
NUP98 Rearrangements in AML: Molecular Mechanisms and Clinical Implications


Cancer Biology and Genetics, Memorial Sloan-Kettering Cancer Center, New York, NY 10065, USA
Onco 2023, 3(3), 147-164; https://doi.org/10.3390/onco3030011
Submission received: 16 May 2023
/
Revised: 5 July 2023
/
Accepted: 11 July 2023
/
Published: 18 July 2023
Abstract
:Simple Summary
NUP98 rearrangements are frequent events in myeloid malignancies, especially in acute myeloid leukemia (AML). AML patients carrying NUP98 fusions show poor response to standard treatments and adverse outcomes. This review focuses on recent progress in understanding the underlying mechanisms of NUP98 fusion driven leukemias and the development of therapeutics against them.
Abstract
NUP98 fusions constitute a small subgroup of AML patients and remain a high-risk AML subtype. There are approximately 30 types of NUP98 fusions identified in AML patients. These patients show resistance to currently available therapies and poor clinical outcomes. NUP98 fusions with different fusion partners have oncogenic transformation potential. This review describes how the NUP98 gene acquires oncogenic properties after rearrangement with multiple partners. In the mechanistic part, the formation of nuclear bodies and dysregulation of the HoxA/Meis1 pathway are highlighted. This review also discusses mutational signatures among NUP98 fusions and their significance in leukemogenesis. It also discusses the clinical implications of NUP98 fusions and their associated mutations in AML patients. Furthermore, it highlights therapeutic vulnerabilities in these leukemias that can be exploited as therapeutic strategies. Lastly, this review discusses the gaps in our knowledge regarding NUP98 fusions in AML, as well as future research opportunities.
Keywords:
leukemia; AML; translocations; fusion genes; NUP98 fusions; NUP98::NSD1; NUP98::KDM5A; FLT3-ITD1. Introduction
Acute myeloid leukemia (AML) is a myeloid malignancy characterized by genomic abnormalities and a high number of blast cells in the bone marrow [1]. Like other cancers, fusion genes are also reported in AML. Fusion genes are formed by chromosome aberrations such as translocations, inversions, deletions, and insertions [2]. Fusion genes can lead to oncogenic transformation by activating oncogenes or inactivating tumor suppressors [3]. Whenever a 3′ oncogene is linked to a strong promoter of a 5′ gene, it becomes overexpressed. In TMPRSS2::ETS fusions in prostate cancer, expression of ETS family transcription factor is driven by TMPRSS2 gene promoter [4]. An oncogene can lose its 3′ UTR microRNA binding site through fusion and lead to higher expression of the oncogene. For example, MYB::NFIB fusion in adenoid cystic carcinomas (ACC) activates critical MYB targets through the loss of the 3′ UTR-regulating microRNA binding site [5]. Similarly, a gene can inactivate its tumor suppressor function in a fusion.
As a result of recent advances in high-throughput sequencing technologies, it has been possible to detect cryptic fusion genes that are usually skipped by conventional karyotyping [6]. For example, one-third of KMT2A fusions in AML are missed by karyotyping and require additional tests like FISH or RT-PCR. These methods fail to identify rare fusion genes [7]. However, next-generation sequencing (NGS) successfully identifies rare fusions in patient samples [8,9]. The discovery of these fusion genes has improved the diagnosis, prognosis, and treatment of cancer patients.
In addition, a malignancy caused by a fusion gene opens the door to targeted therapies. Since fusion genes are exclusive to neoplastic cells and not expressed in healthy cells, they are excellent drug targets for treatment. For instance, imatinib was discovered against the BCR::ABL fusion gene that is expressed in 95 percent of chronic myeloid leukemia (CML) patients [10]. Discovery of fusion genes in AML helps with risk stratification and treatment of AML patients. Approximately 30–40 percent of AML patients carry at least one fusion gene, and NUP98 fusions frequently occur in AML [11,12].
2. NUP98: A Commonly Translocated Gene in AML
Nucleoporin 98 (NUP98) is a gene located on chromosome 11p15 and encodes a precursor protein that results in NUP98 and NUP96 nucleoporins, which are structural components of the nuclear pore complex (NPC) [13,14]. The NPC facilitates the nucleocytoplasmic transport of ions, mRNAs, and proteins. Large molecules are transported via nuclear transport receptors that recognize nuclear export signals (NES) or nuclear localization signals (NLS), while smaller molecules can pass easily through them [15,16].
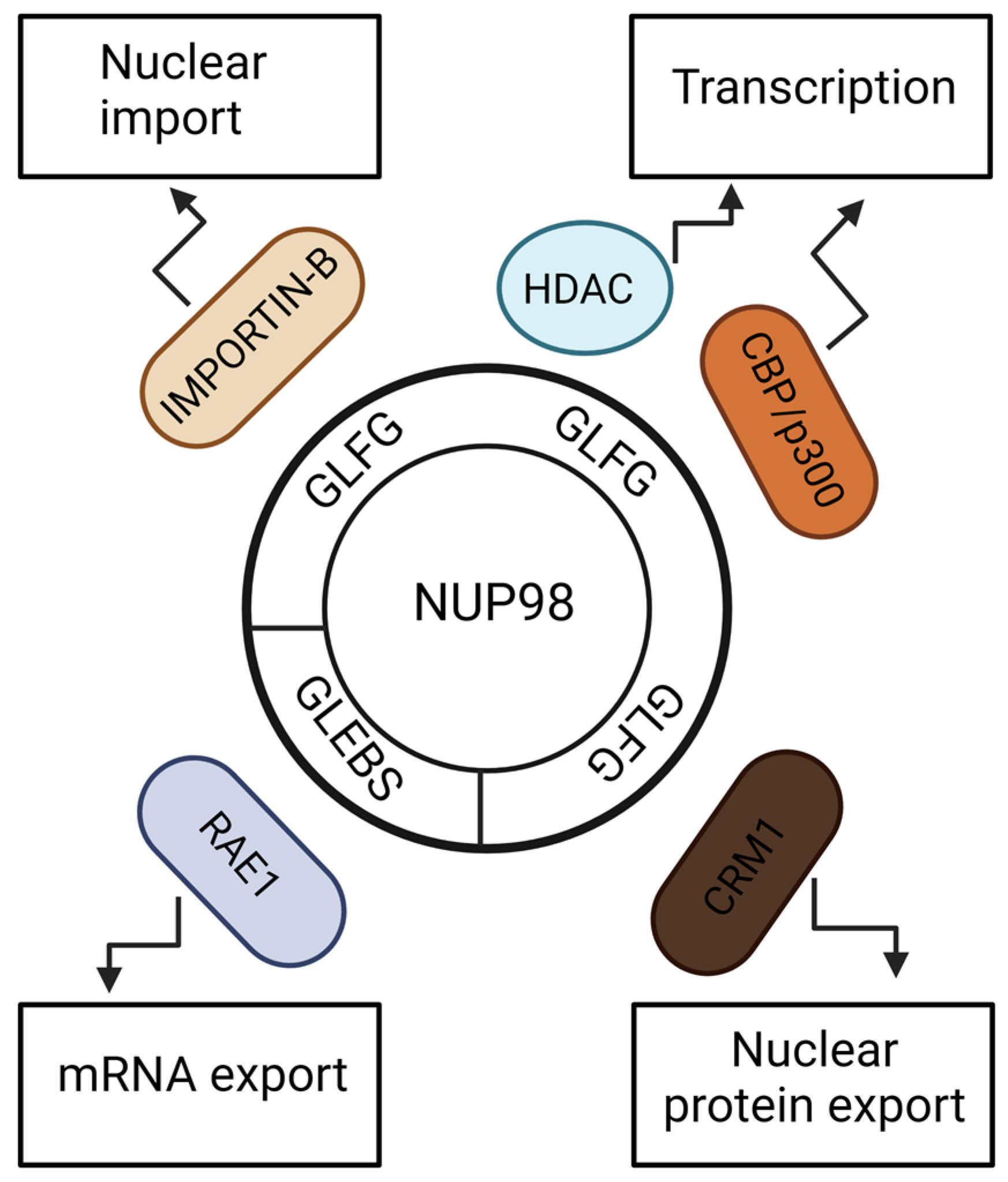
NUP98 is located on both sides of the nuclear pore complex and migrates on and off the NPC [17]. One-third of nucleoporins have phenylalanine–glycine [FG] repeats, but NUP98 has a unique FG repeat signature of Gly-Leu-Phe-Gly (GLFG) repeats [18,19]. In addition to the GLFG repeats, the N terminal part of the NUP98 protein contains a GLE2-binding sequence (GLEBS) motif, and the C terminal part contains an RNA-binding motif. The GLFG repeats interact with Exportin 1 (XPO1) and mediate nuclear protein export (Figure 1) [20]. The GLFG repeats also interact with importin-β family proteins for nuclear import [19]. RNA export factor RAE1 (Gle2) binds to the GLEBS motif to mediate the nuclear export of mRNAs [21,22]. Additionally, NUP98 is involved in the regulation of transcription. NUP98 is a mobile component of NPC and forms nuclear bodies, known as GLFG bodies [23]. The GLFG repeats interact with histone deacetylases (HDACs) and transcriptional co-activators CBP/p300, suggesting involvement of NUP98 in transcriptional regulation [24]. Furthermore, Kalverda showed that altered expression of nucleoplasmic NUP98 affects its target gene expression, supporting its involvement in gene regulation [25].
NUP98 gene alterations have been implicated in several hematological malignancies including AML, chronic myeloid leukemia (CML), juvenile myelomonocytic leukemia (JMML), T-cell acute lymphoblastic leukemia (T-ALL), and myelodysplastic syndrome (MDS) [26]. Notably, NUP98 fusions are majorly reported in myeloid and T-cell malignancies and rarely observed in B-cell malignancies. Around 5% of pediatric AML patients exhibit NUP98 rearrangements [27,28,29,30].
3. Fusion Partners of NUP98 in NUP98-Rearranged AML
The NUP98 gene is rearranged with approximately 30 partners in AML (Table 1). NUP98::NSD1 and NUP98::KDM5A are frequently occurring NUP98 fusions in AML [30]. The NUP98 fusion proteins retain the N-terminal of NUP98, which contains GLFG repeats and GLEBS motif. NUP98 fusion proteins lack the C-terminus portion of NUP98 containing the RNA binding motif. The C-terminus of the fusion protein is contributed by the partner gene [31].
