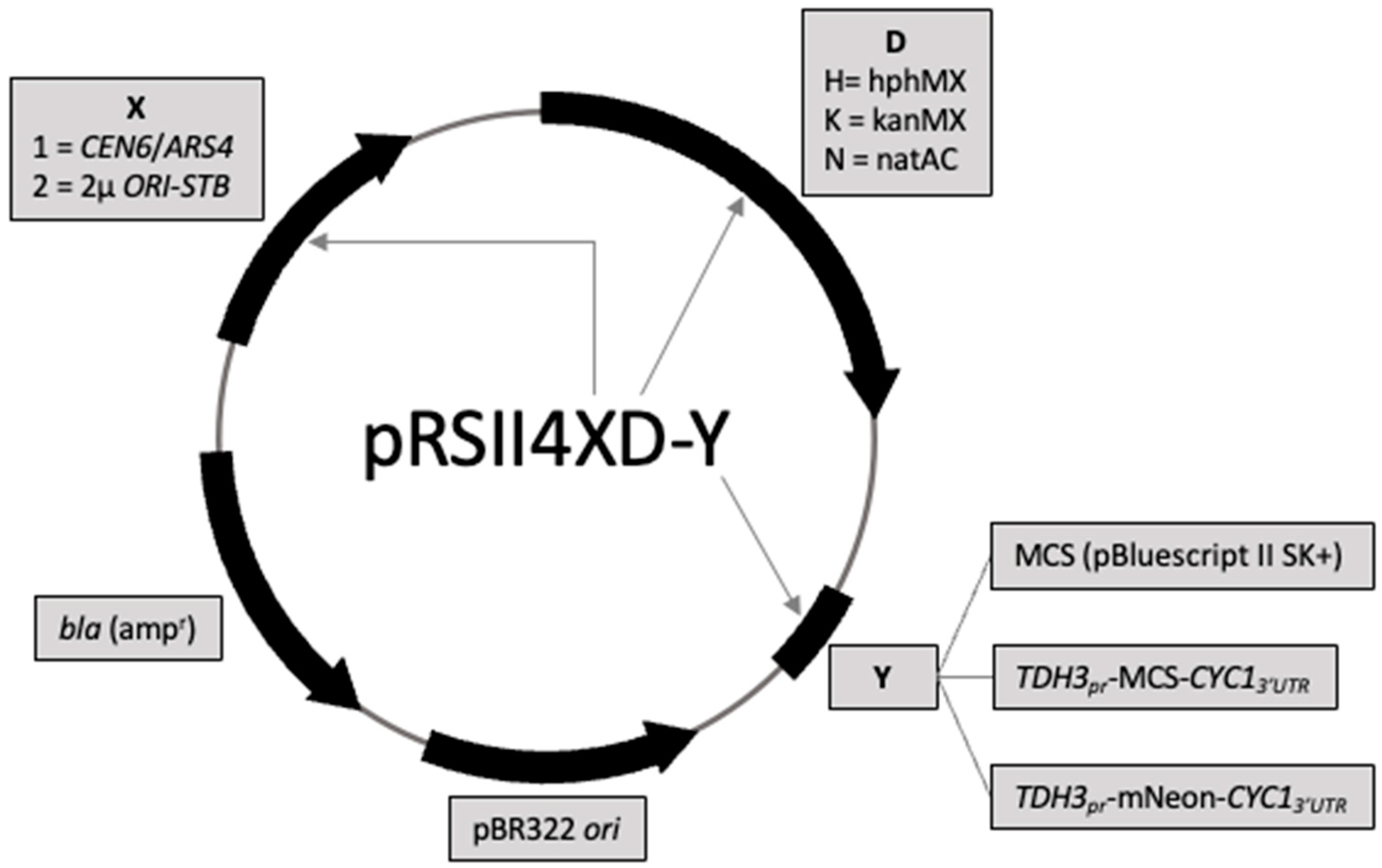{kind=link}
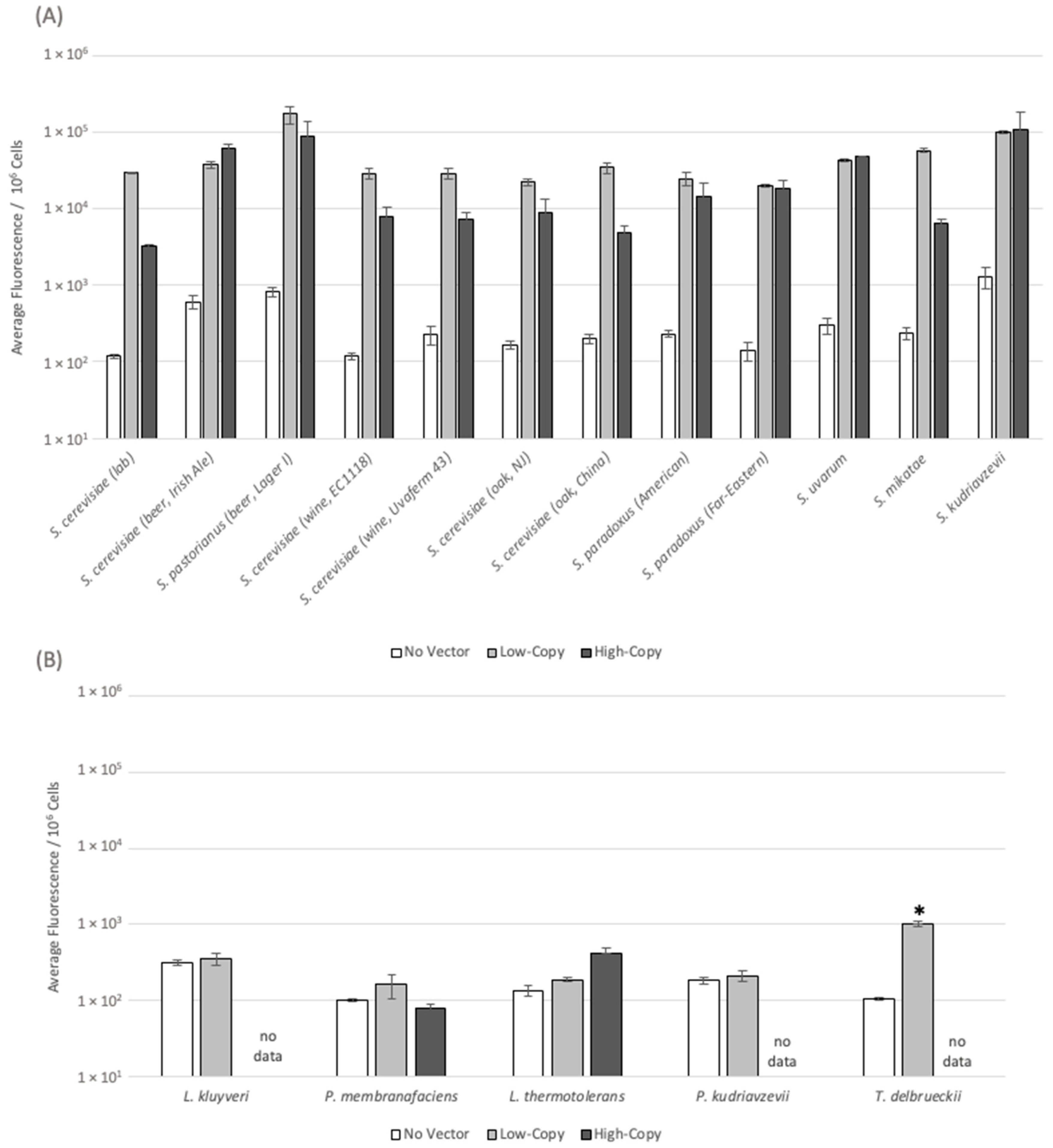{kind=link}
| Strain Code | Aliases | Species | Description | Source |
|---|---|---|---|---|
| DBY12000 | FY2648 | Saccharomyces cerevisiae | Lab strain; prototrophic derivative of S288C, MATa, HAP1+ | [41] |
| DBY17018 | Saccharomyces uvarum | [42] | ||
| DBY17019 | Saccharomyces mikatae | [42] | ||
| DBY17020 | Saccharomyces kudriavzevii | [42] | ||
| DBY17021 | Naumovozyma castellii | [42] | ||
| DBY17022 | Lachancea kluyveri | [42] | ||
| DBY18207 | DBVPG6304 | Saccharomyces paradoxus | American subpopulation; from California | Leonid Kruglyak lab |
| DBY18222 | N-44 | Saccharomyces paradoxus | Far-Eastern subpopulation; from Russia | Leonid Kruglyak lab |
| OYL005 | Saccharomyces cerevisiae | Commercial beer strain; Irish Ale | Omega Yeast Labs | |
| OYL100 | Saccharomyces pastorianus | Commercial beer strain; Lager I | Omega Yeast Labs | |
| PGY12 | BJ20, YJS4581, OS_552 | Saccharomyces cerevisiae | Oak isolate; from Dongling Mountain, Beijing, China | Joseph Schacherer lab |
| PGY34 | YPS1000, YJS168 | Saccharomyces cerevisiae | Oak isolate; from New Jersey, United States | Joseph Schacherer lab |
| PGY68 | Y819 | Saccharomyces cerevisiae | Commercial wine strain; Uvaferm 43 | E&J Gallo Winery |
| PGY83 | Y834 | Saccharomyces cerevisiae | Commercial wine strain; Lalvin EC-1118 | E&J Gallo Winery |
| PGY320 | UCD7 | Pichia membranafaciens | UC-Davis V&E Department | |
| PGY321 | UCD227 | Schizosaccharomyces pombe | Strain FST 40-277; ATCC 2476, NRRL Y-164 | UC-Davis V&E Department |
| PGY323 | UCD601 | Lachancea thermotolerans | Wine isolate; strain Radler 40 | UC-Davis V&E Department |
| PGY325 | UCD751 | Zygosaccharomyces bailii | Strain PS p.194 | UC-Davis V&E Department |
| PGY326 | UCD848 | Metschnikowia pulcherrima | UC-Davis V&E Department | |
