
Utilizing a Metagenome Assembled Genome Approach Revealed Further Insights into Microbially Mediated Heavy-Metal Resistance in Soils from a Former Nuclear Materials Production Facility


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Shotgun Metagenomics and Bioinformatics Processing
2.3. Bioinformatics Analysis to Reconstruct Metagenome Assembled Genomes (MAGs)
- (1)
- Soil from summer with high U contamination: SRR6788480, SRR6788481, and SRR6788482;
- (2)
- Soil from summer with medium U contamination: SRR6788478, SRR6788479, and SRR6788487;
- (3)
- Soil from summer with low U contamination: SRR6788483, SRR6788484, and SRR6788485;
- (4)
- Soil from winter with high U contamination: SRR6788486;
- (5)
- Soil from winter with medium U contamination: SRR6788488;
- (6)
- Soil from winter with low U contamination: SRR6788489.
3. Results and Discussion
3.1. Characteristics of the MAGs Recovered from Shotgun Metagenomes Using the CheckM Pipeline
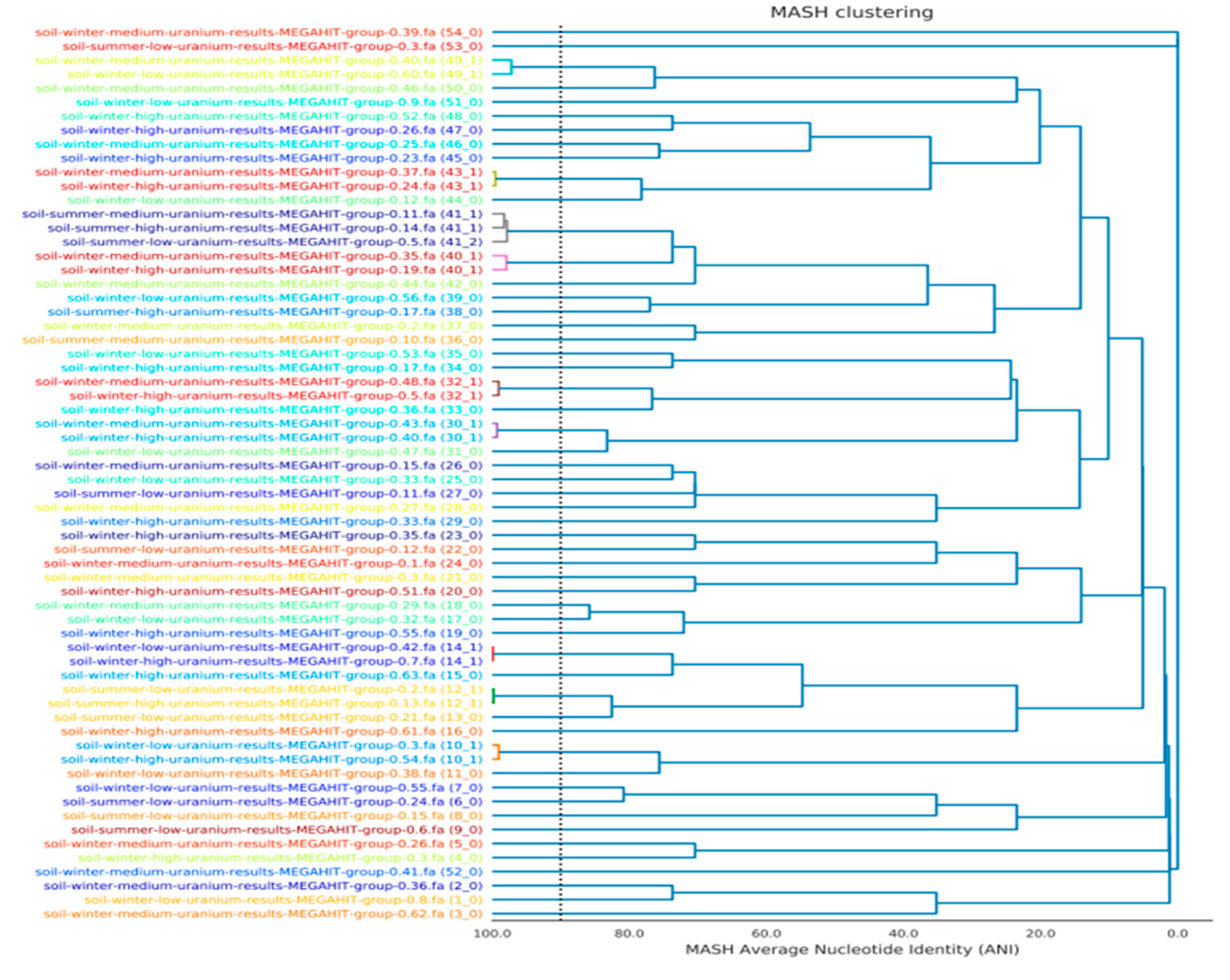