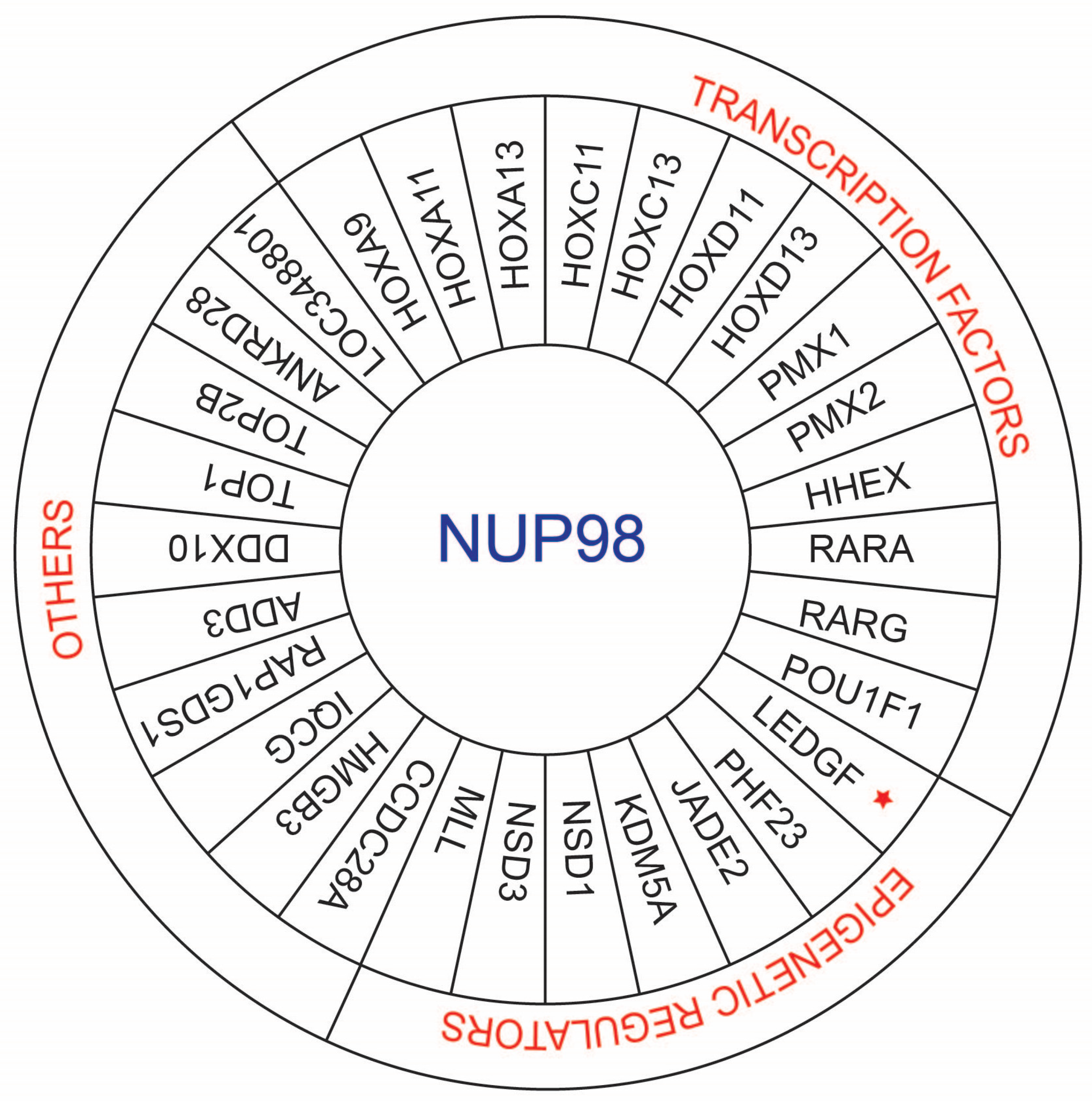
Overall, NUP98 fusions can be divided into three broad parts (Figure 2). The first category includes NUP98 fusions with transcription factors as partners, which can change the expression of target genes through DNA binding domains. The second category is NUP98 fusions with epigenetic modifiers that modify chromatin to change target gene expression. The third category of NUP98 fusions has neither the DNA binding nor chromatin remodeling domain. Transcription factor partners of NUP98 mostly include homeobox genes, including “class I” HOX genes (HOXA9, HOXA11, HOXA13, HOXC11, HOXC13, HOXD11, and HOXD13) and “class II” HOX genes (PMX1, PMX2, HHEX1, and POU1F1) and non-homeobox genes (RARA and RARG) [26]. RARA and RARG are the nuclear receptor (NR) superfamily members [32,33]. The LEDGF (lens epithelium-derived growth factor) gene encodes p75 and p52, which act as transcriptional coactivators [34].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Different fusion partners of NUP98 in AML, its functional category, and the associated FAB subtypes.
Table 1.
Different fusion partners of NUP98 in AML, its functional category, and the associated FAB subtypes.
| Fusion Partner | Functional Category | AML Subtype | Chromosome Rearrangement | Refs. |
|---|---|---|---|---|
| HOXA9 | Transcription factor | M2, M4 | t(7;11)(p15;p15) | [35,36] |
| HOXA11 | Transcription factor | M2 | t(7;11)(p15;p15) | [37] |
| HOXA13 | Transcription factor | M2 | t(7;11)(p15;p15) | [38] |
| HOXC11 | Transcription factor | M1, M2, M5 | t(11;12)(p15;q13) | [39,40] |
| HOXC13 | Transcription factor | M2, M4 | t(11;12)(p15;q13) | [41,42] |
| HOXD11 | Transcription factor | M4 | t(2;11)(q31;p15) | [43] |
| HOXD13 | Transcription factor | t-AML, M4 | t(2;11)(q31;p15) | [44,45] |
| PMX1 | Transcription factor | M2 | t(1;11)(q23;p15) | [46] |
| PMX2 | Transcription factor | t-AML | t(9;11)(q34;p15) | [[47] |
| HHEX | Transcription factor | M1, M2 | t(10;11)(q23;p15) | [48,49] |
| RARA | Transcription factor | M3 or APL | t(11;17) | [32] |
| RARG | Transcription factor | M3 or APL | t(11;12)(p15;q13) | [33,50] |
| POU1F1 | Transcription factor | t-AML | t(3;11)(p11;p15) | [51] |
| LEDGF/PSIP1 | Transcription coactivator | M1, M2 | t(9;11)(p22;p15) | [34,52,53] |
| PHF23 | Epigenetic modifier | M0, M1, M4, M5 | t(11;17)(p15;p13) | [54,55] |
| JADE2/PHF15 | Epigenetic modifier | M3 or APL | t(5;11)(q31;p15) | [56] |
| JARID1A/KDM5A | Epigenetic modifier | M0-M7 | t(11;15)(p15;q35) | [57,58] |
| NSD1 | Epigenetic modifier | M1, M2, M4, M5, M6 | t(5;11)(q35;p15.5) | [27,59,60,61] |
| NSD3 | Epigenetic modifier | M1 | t(8;11)(p11.2;p15) | [62] |
| MLL/KMT2A | Epigenetic modifier | M1, M2 | inv(11)(p15q23) | [63] |
| C6orf80/CCDC28A | Unknown | M7 | t(6;11)(q24.1;p15.5) | [64] |
| HMGB3 | High-mobility group (HMG) protein | t-AML | t(X;11)(q28;p15) | [65] |
| IQCG | Calcium signaling | AML (Unknown) | t(3;11)(q29q13;p15) | [66] |
| RAP1GDS1 | GTPase activity | AML (Unknown) | unknown | [67] |
| ADD3 | Cytoskeletal protein | AML (Unknown) | t(10;11) | [68] |
| DDX10 | RNA helicase | M6 | inv(11)(p15q22) | [69,70] |
| TOP1 | DNA Topoisomerase | M4, M5 | t(11;20)(p15;q11) | [27,71] |
| TOP2B | DNA Topoisomerase | M5 | t(3;11)(p24;p15) | [72] |
| ANKRD28 | Signaling protein | AML (Unknown) | t(3;5;11)(p25;q35;p15) | [73] |
| LOC348801 | Unknown | M2 | t(3;11)(q12;p15) | [74] |
Undifferentiated acute myeloblastic leukemia (M0), acute myeloblastic leukemia with minimal maturation (M1), acute myeloblastic leukemia with maturation (M2), acute promyelocytic leukemia (APL) (M3), acute myelomonocytic leukemia (M4), acute monocytic leukemia (M5), acute erythroid leukemia (AEL) (M6), acute megakaryoblastic leukemia (M7), therapy-related acute myeloid leukemia (t-AML).
NUP98 fusions with epigenetic modifiers typically have plant homeodomain (PHD) domains (PHF23, JADE2, KDM5A, MLL, NSD1, and NSD3) and SET domains (MLL, NSD1, and NSD3) [26,31]. Among the third group, there are a number of partners that have topoisomerase and RNA helicase activities or are involved in signaling activities (Table 1).
AML patients with NUP98 fusions display different French-American-British (FAB) subtypes. PML::RARA fusion is usually a characteristic of acute promyelocytic leukemia (APL) or the M3 subtype of AML [75]. However, certain AML types with NUP98 translocations like NUP98::JADE2, NUP98::RARA, and NUP98::RARG resemble the APL phenotype [32,33,56]. The PML::RARA fusion inhibits RARA target genes that block differentiation at the promyelocyte stage, which leads to APL [76]. This may indicate that the NUP98 rearrangements associated with the APL phenotype prevent the expression of RARA target genes. However, the mechanism of APL transformation and the response to ATRA therapy by these NUP98 fusions is not clearly understood yet. Compared to other NUP98 fusions, the NUP98::KDM5A fusion is enriched in the M6/M7 subtype of AML. NUP98::KDM5A fusion occurs in about 20 percent of acute erythroid leukemia (AEL) cases. NUP98-KDM5A fusion also occurs in approximately 10 percent of pediatric acute megakaryoblastic leukemia (AMKL) cases [57,77]. Although NUP98::NSD1 fusion appears in different AML subtypes, it is more frequent in M4/M5 subtypes [70]. NUP98::HOX fusions mostly occur in undifferentiated or minimally differentiated AML subtypes (Table 1).