| PGY327 | UCD1017 | Hanseniaspora uvarum | Strain 1015-IFI | UC-Davis V&E Department |
| PGY328 | UCD2116 | Pichia kudriavzevii | Candida krusei; from Luna barrel fermentation | UC-Davis V&E Department |
| PGY329 | UCD2221 | Torulaspora delbrueckii | Wine isolate | UC-Davis V&E Department |
| PGY330 | UCD2510 | Saccharomycodes ludwigii | Wine isolate | UC-Davis V&E Department |
| PGY332 | UCD2077 | Brettanomyces bruxellensis | AWRI 1499-like strain | UC-Davis V&E Department |
| Name | Other Name(s) | Host | Maintenance Elements | Selective Elements | Expression Elements | Source | Addgene ID |
|---|---|---|---|---|---|---|---|
| pRSII416 | RB3534 | XL1-B E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Uracil auxotrophic complementation (URA3) | MCS (pBluescript II SK+) | [17] | 35456 |
| pRSII426 | RB3535 | XL1-B E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), Uracil auxotrophic complementation (URA3) | MCS (pBluescript II SK+) | [17] | 35470 |
| pRS416-KanMX-TDH3pr | RB3398 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Uracil auxotrophic complementation (URA3) | TDH3pr-MCS-CYC13′UTR | This Study | |
| pRS416-HphMX-TDH3pr | RB3399 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Uracil auxotrophic complementation (URA3) | TDH3pr-MCS-CYC13′UTR | This Study | |
| pRS416-NatAC-TDH3pr | RB3400 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Uracil auxotrophic complementation (URA3) | TDH3pr-MCS-CYC13′UTR | This Study | |
| p416GPD | p416GPD | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Uracil auxotrophic complementation (URA3) | TDH3pr-MCS-CYC13′UTR | [16] | |
| pKT127-mNeonGreen | DH10B E.coli | E. coli origin (pBR322 ori) | Ampicillin-resistant (bla), G418-resistant (kanMX) | SP6pr-mNeon-ADH13′UTR | [40] | ||
| pRSII41K | PGB74 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), G418-resistant (kanMX) | MCS | This Study | 194522 |