3.2. Statistical Analysis of the MAGs from SRS Uranium-Contaminated Soils
3.3. Assessment of Metabolic Traits of the Winning MAG—Arthrobacter oryzae
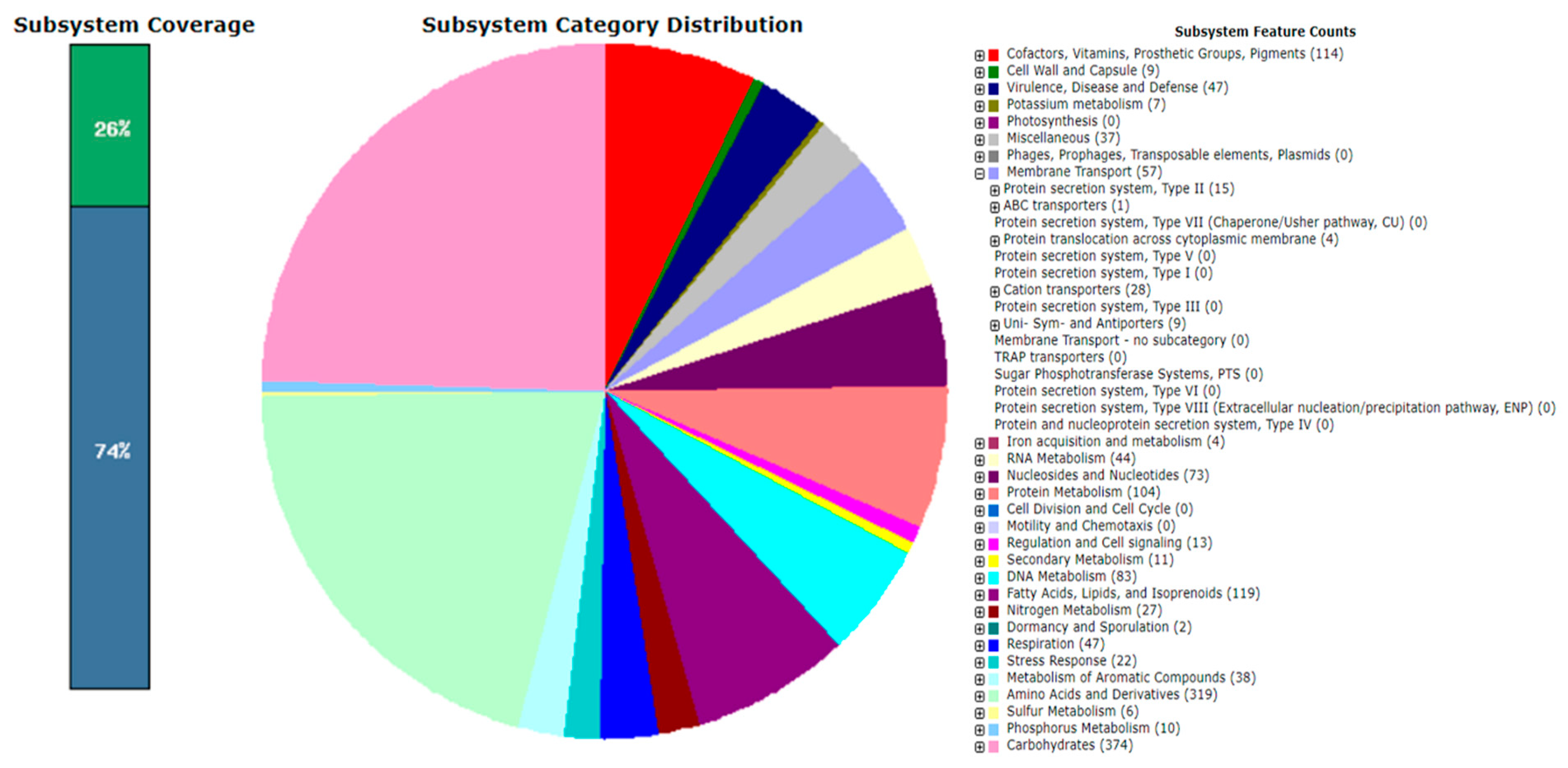
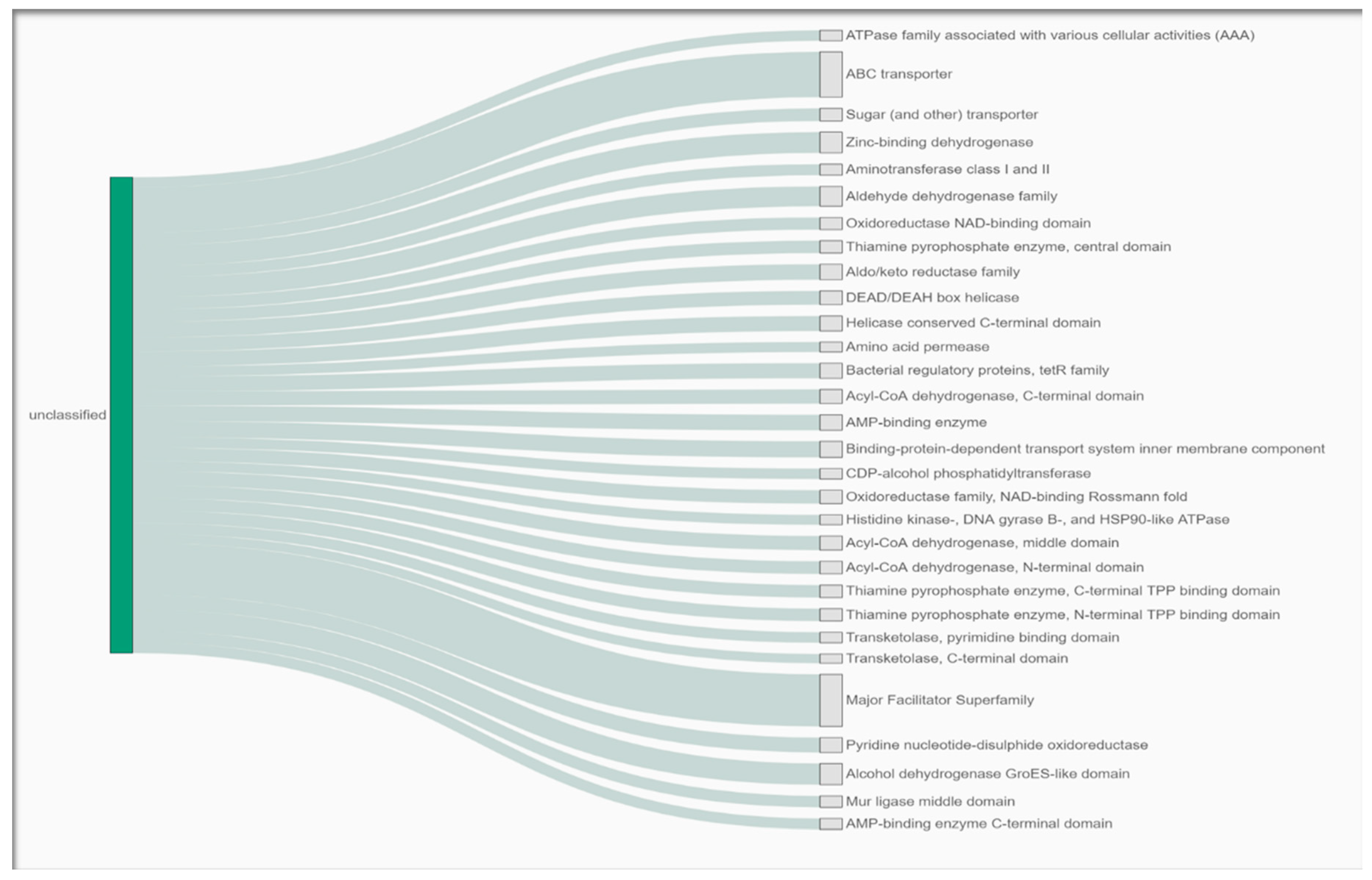
3.4. Gene Functional Analysis of the Arthrobacter oryzae MAG
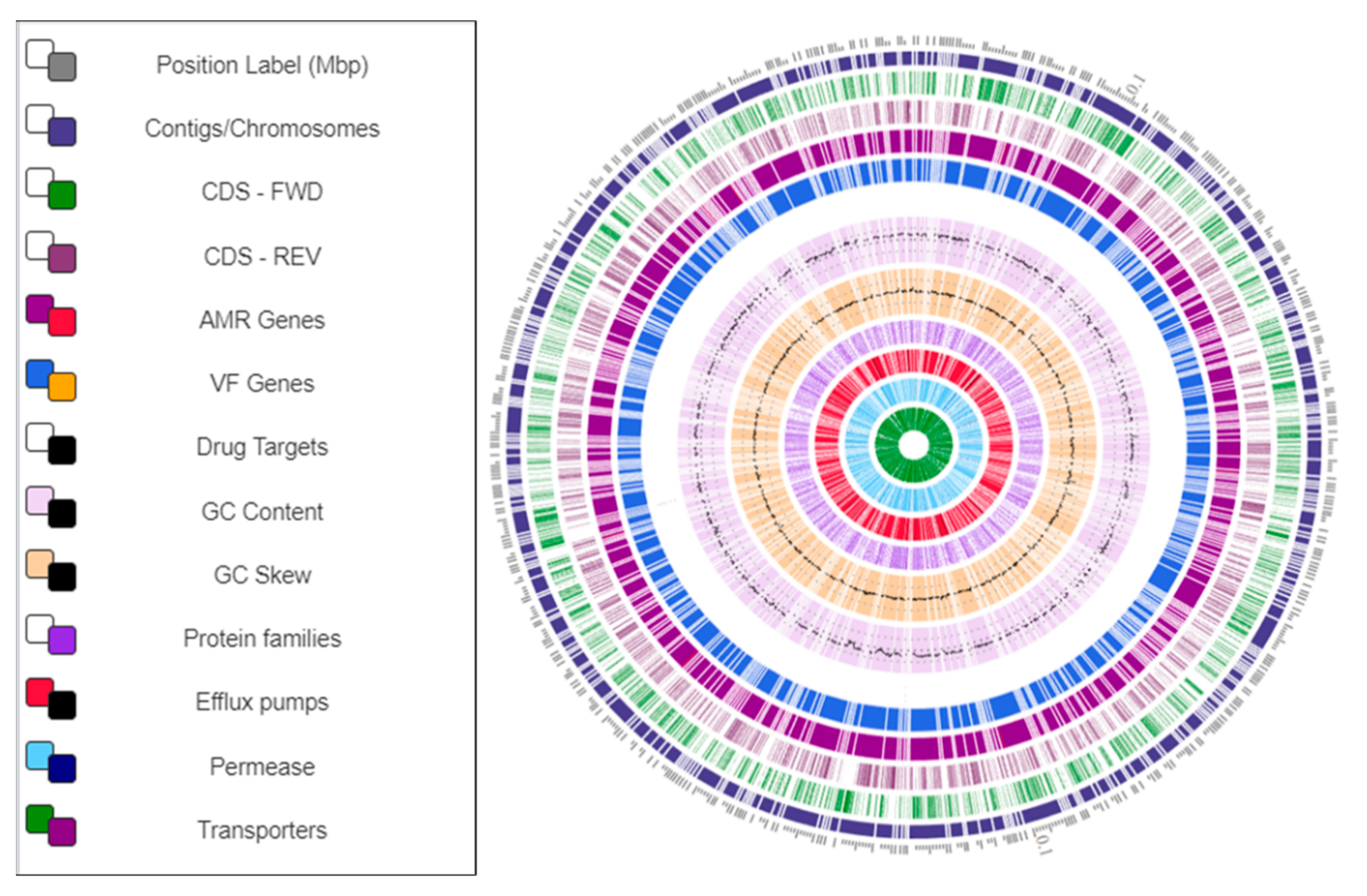
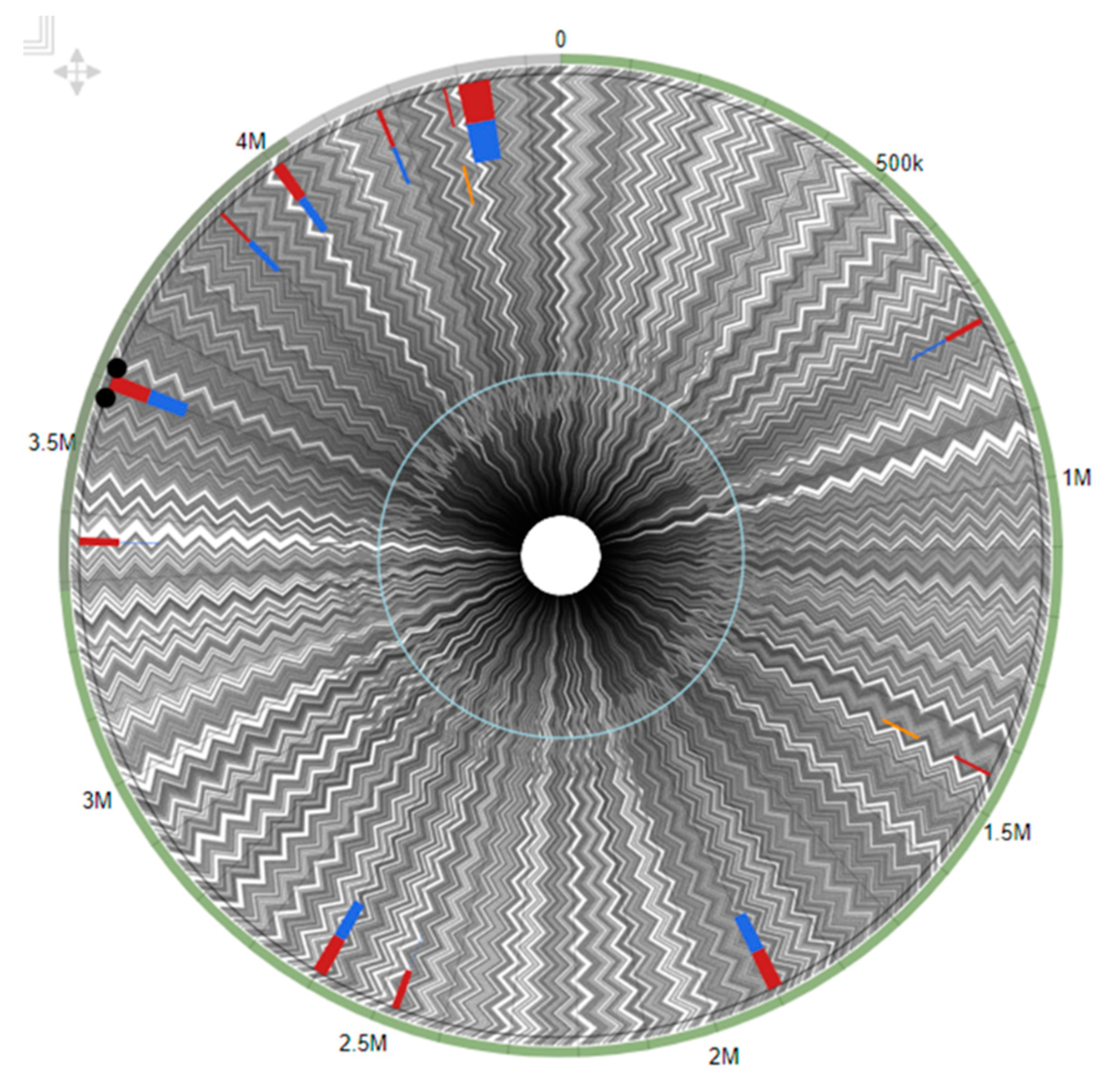
3.5. Genome Map of Arthrobacter oryzae MAG Reconstructed from the SRS Uranium-Contaminated Soil