4. NUP98 Fusions Represent a Poor Prognostic and Chemoresistant AML Subgroup
NUP98 fusions are associated with adverse clinical outcomes in AML. Patients with NUP98 rearrangements, predominantly NUP98::NSD1 fusion, showed poor overall survival [OS] and disease-free survival (DFS) in a pediatric AML cohort [78]. Additionally, more than 70% of NUP98 fusion positive patients were refractory after the induction therapy [27]. In this line, other studies reported induction failure and chemotherapy resistance in pediatric AML patients carrying NUP98::NSD1 fusion [79,80]. Shiba et al. demonstrated that NUP98::NSD1-like patients, with the similar gene expression signature as NUP98::NSD1, confer poor overall survival like NUP98::NSD1 patients. NUP98::HOXA13, DEK::NUP214, MLL::MLLT4 were observed in the NUP98::NSD1-like subgroup [81]. Furthermore, a study by the Children’s Oncology Group (COG) and the European AML study groups demonstrated poor survival and higher relapse risk in NUP98::KDMA+ pediatric AML patients [58]. In a study including acute erythroid leukemia (AEL) patients, NUP98 fusions showed adverse clinical outcomes with estimated OS less than 10 percent [60]. Similarly, NUP98::KDM5A fusion showed unfavorable outcomes in pediatric AMKL patients [77]. A report from the AIEOP AML group, which includes multiple NUP98 fusions, observed worse event-free survival (EFS) and nearly double the relapse rate in NUP98 fusion positive AML patients compared to AML patients without known mutations [28]. Another study conducted by the French ELAM02 study group grouped NUP98 fusions in an adverse subtype together with mutations in WT1, PHF6, and RUNX1. In this study, KMT2A rearrangements were classified as intermediate subtypes, whereas CBF rearrangements, NPM1 mutations, and double CEBPA mutations were classified as favorable subtypes [78]. Similarly, NUP98 fusions confer poor prognosis in the adult AML cohort [70,82]. NUP98 fusions often co-occur with WT1 and FLT3-ITD mutations. Therefore, it is always a question as to how these cooperating mutations affect survival and the response to chemotherapy. Niktoreh et al. found that co-occurrence of NUP98 fusion, WT1, and FLT3-ITD mutations, or any of these two abnormalities, shows significantly low 3-year OS compared to patients with none of these mutations or patients with either one of these mutations [83]. Ostronoff et al. reported that the addition of FLT3 mutations decreases the survival chances of patients with NUP98::NSD1 AML [84]. An interesting study showed that no NUP98::NSD1 relapsed AML patients exhibit FLT3 mutations after chemotherapy, but four out of six exhibit WT1 mutations [79].
5. Mechanism of NUP98 Fusion Mediated AML
The NUP98 fusions mostly retain the N terminus of NUP98 and C terminus of the partner protein [85]. From the N terminus of NUP98, the GLFG repeats play a crucial role in leukemogenesis through recruiting the transcriptional coactivator complex CBP/p300, refs. [24,86] but the GLEBS domain is dispensable for leukemogenesis [87]. A mechanistic question is whether NUP98 or its partner gene plays a key role in leukemogenesis. Various studies have shown that NUP98 fusions lose their transformation properties when either partner is deleted. For example, deletion of NUP98 or the SET domain of NSD1 in NUP98-NSD1 fusion prevents myeloid progenitor immortalization [86]. Further overexpression of neither NUP98 nor its partner protein is sufficient for oncogenic transformation [88,89]. These studies indicate that the fusion protein has unique oncogenic properties in comparison to its associated components.
NUP98 fusions can form distinct nuclear dots, suggesting their involvement in gene regulation [48,90,91]. These GLFG nuclear bodies are distinct from Cajal bodies, PML bodies [in APL], and splicing-factor speckles [23]. NUP98 fusion proteins can bind CRM1 in a distinct manner from wild-type NUP98, thus preventing transcription factors, such as NFAT and NFKB, from being exported from the nucleus. The NUP98::IQCG, NUP98::HOXA9, and NUP98::DDX10 fusion proteins cause nuclear accumulation of P65, which has the potential to activate the NFKB pathway, which may contribute to the development of leukemia mediated by NUP98 fusion proteins [91,92]. Recent studies observe that membraneless organelles are formed within the nucleus through liquid–liquid phase separation (LLPS) that facilitates active transcription [93]. NUP98 fusion oncoproteins have intrinsically disordered FG motifs that create nuclear puncta and promote leukemogenesis through formation of these transcription centers [94,95,96].
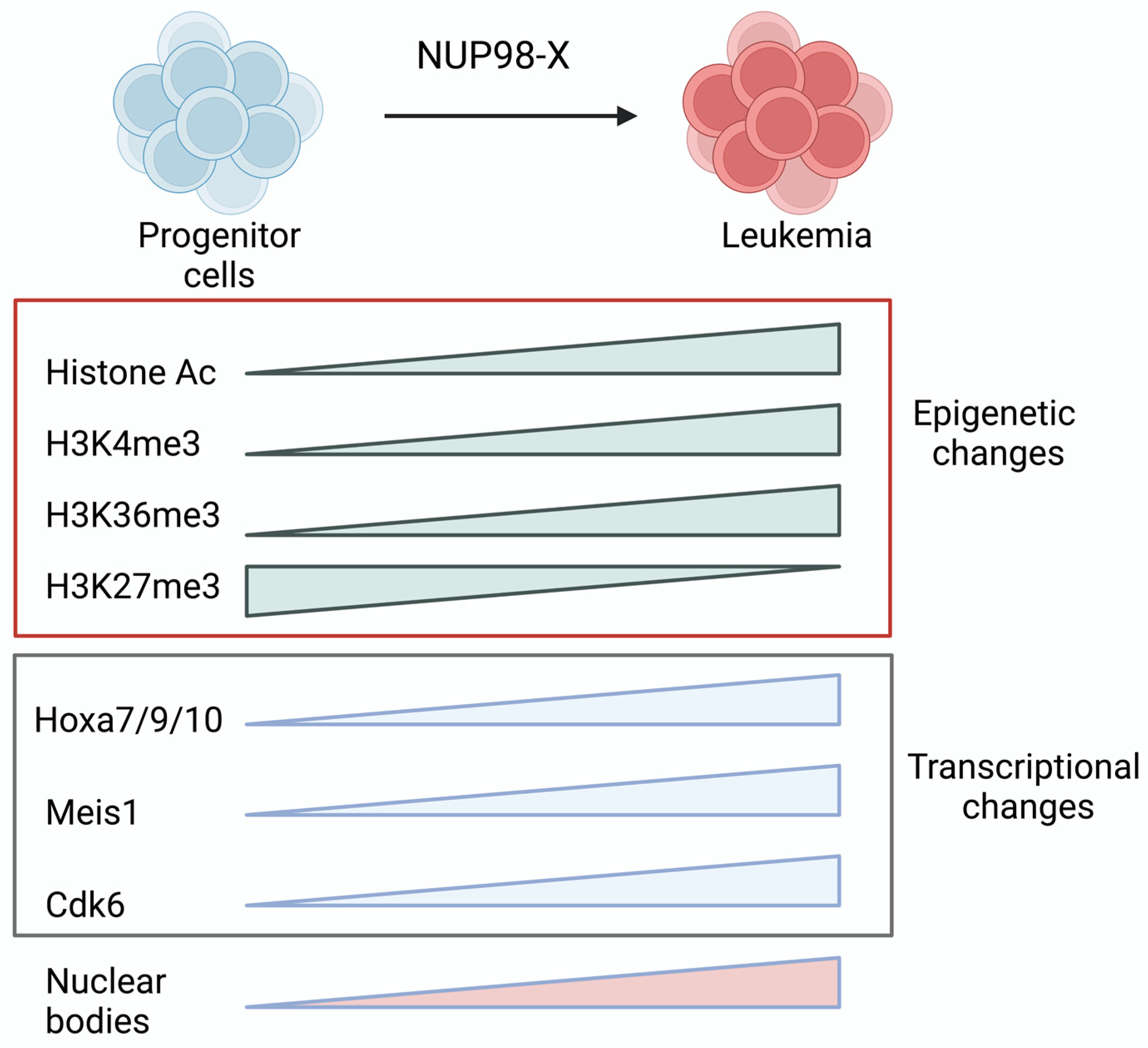
Different NUP98 fusion proteins regulate HOX genes expression to drive leukemogenesis. NUP98 fusions bind near the HOX genes loci and activate their expression through chromatin remodeling. Results from different studies confirm that the HoxA/Meis1 pathway is the major mechanism through which NUP98 oncoproteins drive leukemogenesis. The expression of distal HoxA cluster genes (Hoxa7, Hoxa9, and Hoxa10) and Meis1 are downregulated as hematopoietic stem and progenitor cells differentiate and overexpression of these promote self-renewal [86,97,98]. EZH2, which is part of the polycomb repressive complex 2 (PRC2), silences HOXA genes during differentiation [86]. Cooperation of Hoxa9 with Meis1 causes rapid leukemia induction in mice, indicating a crucial pathway through which leukemogenesis happens [99]. NUP98 fusions activate silenced HoxA cluster genes. While histone acetylation, H3K4, and H3K36 methylation around the HoxA locus confirm active chromatin, H3K27 marks by polycomb repressor complex silence HoxA genes [86,100]. NUP98 fusions prevent the H3K27me3 repressive mark and add few activation marks to induce expression of HoxA genes and Meis1 (Figure 3) [86]. NUP98 fusions acetylate histones through the recruitment of enhancer factors, CREB-binding protein (CBP) and p300, by NUP98 [24,86,101]. NUP98 fusions with a chromatin-modifier partner change the chromatin near the HoxA cluster and Meis1 locus. Histone H3 Lys 36 (H3K36) methylation on the HoxA locus by the SET domain of NUP98::NSD1 activates distal HoxA gene expression and causes bone marrow (BM) immortalization [86]. NSD2 is translocated in multiple myeloma patients and shows its oncogenic activity dependent on dimethylation of histone H3 at lysine 36 (H3K36me2) [102]. Other epigenetic-modifying partners of NUP98, such as PHF23 or KDM5A, dysregulate Hox genes expression through recognition of H3K4me3/2 marks by the plant homeodomain (PHD) finger domain. NSD1 also contains PHD fingers, but it lacks residues that interact with H3K4me3 [101]. Small molecules that inhibit the binding of the PHD domain to H3K4me3 can inhibit leukemogenesis [103,104]. NUP98 fusions with a homeobox partner gene like NUP98::HOXA9, NUP98::HOXA10, and NUP98::HOXD13 cooperate with Meis1 and cause lethal AML [105,106,107]. However, the overexpression of Meis1 does not affect the survival of mice with leukemia induced by NUP98::TOP1 [88]. It indicates that NUP98 fusions might have distinct oncogenic potential. However, it is unclear how NUP98::HOX oncogenes collaborate with MEIS1 as compared to NUP98::HOXA9, as only HOXA9 has a MEIS1 binding site. Additionally, the lack of Hoxa9 does not affect the immortalization properties of an NUP98::HOXA9 fusion oncogene [108]. Based on these findings, there may be redundant functions of Hoxa9 in NUP98 fusion mediated leukemogenesis. Other Hox genes can also drive leukemogenesis, which could be clarified in future studies. NUP98 fusion carrying patients show upregulation of both HOXA and HOXB cluster genes [57,70], but the significance of the upregulation of HOXB cluster genes in NUP98-fusion oncoprotein-driven AML remains unknown.