| pRSII42K | PGB75 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), G418-resistant (kanMX) | MCS | This Study | 194523 |
| pRSII41N | PGB80 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Nourseothricin-resistant (natAC) | MCS | This Study | 194524 |
| pRSII42N | PGB81 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), Nourseothricin-resistant (natAC) | MCS | This Study | 194525 |
| pRSII41H | PGB78 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Hygromycin B-resistant (hphMX) | MCS | This Study | 194526 |
| pRSII42H | PGB79 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), Hygromycin B-resistant (hphMX) | MCS | This Study | 194527 |
| pRSII41K-TDH3pr | PGB94 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), G418-resistant (kanMX) | TDH3pr-MCS-CYC13′UTR | This Study | 194528 |
| pRSII42K-TDH3pr | PGB95 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), G418-resistant (kanMX) | TDH3pr-MCS-CYC13′UTR | This Study | 194529 |
| pRSII41N-TDH3pr | PGB96 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Nourseothricin-resistant (natAC) | TDH3pr-MCS-CYC13′UTR | This Study | 194530 |
| pRSII42N-TDH3pr | PGB97 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), Nourseothricin-resistant (natAC) | TDH3pr-MCS-CYC13′UTR | This Study | 194531 |
| pRSII41H-TDH3pr | PGB99 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Hygromycin B-resistant (hphMX) | TDH3pr-MCS-CYC13’UTR | This Study | 194532 |
| pRSII42H-TDH3pr | PGB98 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), Hygromycin B-resistant (hphMX) | TDH3pr-MCS-CYC13′UTR | This Study | 194533 |
| pRSII41K-TDH3pr-mNeon | PGB100 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), G418-resistant (kanMX) | TDH3pr-mNeon-CYC13′UTR | This Study | 194534 |