3.6. Genomic Islands (GEIs) in the Arthrobacter oryzae MAG
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nolan, J.; Weber, K.A. Natural Uranium Contamination in Major U.S. Aquifers Linked to Nitrate. Environ. Sci. Technol. Lett. 2015, 2, 215–220. [Google Scholar] [CrossRef]
- Vicente-Vicente, L.; Quiros, Y.; Pérez-Barriocanal, F.; López-Novoa, J.M.; López-Hernández, F.J.; Morales, A.I. Nephrotoxicity of uranium: Pathophysiological, diagnostic and therapeutic perspectives. Toxicol. Sci. 2010, 118, 324–347. [Google Scholar] [CrossRef] [PubMed]
- Kurttio, P.; Komulainen, H.; Leino, A.; Salonen, L.; Auvinen, A.; Saha, H. Bone as a Possible Target of Chemical Toxicity of Natural Uranium in Drinking Water. Environ. Health Perspect. 2005, 113, 68–72. [Google Scholar] [CrossRef]
- Olalde, M.; Simon, M.; Mierjeski, A. The Cold War Legacy Lurking in U.S. Groundwater. PROPUBLICA. 2022. Available online: https://www.propublica.org/article/uranium-mills-pollution-cleanup-us (accessed on 11 February 2024).