In addition to transcriptional regulation, NUP98 fusions also facilitate aneuploidy [109]. The anaphase promoting complex/cyclosome (APC/C) plays a key role in the transition from metaphase to anaphase during the cell cycle, and misregulation of this complex can make the cell susceptible to malignant transformation [110]. APC/CCdc20 ubiquitinates securin, leading to its degradation, and activates separase to allow chromosome segregation. Furthermore, the spindle checkpoint protects from improper chromosome segregation by inhibiting APC [111]. NUP98 fusion proteins interact with APC/CCdc20 and mediates aneuploidy through obstructing the interaction of the mitotic checkpoint complex to the APC/CCdc20 and premature securin degradation [109,112].
6. Cooperating Abnormalities in NUP98 Rearranged AML
NUP98 rearranged AML patients show frequent mutations in signal transduction genes (FLT3, NRAS, KRAS, and KIT) and WT1 [113]. Different NUP98 fusions show different co-occurring mutational signatures. For example, NUP98::KDM5A positive AEL cases are often associated with RB1 deletions [60]. A recent report indicated that del(13q) is a frequent event in NUP98::KDM5A AML patients, indicating co-occurrence of NUP98-KDMA fusion with RB1 deletion [30]. FLT3-ITD mutation is a recurring event in NUP98::NSD1 positive AML patients [70,84,114]. FLT3-ITD mutation is also observed in some NUP98::HOXA9 AML patients [114,115]. WT1, NRAS, and KRAS mutations frequently co-occur with NUP98::NSD1 and NUP98::HOXA9 [78,116,117]. Patients with NUP98 rearranged AML also have mutations in other genes, such as ASXL1 and MYC [117,118].
Multiple subtypes of AML (myeloid, erythroid, and megakaryoblastic) exhibit NUP98::KDM5A fusion. NUP98::KDMA positive acute megakaryoblastic leukemia (AMKL) cases usually do not show any additional mutations [57]. It is not yet clear whether different types of AML can be attributed to different origins of cells or the presence or absence of specific additional genetic changes. Mice transplanted with hematopoietic stem and progenitor cells (HSPCs) expressing NUP98::KDM5A fusion oncoprotein develop a myeloid leukemia phenotype [60]. This suggests that additional alterations, such as RB1 deletion, are required for the development of erythroid leukemia, thus explaining why RB1 deletion occurs concurrently with NUP98::KDM5A positive AEL. There is, however, a need for more studies to clarify how the same fusion can lead to different types of AML.
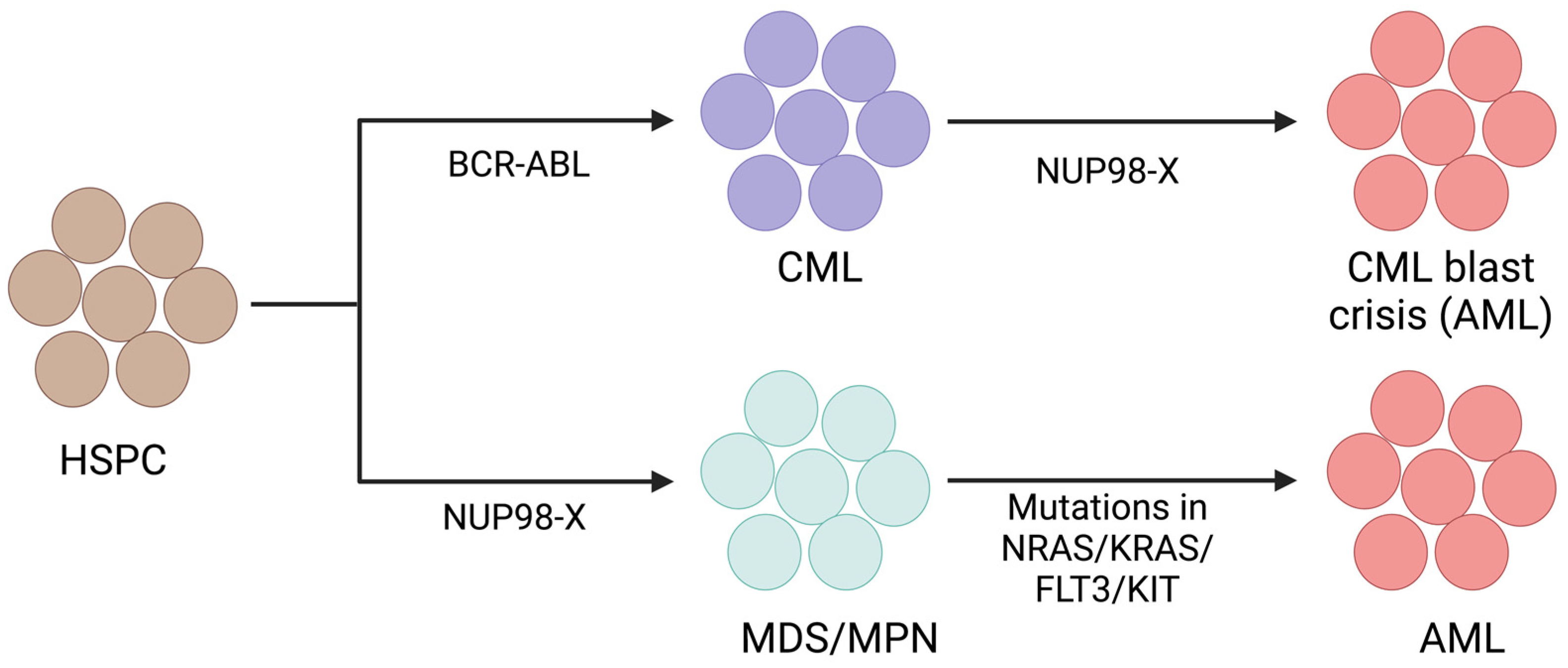
One genetic alteration alone does not cause AML; at least two types of genetic alterations must be present for the disease to manifest. While Class-I mutations provide proliferative advantage to cells, Class-II mutations impair differentiation [119]. Class-I mutations include mutations in proliferative genes like FLT3, RAS, or KIT, but Class-II mutations include different translocations like MLL rearrangements, RUNX1::ETO, and PML::RARα fusion [119,120]. Several studies have demonstrated that BCR::ABL positive CML progresses to AML (CML blast crisis) through the acquisition of the NUP98::HOXA9 fusion gene (Figure 4) [121,122]. Interestingly, another study observed cooperation of NUP98::HOXA9 with BCR::ABL for causing AML with features of a CML blast crisis in a murine model [123]. Additionally, NUP98::HOXA13 and NUP98::HOXA11 were also reported in a CML blast crisis [37,124]. Another intriguing study observed the appearance of NUP98::DDX10 fusion as a resistance mechanism to imatinib in CML and caused a blast crisis [125]. Fusions involving NUP98 induce HOX genes expression and stem cell renewal, which are considered Class-II mutations. NUP98 fusions often show mutations in signaling genes like FLT3, NRAS, and KIT, indicating the requirement of Class-I mutations for NUP98 fusion mediated AML development [113,116]. Different murine model studies have also shown that NUP98 fusions alone induce long-latency myeloid disease, but when they collaborate with different proliferative events like FLT3 or NRAS mutations, they induce a lethal short-latency AML phenotype [126,127,128,129,130]. Retroviral insertional mutagenesis is an excellent tool to identify cooperative events for carcinogenesis [131]. A retroviral insertional mutagenesis study using NUP98::HOXD13 fusion showed insertional events near Meis1, several signal transduction genes, and cell-cycle genes [132]. Similarly, another study observed spontaneous mutations in Nras and Kras in NUP98::HOXD13 mice with the AML phenotype, but these mutations were absent in NUP98::HOXD13 mice with the MDS phenotype [133]. Our study demonstrated that the addition of the NRAS p.G12D mutation further elevates the expression of distal Hoxa genes in NUP98::NSD1 immortalized cells [128]. Overall, the above studies support why signaling mutations are more frequent with NUP98 rearrangements in AML (Figure 4). However, additional future studies are required to understand and clarify the clonal evolution pattern of NUP98 fusion mediated AML. Furthermore, the future studies can explain the mechanistic significance of the co-occurrence of NUP98::NSD1 fusion with WT1 mutation and NUP98::KDM5A fusion with deletion of RB1.