| pRSII42K-TDH3pr-mNeon | PGB101 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), G418-resistant (kanMX) | TDH3pr-mNeon-CYC13′UTR | This Study | 194535 |
| pRSII41N-TDH3pr-mNeon | PGB102 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Nourseothricin-resistant (natAC) | TDH3pr-mNeon-CYC13′UTR | This Study | 194536 |
| pRSII42N-TDH3pr-mNeon | PGB103 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), Nourseothricin-resistant (natAC) | TDH3pr-mNeon-CYC13′UTR | This Study | 194537 |
| pRSII41H-TDH3pr-mNeon | PGB105 | TOP10 E.coli | E. coli origin (pBR322 ori), Low-copy (CEN6/ARS4) | Ampicillin-resistant (bla), Hygromycin B-resistant (hphMX) | TDH3pr-mNeon-CYC13′UTR | This Study | 194538 |
| pRSII42H-TDH3pr-mNeon | PGB104 | TOP10 E.coli | E. coli origin (pBR322 ori), High-copy (2µ ORI-STB) | Ampicillin-resistant (bla), Hygromycin B-resistant (hphMX) | TDH3pr-mNeon-CYC13′UTR | This Study | 194539 |
| Yeast | Transformation Efficiency † ± (SD) [CFU/106 cells/µg DNA] | Efficiency Score ‡ |
|---|---|---|
| S. cerevisiae (lab) | 3984.09 (±497.32) | VERY HIGH |
| S. kudriavzevii | 1094.97 (±426.13) | VERY HIGH |
| S. mikatae | 535.73 (±356.49) | HIGH |
| L. thermotolerans | 434.72 (±302.61) | HIGH |
| S. cerevisiae (wine, EC1118) | 257.85 (±180.42) | HIGH |
| S. cerevisiae (oak, China) | 202.11 (±197.35) | HIGH |
| L. kluyveri | 126.56 (±110.13) | HIGH |
| S. cerevisiae (oak, NJ) | 109.23 (±94.85) | HIGH |
| S. cerevisiae (wine, Uvaferm 43) | 99.01 (±75.99) | MEDIUM |
| S. paradoxus (Far-Eastern subpopulation) | 61.91 (±2.76) | MEDIUM |
| T. delbrueckii | 57.96 (±36.28) | MEDIUM |
| S. uvarum | 53.10 (±17.25) | MEDIUM |
| S. paradoxus (American subpopulation) | 36.13 (±36.57) | MEDIUM |
| S. pastorianus (beer, Lager I) | 6.47 (±8.15) | LOW |
| S. cerevisiae (beer, Irish Ale) | 3.36 (±3.41) | LOW |
| P. kudriavzevii | 0.39 (±0.34) | LOW |