- Punshon, T.; Gaines, K.F.; Jenkins, R.K. Bioavailability and Trophic Transfer of Sediment-Bound Ni and U in a Southeastern Wetland System. Arch. Environ. Contam. Toxicol. 2003, 44, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Sowder, A.G.; Bertsch, P.M.; Morris, P.J. Partitioning and availability of uranium and nickel in contaminated riparian sediments. J. Environ. Qual. 2003, 32, 885–898. [Google Scholar] [CrossRef]
- Banala, U.K.; Indradyumna Das, N.P.; Toleti, S.R. Microbial interactions with uranium: Towards an effective bioremediation approach. Environ. Technol. Innov. 2021, 21, 101254. [Google Scholar] [CrossRef]
- Yan, X.; Luo, X.; Zhao, M. Metagenomic analysis of microbial community in uranium-contaminated soil. Appl. Microbiol. Biotechnol. 2016, 100, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Merroun, M.L.; Selenska-Pobell, S. Bacterial interactions with uranium: An environmental perspective. J. Contam. Hydrol. 2008, 102, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Pathak, A.; Rathore, R.S.; Prakash, O.; Singh, R.; Jaswal, R.; Seaman, J.; Chauhan, A. Proteogenomic Analysis of Burkholderia Species Strains 25 and 46 Isolated from Uraniferous Soils Reveals Multiple Mechanisms to Cope with Uranium Stress. Cells 2018, 7, 269. [Google Scholar] [CrossRef]
- Pathak, A.; Chauhan, A.; Stothard, P.; Green, S.; Maienschein-Cline, M.; Jaswal, R.; Seaman, J. Genome-centric evaluation of Burkholderia sp. strain SRS-W-2-2016 resistant to high concentrations of uranium and nickel isolated from the Savannah River Site (SRS), USA. Genom. Data 2017, 12, 62–68. [Google Scholar] [CrossRef]
- Pathak, A.; Jaswal, R.; Xu, X.; White, J.R.; Edwards, B.; Hunt, J.; Brooks, S.; Rathore, R.S.; Agarwal, M.; Chauhan, A. Characterization of Bacterial and Fungal Assemblages from Historically Contaminated Metalliferous Soils Using Metagenomics Coupled with Diffusion Chambers and Microbial Traps. Front. Microbiol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Jaswal, R.; Pathak, A.; Edwards, B., III; Lewis, R., III; Seaman, J.C.; Stothard, P.; Krivushin, K.; Blom, J.; Rupp, O.; Chauhan, A. Metagenomics-Guided Survey, Isolation, and Characterization of Uranium Resistant Microbiota from the Savannah River Site, USA. Genes 2019, 10, 325. [Google Scholar] [CrossRef]
- Chauhan, A.; Pathak, A.; Jaswal, R.; Edwards, B., III; Chappell, D.; Ball, C.; Garcia-Sillas, R.; Stothard, P.; Seaman, J. Physiological and Comparative Genomic Analysis of Arthrobacter sp. SRS-W-1-2016 Provides Insights on Niche Adaptation for Survival in Uraniferous Soils. Genes 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Katsenovich, Y.; Carvajal, D.; Guduru, R.; Lagos, L.; Li, C.-Z. Assessment of the resistance to uranium (vi) exposure by Arthrobacter sp. Isolated from hanford site soil. Geomicrobiol. J. 2013, 30, 120–130. [Google Scholar] [CrossRef]
- Suzuki, Y.; Banfield, J.F. Resistance to, and Accumulation of, Uranium by Bacteria from a Uranium-Contaminated Site. Geomicrobiol. J. 2004, 21, 113–121. [Google Scholar] [CrossRef]
- Bader, M.; Müller, K.; Foerstendorf, H.; Schmidt, M.; Simmons, K.; Swanson, J.S.; Reed, D.T.; Stumpf, T.; Cherkouk, A. Comparative analysis of uranium bioassociation with halophilic bacteria and archaea. PLoS ONE 2018, 13, e0190953. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.C.; Chapman, S.W. Bacteria that masquerade as fungi: Actinomycosis/nocardia. Proc. Am. Thorac. Soc. 2010, 7, 216–221. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, M.; Yang, J. Recovering metagenome-assembled genomes from shotgun metagenomic sequencing data: Methods, applications, challenges, and opportunities. Microbiol. Res. 2022, 260, 127023. [Google Scholar] [CrossRef] [PubMed]