7. Therapeutic Strategies to Treat NUP98 Fusion Positive AML Patients
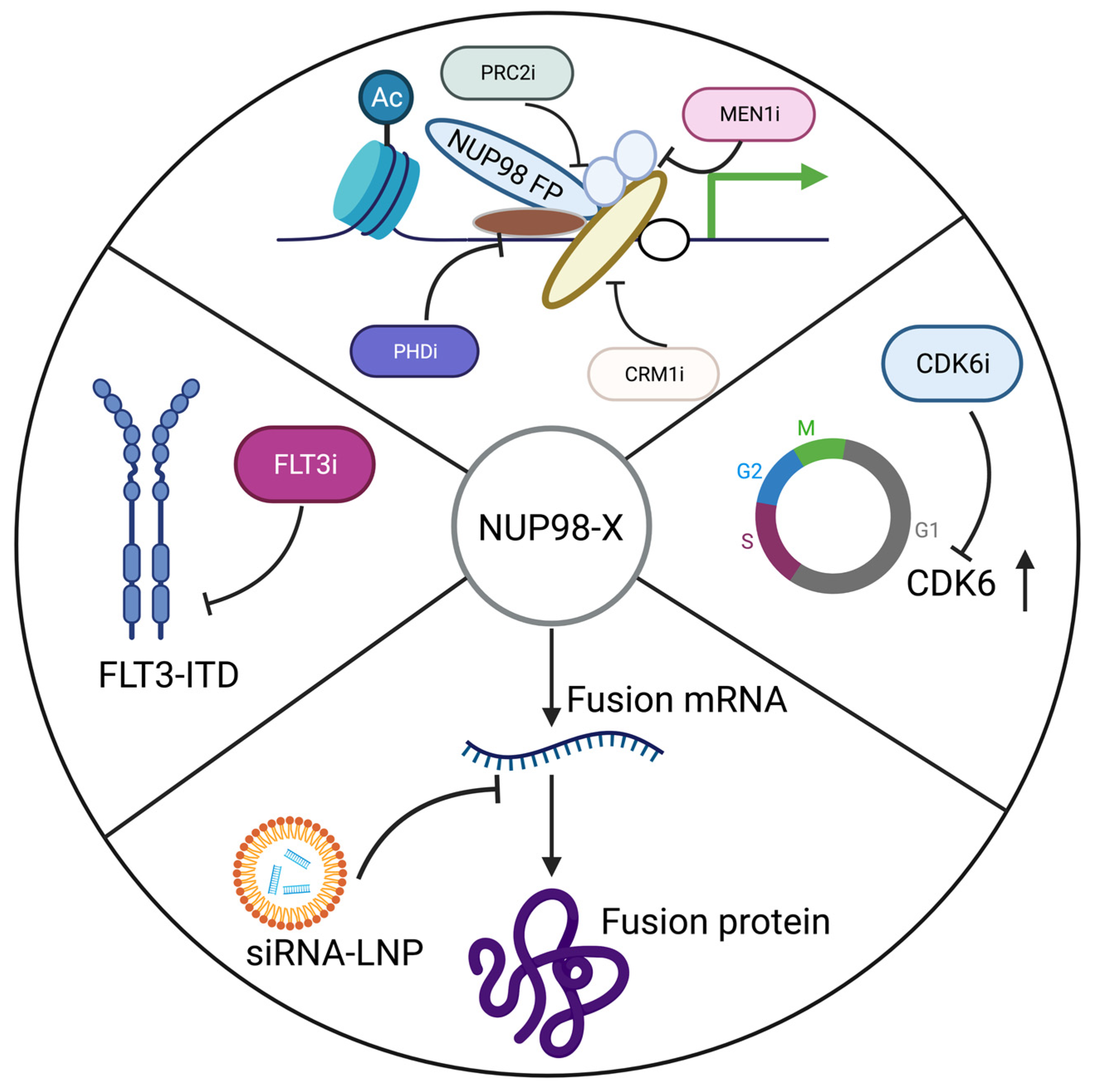
As discussed above, AML patients carrying a NUP98 fusion show low response to conventional therapies. Therefore, we should explore more targeted therapies for this subset of AML patients. Acute promyelocytic leukemia (APL), a subtype of AML that express PML::RARα fusion, is now the most curable AML through targetable degradation of fusion protein using all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) [134]. Therefore, eliminating the NUP98 chimeric protein or its potential downstream target can be developed as a curative treatment option for AML patients carrying NUP98 fusions. There are different ways to target chimeric oncoproteins. In order to eliminate AML, the oncoprotein itself can be targeted directly, or downstream targets of this fusion oncogene or its interactors can be targeted [135]. Several approaches have been proposed for treating NUP98 rearranged AML (Figure 5).
Because these fusion genes are expressed only in leukemia cells and not in healthy cells, siRNAs targeting fusion junctions can be designed specifically to target these leukemia cells. Using the patient-derived xenograft (PDX) model, we showed that siRNA lipid nanoparticle formulations can be used as a therapeutic strategy against NUP98::NSD1 leukemia [128]. This approach is validated against BCR::ABL, TCF3::PBX1, and RUNX1::ETO fusion oncogenes [136,137,138]. However, a same fusion protein with a different breakpoint can appear as a resistance mechanism to this kind of therapy.
CDK6, which is required for G1- to S-phase transition, is upregulated by different NUP98 fusion oncoproteins. NUP98 fusion positive human and mouse samples treated with palbociclib, a CDK4/CDK6 inhibitor, show a reduction in leukemia growth [67,139]. Different FDA-approved CDK 4/6 inhibitors, which are palbociclib, ribociclib, and abemaciclib, are used for the treatment of metastatic breast-cancer patients [140].
Previously, it was demonstrated that the menin (MEN1)-MLL interaction is critical for MLL-rearranged and NPMc-mutant leukemias, and small molecules disrupting this interaction are effective against these leukemias [141,142]. Disruption of this interaction downregulates leukemic gene expression. Different menin inhibitors (SDNX-5613, JNJ-75276617, BMF-219, DSP 5336, and KO-539) are currently in early phase clinical trials [143]. A recent study shows that menin–MLL interaction is critical in NUP98-rearranged AML. VTP50469, an inhibitor of menin–MLL interaction, abrogates leukemogenesis and upregulates differentiation in leukemic cells carrying the NUP98 fusion. Inhibition of menin–MLL interaction downregulates the expression of Meis1 [144]. Overall, leukemias with KMT2A rearrangements, NUP98 rearrangements, or NPM1 mutations depend upon the HOXA/MEIS1 pathway and are susceptible to menin inhibition [145]. These genotypes account for the majority of patients with AML, indicating that menin inhibitors may have a beneficial effect on a large proportion of AML patients [145]. A recent clinical trial report for revumenib (SNDX-5613), a potent oral menin inhibitor, showed that it was effective in treating relapsed or refractory AML with KMT2A rearrangements or NPM1 mutations [146]. Even though the report indicates promising clinical efficacy, minimal toxicity, and good tolerance for revumenib, clinical resistance could result from mutations in menin that prevent the drug from binding to the target [147].
The NUP98 fusions interact with XPO1; therefore, XPO1 inhibitors may be effective against NUP98 leukemia [26]. Oka et al. found that Crm1 recruits Nup98-Hoxa9 to Hox cluster genes to drive its expression [148]. Selinexor, an XPO1 inhibitor, has been approved for the treatment of multiple myeloma [26].
Ren et al. showed that the PRC2-KDM5B axis is crucial for NUP98::NSD1 AML. An inhibitor of polycomb repressive complex 2 (PRC2), UNC1999, reduced the leukemic burden in NUP98::NSD1-bearing mice and improved their survival [149]. PRC2 forms silenced chromatin through H3K27me3. The enhancer of zest homolog 2 (EZH2) is a catalytic subunit of PRC2, so EZH2 inhibitors can also be tested against NUP98 fusion AML [150]. Tazemetostat, an FDA-approved EZH2 inhibitor, is currently used for the treatment of epithelioid sarcoma and follicular lymphoma patients [151]. Another study shows that NUP98::PHF23 and NUP98::JARID1A leukemias are sensitive to disulfiram or a small molecule that inhibits binding of the PHF23 plant homeodomain (PHD) motif to H3K4me3, which is essential for HOXA and MEIS1 aberrant expression [103,104].
Since NUP98 fusions are frequently associated with other kinase mutations, primarily FLT3 mutations, kinase inhibitors present an interesting aspect if they are capable of effectively eradicating these leukemic cells. Leukemic cells that express NUP98::NSD1 and FLT3-ITD are effectively inhibited by a potent FLT3 inhibitor [129]. However, it has been demonstrated that leukemic clones with kinase mutations disappear after chemotherapy [79]. Therefore, kinase inhibitors might alone be insufficient to eliminate major leukemic clones, but they should be used in combination with other inhibitors targeting NUP98 fusions. The BCL2 inhibitor navitoclax and SRC/ABL inhibitor dasatinib show synergistic effects against AML cells co-expressing NUP98::NSD1 and FLT3-ITD [152].
In these AML patients, allogeneic hematopoietic stem-cell transplantation (allo-HSCT) is an effective treatment method to prevent relapse. In different studies, relapse was observed despite allo-HSCT [58,116]. According to a study, allo-HSCT in the first complete remission (CR1) is more effective than HSCT in the second complete remission (CR2) for AML patients with NUP98::HOXA9 fusion [153]. These studies indicate that allo-HSCT is partially effective for NUP98 rearranged AML patients.
Future research may lead to the development of other treatment options for NUP98 rearranged AML. Since fusion oncoproteins are exclusively expressed on cancer cells, novel ways to target fusion oncoprotein can be explored. The proteolysis-targeting chimeras (PROTACs) technology can be used to target and degrade the fusion oncoprotein and can be developed as a potential treatment approach for NUP98 fusion positive AML patients [154]. As an ideal source of neoantigens, fusion genes can be exploited for the development of immunotherapy, such as the development of adoptive cell therapy, vaccines, and checkpoint blockade therapy [155,156]. The benefit of checkpoint blockade therapy for patients with an NUP98 rearrangement remains undetermined. The identification of unique cell-surface proteins in the NUP98 fusion subtype AML, which are not expressed in healthy counterparts, may be used for developing chimeric antigen receptor (CAR) T-cell therapy. A recent study demonstrated that CD123 is highly expressed in NUP98::NSD1 positive AML patients and can be exploited as an therapeutic target [157].
8. Concluding Remarks
In recent years, new fusion partners of NUP98 have been discovered in patients with AML.. Although many mechanisms are proposed to explain the leukemogenic activity of NUP98 fusion proteins, HOXA/MES1 appears to be the major player. There is remarkable progress in developing disease models to understand the oncogenic function of NUP98 fusion proteins. These models have been instrumental in validating therapeutic targets and assessing drug responses. Furthermore, the development of patient-derived xenograft (PDX) models of NUP98 fusions recapitulates the disease well and is an effective method to track patient cells’ response to different drugs. We currently have PDX models for only a few NUP98 rearrangements, but we will need to establish PDX models for all common NUP98 rearrangements in the near future [26,158,159]. In different NUP98 fusion mouse models, the siRNA-LNP formulations, CDK6 inhibitor, and menin inhibitor show promising antileukemic activity.