| P. membranafaciens | 0.35 (±0.60) | LOW |
| M. pulcherrima | 0.10 (±0.17) | LOW |
| N. castelli | 0 | NONE |
| H. uvarum | 0 | NONE |
| Z. bailii | 0 | NONE |
| B. bruxellensis | 0 | NONE |
| S. pombe | 0 | NONE |
| S. ludwigii | 0 | NONE |
| Viable Re-Streaks (Maximum of 5) | ||
|---|---|---|
| Yeast | Low Copy | High Copy |
| S. cerevisiae (lab) | 5 | 5 |
| L. thermotolerans | 5 | 5 |
| S. mikatae | 5 | 5 |
| S. cerevisiae (wine, EC1118) | 5 | 5 |
| S. kudriavzevii | 5 | 5 |
| S. cerevisiae (oak, China) | 5 | 5 |
| S. cerevisiae (oak, NJ) | 5 | 5 |
| S. cerevisiae (wine, Uvaferm 43) | 5 | 5 |
| S. paradoxus (Far-Eastern) | 5 | 5 |
| S. uvarum | 5 | 5 |
| S. paradoxus (American) | 5 | 5 |
| S. pastorianus (beer, Lager I) | 5 | 5 |
| S. cerevisiae (beer, Irish Ale) | 5 | 5 |
| P. membranafaciens | 5 | 5 |
| T. delbrueckii | 5 | 1 |
| P. kudriavzevii | 5 | 1 |
| L. kluyveri | 5 | not tested |
| M. pulcherrima | 5 | not tested |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, J.R.; Sislak, C.D.; Fernandez Mendoza, P.; Carmichael, L.; Lewis, A.G.; Chen, A.; Jiang, G.Z.; Gibney, P.A. Auxotrophy-Independent Plasmid Shuttle Vectors for Applications in Diverse Yeasts. Appl. Microbiol. 2024, 4, 453-469. https://doi.org/10.3390/applmicrobiol4010031
Smith JR, Sislak CD, Fernandez Mendoza P, Carmichael L, Lewis AG, Chen A, Jiang GZ, Gibney PA. Auxotrophy-Independent Plasmid Shuttle Vectors for Applications in Diverse Yeasts. Applied Microbiology. 2024; 4(1):453-469. https://doi.org/10.3390/applmicrobiol4010031
Chicago/Turabian StyleSmith, Jeremy R., Christine D. Sislak, Pedro Fernandez Mendoza, Laurin Carmichael, Alisha G. Lewis, Anqi Chen, Glycine Z. Jiang, and Patrick A. Gibney. 2024. "Auxotrophy-Independent Plasmid Shuttle Vectors for Applications in Diverse Yeasts" Applied Microbiology 4, no. 1: 453-469. https://doi.org/10.3390/applmicrobiol4010031