- Jaswal, R.; Pathak, A.; Chauhan, A. Metagenomic Evaluation of Bacterial and Fungal Assemblages Enriched within Diffusion Chambers and Microbial Traps Containing Uraniferous Soils. Microorganisms 2019, 6, 324. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Saheb Kashaf, S.; Almeida, A.; Segre, J.A.; Finn, R.D. Recovering prokaryotic genomes from host-associated, short-read shotgun metagenomic sequencing data. Nat. Protoc. 2021, 16, 2520–2541. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Olm, M.; Brown, C.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Surachat, K.; Kantachote, D.; Deachamag, P.; Wonglapsuwan, M. In silico genomic analysis of Rhodopseudomonas palustris strains revealed potential biocontrol agents and crop yield enhancers. Biol. Control 2022, 176, 105085. [Google Scholar] [CrossRef]
- Guan, L. Structure and mechanism of membrane transporters. Sci. Rep. 2022, 12, 13248. [Google Scholar] [CrossRef]
- Akhtar, A.A.; Turner, D.P.J. The role of bacterial ATP-binding cassette (ABC) transporters in pathogenesis and virulence: Therapeutic and vaccine potential. Microb. Pathog. 2022, 171, 105734. [Google Scholar] [CrossRef] [PubMed]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Hemme, C.; Deng, Y.; Gentry, T.; Fields, M.W.; Wu, L.; Barua, S.; Barry, K.; Tringe, S.G.; Watson, D.B.; He, Z.; et al. Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J. 2010, 4, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Brettin, T.; Davis, J.J.; Gerdes, S.; Kenyon, R.; Machi, D.; Mao, C.; Olson, R.; Overbeek, R.; Pusch, G.D.; et al. Assembly, Annotation, and Comparative Genomics in PATRIC, the All-Bacterial Bioinformatics Resource Center. Methods Mol. Biol. 2018, 1704, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kaushik, V.; Kulshrestha, M.; Tiwari, V. Different Efflux Pump Systems in Acinetobacter baumannii and Their Role in Multidrug Resistance. Adv. Exp. Med. Biol. 2023, 1370, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Rafeeq, H.; Afsheen, N.; Rafique, S.; Arshad, A.; Intisar, M.; Hussain, A.; Bilal, M.; Iqbal, H.M. Genetically engineered microorganisms for environmental remediation. Chemosphere 2023, 310, 136751. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Cho, A.; Hong, S.G.; Choi, H.G.; Kim, O.S. Draft Genome Sequence of Arthrobacter oryzae TNBS02, a Bacterium Containing Heavy Metal Resistance Genes, Isolated from Soil of Antarctica. Microbiol. Resour. Announc. 2019, 8, e01501-18. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Leong, H.W. Computational methods for predicting genomic islands in microbial genomes. Comput. Struct. Biotechnol. J. 2016, 14, 200–206. [Google Scholar] [CrossRef]
- Langille, M.; Hsiao, W.; Brinkman, F. Detecting genomic islands using bioinformatics approaches. Nat. Rev. Microbiol. 2010, 8, 373–382. [Google Scholar] [CrossRef]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef]
- Li, L.; Meng, D.; Yin, H.; Zhang, T.; Liu, Y. Genome-resolved metagenomics provides insights into the ecological roles of the keystone taxa in heavy-metal-contaminated soils. Front. Microbiol. 2023, 14, 1203164. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, L.; Xu, K.; Li, K.; Ren, H. Revealing taxon-specific heavy metal-resistance mechanisms in denitrifying phosphorus removal sludge using genome-centric etaproteomics. Microbiome 2021, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Jaswal, R.; Xu, X.; Chauhan, A. Draft Genome Sequence of Serratia sp. Strain S1B, Isolated from Mercury-Contaminated Soil. Genome Announc. 2018, 6, e00534-18. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome | Marker Lineage | Completeness | Contamination | Strain_Heterogeneity | Length | N50 |
|---|---|---|---|---|---|---|
| soil-summer-high-uranium-results-MEGAHIT-group-0.13.fa | p__Actinobacteria (UID1454) | 95.73 | 1.42 | 33.33 | 3,083,233 | 58,255 |
| soil-summer-low-uranium-results-MEGAHIT-group-0.12.fa | k__Bacteria (UID1452) | 92.75 | 2.42 | 50 | 3,127,523 | 20,566 |