Future studies may focus on discovering the new therapeutic vulnerabilities of NUP98 fusions and developing immunotherapies for these patients. Further research into the shared and unique mechanisms of leukemogenic transformation among NUP98 fusion oncoproteins is necessary to clarify whether a single agent can be used for the treatment of all AML patients with different NUP98 rearrangements.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The figures were prepared using Biorender and Adobe illustrator.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Taniue, K.; Akimitsu, N. Fusion Genes and RNAs in Cancer Development. Non-Coding RNA 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.J.; Rohan, S.; Kao, J.; Kitabayashi, N.; Mathew, S.; Chen, Y.-T. Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: Frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod. Pathol. 2007, 20, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, M.; Andrén, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef]
- Annala, M.J.; Parker, B.C.; Zhang, W.; Nykter, M. Fusion genes and their discovery using high throughput sequencing. Cancer Lett. 2013, 340, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Engvall, M.; Cahill, N.; Jonsson, B.-I.; Höglund, M.; Hallböök, H.; Cavelier, L. Detection of leukemia gene fusions by targeted RNA-sequencing in routine diagnostics. BMC Med. Genom. 2020, 13, 106. [Google Scholar] [CrossRef]
- Heydt, C.; Wölwer, C.B.; Velazquez Camacho, O.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; et al. Detection of gene fusions using targeted next-generation sequencing: A comparative evaluation. BMC Med. Genom. 2021, 14, 62. [Google Scholar] [CrossRef]
- Padella, A.; Simonetti, G.; Paciello, G.; Giotopoulos, G.; Baldazzi, C.; Righi, S.; Ghetti, M.; Stengel, A.; Guadagnuolo, V.; De Tommaso, R.; et al. Novel and Rare Fusion Transcripts Involving Transcription Factors and Tumor Suppressor Genes in Acute Myeloid Leukemia. Cancers 2019, 11, 1951. [Google Scholar] [CrossRef] [Green Version]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Stengel, A.; Shahswar, R.; Haferlach, T.; Walter, W.; Hutter, S.; Meggendorfer, M.; Kern, W.; Haferlach, C. Whole transcriptome sequencing detects a large number of novel fusion transcripts in patients with AML and MDS. Blood Adv. 2020, 4, 5393–5401. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Kasper, L.H.; Mantcheva, R.T.; Mantchev, G.T.; Springett, M.J.; van Deursen, J.M. Disruption of the FG nucleoporin NUP98 causes selective changes in nuclear pore complex stoichiometry and function. Proc. Natl. Acad. Sci. USA 2001, 98, 3191–3196. [Google Scholar] [CrossRef] [PubMed]
- Fontoura, B.M.; Blobel, G.; Matunis, M.J. A conserved biogenesis pathway for nucleoporins: Proteolytic processing of a 186-kilodalton precursor generates Nup98 and the novel nucleoporin, Nup96. J. Cell Biol. 1999, 144, 1097–1112. [Google Scholar] [CrossRef]
- Paci, G.; Caria, J.; Lemke, E.A. Cargo transport through the nuclear pore complex at a glance. J. Cell Sci. 2021, 134, jcs247874. [Google Scholar] [CrossRef] [PubMed]
- Nofrini, V.; Di Giacomo, D.; Mecucci, C. Nucleoporin genes in human diseases. Eur. J. Hum. Genet. EJHG 2016, 24, 1388–1395. [Google Scholar] [CrossRef] [Green Version]
- Griffis, E.R.; Xu, S.; Powers, M.A. Nup98 localizes to both nuclear and cytoplasmic sides of the nuclear pore and binds to two distinct nucleoporin subcomplexes. Mol. Biol. Cell 2003, 14, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Chatel, G.; Desai, S.H.; Mattheyses, A.L.; Powers, M.A.; Fahrenkrog, B. Domain topology of nucleoporin Nup98 within the nuclear pore complex. J. Struct. Biol. 2012, 177, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, M.; Asakawa, H.; Hiraoka, Y.; Haraguchi, T. Nucleoporin Nup98: A gatekeeper in the eukaryotic kingdoms. Genes Cells Devoted Mol. Cell. Mech. 2010, 15, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Asally, M.; Yasuda, Y.; Ogawa, Y.; Tachibana, T.; Yoneda, Y. The mobile FG nucleoporin Nup98 is a cofactor for Crm1-dependent protein export. Mol. Biol. Cell 2010, 21, 1885–1896. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.E.; Fornerod, M.; Kasper, L.H.; van Deursen, J.M. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J. Cell Biol. 1999, 145, 237–254. [Google Scholar] [CrossRef]
- Ren, Y.; Seo, H.S.; Blobel, G.; Hoelz, A. Structural and functional analysis of the interaction between the nucleoporin NUP98 and the mRNA export factor Rae1. Proc. Natl. Acad. Sci. USA 2010, 107, 10406–10411. [Google Scholar] [CrossRef] [PubMed]
- Griffis, E.R.; Altan, N.; Lippincott-Schwartz, J.; Powers, M.A. NUP98 is a mobile nucleoporin with transcription-dependent dynamics. Mol. Biol. Cell 2002, 13, 1282–1297. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Brindle, P.K.; Schnabel, C.A.; Pritchard, C.E.; Cleary, M.L.; van Deursen, J.M. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell Biol. 1999, 19, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Kalverda, B.; Pickersgill, H.; Shloma, V.V.; Fornerod, M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 2010, 140, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Michmerhuizen, N.L.; Klco, J.M.; Mullighan, C.G. Mechanistic insights and potential therapeutic approaches for NUP98-rearranged hematologic malignancies. Blood 2020, 136, 2275–2289. [Google Scholar] [CrossRef]
- Struski, S.; Lagarde, S.; Bories, P.; Puiseux, C.; Prade, N.; Cuccuini, W.; Pages, M.P.; Bidet, A.; Gervais, C.; Lafage-Pochitaloff, M.; et al. NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 2017, 31, 565–572. [Google Scholar] [CrossRef]
- Bisio, V.; Zampini, M.; Tregnago, C.; Manara, E.; Salsi, V.; Di Meglio, A.; Masetti, R.; Togni, M.; Di Giacomo, D.; Minuzzo, S.; et al. NUP98-fusion transcripts characterize different biological entities within acute myeloid leukemia: A report from the AIEOP-AML group. Leukemia 2017, 31, 974–977. [Google Scholar] [CrossRef]
- Bertrums, E.J.M.; Smith, J.L.; Ries, R.E.; Alonzo, T.A.; Ostronoff, F.; Kaspers, G.J.L.; Hasle, H.; Zwaan, C.M.; Hirsch, B.A.; Raimondi, S.C.; et al. The Molecular Characteristics and Clinical Relevance of NUP98-Other Translocations in Pediatric Acute Myeloid Leukemia. Blood 2020, 136, 36–37. [Google Scholar] [CrossRef]
- Bertrums, E.J.M.; Smith, J.L.; Harmon, L.; Ries, R.E.; Wang, Y.J.; Alonzo, T.A.; Menssen, A.J.; Chisholm, K.M.; Leonti, A.R.; Tarlock, K.; et al. Comprehensive molecular and clinical characterization of NUP98 fusions in pediatric acute myeloid leukemia. Haematologica 2023. [Google Scholar] [CrossRef]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.H.; Yang, M.C.; Wang, F.; Lou, Y.J.; Jin, J.; Li, K.; Zhang, S.Z. Identification of a novel NUP98-RARA fusion transcript as the 14th variant of acute promyelocytic leukemia. Am. J. Hematol. 2020, 95, E184–E186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Such, E.; Cervera, J.; Valencia, A.; Barragán, E.; Ibañez, M.; Luna, I.; Fuster, Ó.; Perez-Sirvent, M.L.; Senent, L.; Sempere, A.; et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood 2011, 117, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Gallego Hernanz, M.P.; Torregrosa Diaz, J.M.; Sorel, N.; Bobin, A.; Dindinaud, E.; Bouyer, S.; Desmier, D.; Brizard, F.; Leleu, X.; Maillard, N.; et al. Long-term molecular remission in a patient with acute myeloid leukemia harboring a new NUP98-LEDGF rearrangement. Cancer Med. 2019, 8, 1765–1770. [Google Scholar] [CrossRef]
- Huang, S.Y.; Tang, J.L.; Liang, Y.J.; Wang, C.H.; Chen, Y.C.; Tien, H.F. Clinical, haematological and molecular studies in patients with chromosome translocation t(7;11): A study of four Chinese patients in Taiwan. Br. J. Haematol. 1997, 96, 682–687. [Google Scholar] [CrossRef]
- Lahortiga, I.; Belloni, E.; Vázquez, I.; Agirre, X.; Larrayoz, M.J.; Vizmanos, J.L.; Valgañón, M.; Zudaire, I.; Sáez, B.; Mateos, M.C.; et al. NUP98 is fused to HOXA9 in a variant complex t(7;11;13;17) in a patient with AML-M2. Cancer Genet. Cytogenet. 2005, 157, 151–156. [Google Scholar] [CrossRef]
- Suzuki, A.; Ito, Y.; Sashida, G.; Honda, S.; Katagiri, T.; Fujino, T.; Nakamura, T.; Ohyashiki, K. t(7;11)(p15;p15) Chronic myeloid leukaemia developed into blastic transformation showing a novel NUP98/HOXA11 fusion. Br. J. Haematol. 2002, 116, 170–172. [Google Scholar] [CrossRef]
- Taketani, T.; Taki, T.; Ono, R.; Kobayashi, Y.; Ida, K.; Hayashi, Y. The chromosome translocation t(7;11)(p15;p15) in acute myeloid leukemia results in fusion of the NUP98 gene with a HOXA cluster gene, HOXA13, but not HOXA9. Genes Chromosomes Cancer 2002, 34, 437–443. [Google Scholar] [CrossRef]
- Taketani, T.; Taki, T.; Shibuya, N.; Kikuchi, A.; Hanada, R.; Hayashi, Y. Novel NUP98-HOXC11 Fusion Gene Resulted from a Chromosomal Break within Exon 1 of HOXC11 in Acute Myeloid Leukemia with t(11;12)(p15;q13)1. Cancer Res. 2002, 62, 4571–4574. [Google Scholar]
- Gu, B.W.; Wang, Q.; Wang, J.M.; Xue, Y.Q.; Fang, J.; Wong, K.F.; Chen, B.; Shi, Z.Z.; Shi, J.Y.; Bai, X.T.; et al. Major form of NUP98/HOXC11 fusion in adult AML with t(11;12)(p15;q13) translocation exhibits aberrant trans-regulatory activity. Leukemia 2003, 17, 1858–1864. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulos, I.; Isaksson, M.; Billström, R.; Strömbeck, B.; Mitelman, F.; Johansson, B. Fusion of the NUP98 gene and the homeobox gene HOXC13 in acute myeloid leukemia with t(11;12)(p15;q13). Genes Chromosomes Cancer 2003, 36, 107–112. [Google Scholar] [CrossRef]
- Tosić, N.; Stojiljković, M.; Colović, N.; Colović, M.; Pavlović, S. Acute myeloid leukemia with NUP98-HOXC13 fusion and FLT3 internal tandem duplication mutation: Case report and literature review. Cancer Genet. Cytogenet. 2009, 193, 98–103. [Google Scholar] [CrossRef]
- Taketani, T.; Taki, T.; Shibuya, N.; Ito, E.; Kitazawa, J.; Terui, K.; Hayashi, Y. The HOXD11 Gene Is Fused to the NUP98 Gene in Acute Myeloid Leukemia with t(2;11)(q31;p15)1. Cancer Res. 2002, 62, 33–37. [Google Scholar] [PubMed]
- Raza-Egilmez, S.Z.; Jani-Sait, S.N.; Grossi, M.; Higgins, M.J.; Shows, T.B.; Aplan, P.D. NUP98-HOXD13 gene fusion in therapy-related acute myelogenous leukemia. Cancer Res. 1998, 58, 4269–4273. [Google Scholar] [PubMed]
- Arai, Y.; Kyo, T.; Miwa, H.; Arai, K.; Kamada, N.; Kita, K.; Ohki, M. Heterogeneous fusion transcripts involving the NUP98 gene and HOXD13 gene activation in a case of acute myeloid leukemia with the t(2;11)(q31;p15) translocation. Leukemia 2000, 14, 1621–1629. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Yamazaki, Y.; Hatano, Y.; Miura, I. NUP98 is fused to PMX1 homeobox gene in human acute myelogenous leukemia with chromosome translocation t(1;11)(q23;p15). Blood 1999, 94, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Gervais, C.; Mauvieux, L.; Perrusson, N.; Hélias, C.; Struski, S.; Leymarie, V.; Lioure, B.; Lessard, M. A new translocation t(9;11)(q34;p15) fuses NUP98 to a novel homeobox partner gene, PRRX2, in a therapy-related acute myeloid leukemia. Leukemia 2005, 19, 145–148. [Google Scholar] [CrossRef]
- Jankovic, D.; Gorello, P.; Liu, T.; Ehret, S.; La Starza, R.; Desjobert, C.; Baty, F.; Brutsche, M.; Jayaraman, P.-S.; Santoro, A.; et al. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood 2008, 111, 5672–5682. [Google Scholar] [CrossRef]
- Sorel, N.; Raimbault, A.; Brizard, F.; Depaire, T.; Pierini, V.; Dupraz, C.; Millot, F.; Mecucci, C.; Chomel, J.C. Identification and genetic characterization of a NUP98-HHEX molecular rearrangement in a pediatric acute myeloid leukemia. Leuk. Lymphoma 2021, 62, 3531–3535. [Google Scholar] [CrossRef]
- Tao, S.; Song, L.; Deng, Y.; Chen, Y.; Shi, Y.; Gan, Y.; Deng, Z.; Ding, B.; He, Z.; Wang, C.; et al. Acute Myeloid Leukemia with NUP98-RARG Gene Fusion Similar to Acute Promyelocytic Leukemia: Case Report and Literature Review. OncoTargets Ther. 2020, 13, 10559–10566. [Google Scholar] [CrossRef]
- Lisboa, S.; Cerveira, N.; Bizarro, S.; Correia, C.; Vieira, J.; Torres, L.; Mariz, J.M.; Teixeira, M.R. POU1F1 is a novel fusion partner of NUP98 in acute myeloid leukemia with t(3;11)(p11;p15). Mol. Cancer 2013, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, H.G.; Hong, J.; Aplan, P.D.; Tcheurekdjian, L.; Forman, S.J.; Slovak, M.L. t(9;11)(p22;p15) in acute myeloid leukemia results in a fusion between NUP98 and the gene encoding transcriptional coactivators p52 and p75-lens epithelium-derived growth factor (LEDGF). Cancer Res. 2000, 60, 6227–6229. [Google Scholar]
- Hussey, D.J.; Moore, S.; Nicola, M.; Dobrovic, A. Fusion of the NUP98 gene with the LEDGF/p52 gene defines a recurrent acute myeloid leukemia translocation. BMC Genet. 2001, 2, 20. [Google Scholar] [CrossRef]
- Reader, J.C.; Meekins, J.S.; Gojo, I.; Ning, Y. A novel NUP98-PHF23 fusion resulting from a cryptic translocation t(11;17)(p15;p13) in acute myeloid leukemia. Leukemia 2007, 21, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Togni, M.; Masetti, R.; Pigazzi, M.; Astolfi, A.; Zama, D.; Indio, V.; Serravalle, S.; Manara, E.; Bisio, V.; Rizzari, C.; et al. Identification of the NUP98-PHF23 fusion gene in pediatric cytogenetically normal acute myeloid leukemia by whole-transcriptome sequencing. J. Hematol. Oncol. 2015, 8, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-K.; Chan, H.-Y.; Yung, Y.-L.; Wan, T.S.K.; Leung, A.W.K.; Li, C.-K.; Tian, K.; Chan, N.P.H.; Cheung, J.S.; Ng, M.H.L. A novel NUP98-JADE2 fusion in a patient with acute myeloid leukemia resembling acute promyelocytic leukemia. Blood Adv. 2022, 6, 410–415. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.D.E.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; van Galen, J.F.; Berna Beverloo, H.; Baruchel, A.; Trka, J.; Reinhardt, D.; Sonneveld, E.; Zimmermann, M.; et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 2013, 27, 2280–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noort, S.; Wander, P.; Alonzo, T.A.; Smith, J.; Ries, R.E.; Gerbing, R.B.; Dolman, M.E.M.; Locatelli, F.; Reinhardt, D.; Baruchel, A.; et al. The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 2021, 106, 630–634. [Google Scholar] [CrossRef]
- Jaju, R.J.; Fidler, C.; Haas, O.A.; Strickson, A.J.; Watkins, F.; Clark, K.; Cross, N.C.P.; Cheng, J.-F.; Aplan, P.D.; Kearney, L.; et al. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 2001, 98, 1264–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobucci, I.; Wen, J.; Meggendorfer, M.; Choi, J.K.; Shi, L.; Pounds, S.B.; Carmichael, C.L.; Masih, K.E.; Morris, S.M.; Lindsley, R.C.; et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat. Genet. 2019, 51, 694–704. [Google Scholar] [CrossRef]
- Wang, T.; Ni, J.B.; Wang, X.Y.; Dai, Y.; Ma, X.L.; Su, Y.C.; Gao, Y.Y.; Chen, X.; Yuan, L.L.; Liu, H.X. [Genetic characteristics and clinical outcomes of pediatric acute myeloid leukemia with NUP98-NSD1 fusion gene]. Zhonghua Yi Xue Za Zhi 2019, 99, 2820–2825. [Google Scholar] [CrossRef] [PubMed]
- Rosati, R.; La Starza, R.; Veronese, A.; Aventin, A.; Schwienbacher, C.; Vallespi, T.; Negrini, M.; Martelli, M.F.; Mecucci, C. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 2002, 99, 3857–3860. [Google Scholar] [CrossRef] [Green Version]
- Kaltenbach, S.; Soler, G.; Barin, C.; Gervais, C.; Bernard, O.A.; Penard-Lacronique, V.; Romana, S.P. NUP98-MLL fusion in human acute myeloblastic leukemia. Blood 2010, 116, 2332–2335. [Google Scholar] [CrossRef]
- Tosi, S.; Ballabio, E.; Teigler-Schlegel, A.; Boultwood, J.; Bruch, J.; Harbott, J. Characterization of 6q abnormalities in childhood acute myeloid leukemia and identification of a novel t(6;11)(q24.1;p15.5) resulting in a NUP98-C6orf80 fusion in a case of acute megakaryoblastic leukemia. Genes Chromosomes Cancer 2005, 44, 225–232. [Google Scholar] [CrossRef]
- Petit, A.; Ragu, C.; Della-Valle, V.; Mozziconacci, M.J.; Lafage-Pochitaloff, M.; Soler, G.; Schluth, C.; Radford, I.; Ottolenghi, C.; Bernard, O.A.; et al. NUP98–HMGB3: A novel oncogenic fusion. Leukemia 2010, 24, 654–658. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Zhu, Y.-J.; Gu, B.-W.; Cai, X.; Bai, X.-T.; Yuan, H.-Y.; Zhu, J.; Chen, Z.; Xue, Y.-Q.; Chen, S.-J. A New Fusion Gene NUP98-IQCG Identified in an Acute T/Myeloid Leukemia with t(3;11)(q29q13;p15) Translocation. Blood 2007, 110, 1828. [Google Scholar] [CrossRef]
- Umeda, M.; Michmerhuizen, N.; Ma, J.; Westover, T.; Walsh, M.P.; Song, G.; Mecucci, C.; Giacomo, D.D.; Locatelli, F.; Masetti, R.; et al. AML-283 The Genetic Landscape of NUP98-Rearranged Pediatric Leukemia. Clin. Lymphoma Myeloma Leuk. 2022, 22, S233. [Google Scholar] [CrossRef]
- Bisio, V.; Pigazzi, M.; Manara, E.; Masetti, R.; Togni, M.; Astolfi, A.; Mecucci, C.; Zappavigna, V.; Salsi, V.; Merli, P.; et al. NUP98 Fusion Proteins Are Recurrent Aberrancies in Childhood Acute Myeloid Leukemia: A Report from the AIEOP AML-2001-02 Study Group. Blood 2014, 124, 1025. [Google Scholar] [CrossRef]