| soil-summer-low-uranium-results-MEGAHIT-group-0.21.fa | p__Actinobacteria (UID1454) | 94.44 | 3.16 | 14.29 | 2,333,606 | 75,210 |
| soil-summer-low-uranium-results-MEGAHIT-group-0.24.fa | p__Euryarchaeota (UID3) | 99.2 | 1.6 | 0 | 2,661,188 | 139,475 |
| soil-summer-low-uranium-results-MEGAHIT-group-0.3.fa | p__Euryarchaeota (UID49) | 98.53 | 2.61 | 0 | 2,895,156 | 27,231 |
| soil-summer-low-uranium-results-MEGAHIT-group-0.6.fa | k__Archaea (UID2) | 91.75 | 2.91 | 0 | 2,256,961 | 33,599 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.17.fa | c__Betaproteobacteria (UID3971) | 95.79 | 3.48 | 36.36 | 4,120,679 | 30,345 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.23.fa | k__Bacteria (UID3187) | 91.88 | 5.27 | 12.5 | 8,053,684 | 54,917 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.33.fa | p__Bacteroidetes (UID2591) | 94.88 | 2.64 | 37.5 | 3,768,066 | 14,449 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.35.fa | k__Bacteria (UID1452) | 91.36 | 4.63 | 16.67 | 2,805,076 | 12,991 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.40.fa | c__Gammaproteobacteria (UID4202) | 92.77 | 3.83 | 23.81 | 3,661,217 | 32,786 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.5.fa | c__Alphaproteobacteria (UID3305) | 96.3 | 3.07 | 30 | 4,186,502 | 26,896 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.51.fa | k__Bacteria (UID1452) | 91.67 | 1.57 | 0 | 4,442,876 | 15,948 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.52.fa | k__Bacteria (UID3187) | 90.17 | 3.47 | 16.67 | 4,854,587 | 19,436 |
| soil-winter-high-uranium-results-MEGAHIT-group-0.61.fa | k__Bacteria (UID3187) | 92.07 | 9.96 | 31.82 | 5,211,487 | 10,544 |
| soil-winter-low-uranium-results-MEGAHIT-group-0.12.fa | k__Bacteria (UID3187) | 97.15 | 2.56 | 0 | 6,711,185 | 35,231 |
| soil-winter-low-uranium-results-MEGAHIT-group-0.38.fa | k__Bacteria (UID203) | 91.23 | 5.5 | 75 | 5,025,612 | 9893 |
| soil-winter-low-uranium-results-MEGAHIT-group-0.47.fa | c__Gammaproteobacteria (UID4202) | 94.57 | 8.5 | 20 | 3,319,549 | 31,643 |
| soil-winter-low-uranium-results-MEGAHIT-group-0.56.fa | k__Bacteria (UID1452) | 97.73 | 9.05 | 7.69 | 4,126,511 | 52,840 |
| soil-winter-low-uranium-results-MEGAHIT-group-0.9.fa | k__Bacteria (UID3187) | 94.02 | 6.41 | 0 | 8,036,188 | 19,973 |
| soil-winter-medium-uranium-results-MEGAHIT-group-0.2.fa | k__Bacteria (UID3187) | 95.22 | 6.09 | 0 | 6,095,384 | 47,944 |
| soil-winter-medium-uranium-results-MEGAHIT-group-0.3.fa | k__Bacteria (UID1452) | 91.09 | 3.47 | 75 | 2,893,218 | 16,581 |
| soil-winter-medium-uranium-results-MEGAHIT-group-0.37.fa | k__Bacteria (UID3187) | 94.87 | 4.7 | 16.67 | 6,098,353 | 118,469 |
| soil-winter-medium-uranium-results-MEGAHIT-group-0.39.fa | k__Bacteria (UID1452) | 91 | 1.39 | 0 | 2,674,289 | 7171 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kommu, N.; Stothard, P.; Chukwujindu, C.; Pathak, A.; Chauhan, A. Utilizing a Metagenome Assembled Genome Approach Revealed Further Insights into Microbially Mediated Heavy-Metal Resistance in Soils from a Former Nuclear Materials Production Facility. Appl. Microbiol. 2024, 4, 376-389. https://doi.org/10.3390/applmicrobiol4010026
Kommu N, Stothard P, Chukwujindu C, Pathak A, Chauhan A. Utilizing a Metagenome Assembled Genome Approach Revealed Further Insights into Microbially Mediated Heavy-Metal Resistance in Soils from a Former Nuclear Materials Production Facility. Applied Microbiology. 2024; 4(1):376-389. https://doi.org/10.3390/applmicrobiol4010026
Chicago/Turabian StyleKommu, Navya, Paul Stothard, Christian Chukwujindu, Ashish Pathak, and Ashvini Chauhan. 2024. "Utilizing a Metagenome Assembled Genome Approach Revealed Further Insights into Microbially Mediated Heavy-Metal Resistance in Soils from a Former Nuclear Materials Production Facility" Applied Microbiology 4, no. 1: 376-389. https://doi.org/10.3390/applmicrobiol4010026