- Arai, Y.; Hosoda, F.; Kobayashi, H.; Arai, K.; Hayashi, Y.; Kamada, N.; Kaneko, Y.; Ohki, M. The inv(11)(p15q22) chromosome translocation of de novo and therapy-related myeloid malignancies results in fusion of the nucleoporin gene, NUP98, with the putative RNA helicase gene, DDX10. Blood 1997, 89, 3936–3944. [Google Scholar] [CrossRef] [PubMed]
- Hollink, I.H.I.M.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T.C.J.M.; Pratcorona, M.; Abbas, S.; Kuipers, J.E.; van Galen, J.F.; Beverloo, H.B.; Sonneveld, E.; Kaspers, G.-J.J.L.; et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011, 118, 3645–3656. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Xue, Y.; Chen, Z.; Guo, Y.; Wu, Y.; Pan, J. Generation of the NUP98-TOP1 fusion transcript by the t(11;20) (p15;q11) in a case of acute monocytic leukemia. Cancer Genet. Cytogenet. 2003, 140, 153–156. [Google Scholar] [CrossRef]
- Nebral, K.; Schmidt, H.H.; Haas, O.A.; Strehl, S. NUP98 Is Fused to Topoisomerase (DNA) IIβ 180 kDa (TOP2B) in a Patient with Acute Myeloid Leukemia with a New t(3;11)(p24;p15). Clin. Cancer Res. 2005, 11, 6489–6494. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Yagasaki, F.; Okamura, D.; Maeda, T.; Sugahara, Y.; Jinnai, I.; Bessho, M. A novel gene, ANKRD28 on 3p25, is fused with NUP98 on 11p15 in a cryptic 3-way translocation of t(3;5;11)(p25;q35;p15) in an adult patient with myelodysplastic syndrome/acute myelogenous leukemia. Int. J. Hematol. 2007, 86, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Gorello, P.; Brandimarte, L.; La Starza, R.; Pierini, V.; Bury, L.; Rosati, R.; Martelli, M.F.; Vandenberghe, P.; Wlodarska, I.; Mecucci, C. t(3;11)(q12;p15)/NUP98-LOC348801 fusion transcript in acute myeloid leukemia. Haematologica 2008, 93, 1398–1401. [Google Scholar] [CrossRef] [Green Version]
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.Á.; Barragán, E.; Cervera, J. Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single PML-RARA Fusion Gene. Cancers 2020, 12, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurnari, C.; Voso, M.T.; Girardi, K.; Mastronuzzi, A.; Strocchio, L. Acute Promyelocytic Leukemia in Children: A Model of Precision Medicine and Chemotherapy-Free Therapy. Int. J. Mol. Sci. 2021, 22, 642. [Google Scholar] [CrossRef]
- De Rooij, J.D.E.; Masetti, R.; van den Heuvel-Eibrink, M.M.; Cayuela, J.-M.; Trka, J.; Reinhardt, D.; Rasche, M.; Sonneveld, E.; Alonzo, T.A.; Fornerod, M.; et al. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: A retrospective intergroup study. Blood 2016, 127, 3424–3430. [Google Scholar] [CrossRef] [PubMed]
- Marceau-Renaut, A.; Duployez, N.; Ducourneau, B.; Labopin, M.; Petit, A.; Rousseau, A.; Geffroy, S.; Bucci, M.; Cuccuini, W.; Fenneteau, O.; et al. Molecular Profiling Defines Distinct Prognostic Subgroups in Childhood AML: A Report From the French ELAM02 Study Group. HemaSphere 2018, 2, e31. [Google Scholar] [CrossRef]
- McNeer, N.A.; Philip, J.; Geiger, H.; Ries, R.E.; Lavallée, V.-P.; Walsh, M.; Shah, M.; Arora, K.; Emde, A.-K.; Robine, N.; et al. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 2019, 33, 1934–1943. [Google Scholar] [CrossRef]
- Shimada, A.; Iijima-Yamashita, Y.; Tawa, A.; Tomizawa, D.; Yamada, M.; Norio, S.; Watanabe, T.; Taga, T.; Iwamoto, S.; Terui, K.; et al. Risk-stratified therapy for children with FLT3-ITD-positive acute myeloid leukemia: Results from the JPLSG AML-05 study. Int. J. Hematol. 2018, 107, 586–595. [Google Scholar] [CrossRef]
- Shiba, N.; Ichikawa, H.; Taki, T.; Park, M.J.; Jo, A.; Mitani, S.; Kobayashi, T.; Shimada, A.; Sotomatsu, M.; Arakawa, H.; et al. NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 2013, 52, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Raess, P.W.; Dunlap, J.; Hoyos, C.M.; Li, H.; Li, P.; Swords, R.; Olson, S.B.; Yang, F.; Anekpuritanang, T.; et al. Adult acute myeloid leukemia patients with NUP98 rearrangement have frequent cryptic translocations and unfavorable outcome. Leuk. Lymphoma 2022, 63, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Niktoreh, N.; Walter, C.; Zimmermann, M.; von Neuhoff, C.; von Neuhoff, N.; Rasche, M.; Waack, K.; Creutzig, U.; Hanenberg, H.; Reinhardt, D. Mutated WT1, FLT3-ITD, and NUP98-NSD1 Fusion in Various Combinations Define a Poor Prognostic Group in Pediatric Acute Myeloid Leukemia. J. Oncol. 2019, 2019, 1609128. [Google Scholar] [CrossRef] [Green Version]
- Ostronoff, F.; Othus, M.; Gerbing, R.B.; Loken, M.R.; Raimondi, S.C.; Hirsch, B.A.; Lange, B.J.; Petersdorf, S.; Radich, J.; Appelbaum, F.R.; et al. NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: A COG and SWOG report. Blood 2014, 124, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.M.; Hetzer, M.W. The role of NUP98 in transcription regulation in healthy and diseased cells. Trends Cell Biol. 2013, 23, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Yung, E.; Sekulovic, S.; Argiropoulos, B.; Lai, C.K.; Leung, M.; Berg, T.; Vollett, S.; Chang, V.C.; Wan, A.; Wong, S.; et al. Delineating domains and functions of NUP98 contributing to the leukemogenic activity of NUP98-HOX fusions. Leuk. Res. 2011, 35, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, R.M.; Aplan, P.D.; Humphries, R.K. NUP98-topoisomerase I acute myeloid leukemia-associated fusion gene has potent leukemogenic activities independent of an engineered catalytic site mutation. Blood 2004, 104, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
- Hirose, K.; Abramovich, C.; Argiropoulos, B.; Humphries, R.K. Leukemogenic properties of NUP98-PMX1 are linked to NUP98 and homeodomain sequence functions but not to binding properties of PMX1 to serum response factor. Oncogene 2008, 27, 6056–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, M.; Zhang, Q.; Liu, P.; Huang, J.; Wang, Y.; Chen, S. Inhibition of the nuclear export of p65 and IQCG in leukemogenesis by NUP98-IQCG. Front. Med. 2016, 10, 410–419. [Google Scholar] [CrossRef]
- Takeda, A.; Sarma, N.J.; Abdul-Nabi, A.M.; Yaseen, N.R. Inhibition of CRM1-mediated nuclear export of transcription factors by leukemogenic NUP98 fusion proteins. J. Biol. Chem. 2010, 285, 16248–16257. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.M.; Zhang, Q.Y.; Wang, Y.Y.; Liu, P.; Ren, R.B.; Huang, J.Y.; Chen, L.T.; Xi, X.D.; Chen, Z.; Chen, S.J. Human NUP98-IQCG fusion protein induces acute myelomonocytic leukemia in mice by dysregulating the Hox/Pbx3 pathway. Leukemia 2016, 30, 1590–1593. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.-H.; Keeley, D.P.; et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef]
- Chandra, B.; Michmerhuizen, N.L.; Shirnekhi, H.K.; Tripathi, S.; Pioso, B.J.; Baggett, D.W.; Mitrea, D.M.; Iacobucci, I.; White, M.R.; Chen, J.; et al. Phase Separation Mediates NUP98 Fusion Oncoprotein Leukemic Transformation. Cancer Discov. 2022, 12, 1152–1169. [Google Scholar] [CrossRef] [PubMed]
- Terlecki-Zaniewicz, S.; Humer, T.; Eder, T.; Schmoellerl, J.; Heyes, E.; Manhart, G.; Kuchynka, N.; Parapatics, K.; Liberante, F.G.; Müller, A.C.; et al. Biomolecular condensation of NUP98 fusion proteins drives leukemogenic gene expression. Nat. Struct. Mol. Biol. 2021, 28, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.J.; Christensen, J.; Fong, S.; Hu, Y.L.; Weissman, I.; Sauvageau, G.; Humphries, R.K.; Largman, C. Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 2005, 106, 3988–3994. [Google Scholar] [CrossRef] [Green Version]
- Pineault, N.; Helgason, C.D.; Lawrence, H.J.; Humphries, R.K. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp. Hematol. 2002, 30, 49–57. [Google Scholar] [CrossRef]
- Kroon, E.; Krosl, J.; Thorsteinsdottir, U.; Baban, S.; Buchberg, A.M.; Sauvageau, G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998, 17, 3714–3725. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Wang, G.G.; Song, J.; Wang, Z.; Dormann, H.L.; Casadio, F.; Li, H.; Luo, J.L.; Patel, D.J.; Allis, C.D. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009, 459, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. NUP98-PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guo, Y.; Gough, S.M.; Zhang, J.; Vann, K.R.; Li, K.; Cai, L.; Shi, X.; Aplan, P.D.; Wang, G.G.; et al. Mechanistic insights into chromatin targeting by leukemic NUP98-PHF23 fusion. Nat. Commun. 2020, 11, 3339. [Google Scholar] [CrossRef]
- Kroon, E.; Thorsteinsdottir, U.; Mayotte, N.; Nakamura, T.; Sauvageau, G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J. 2001, 20, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Pineault, N.; Abramovich, C.; Humphries, R.K. Transplantable cell lines generated with NUP98-Hox fusion genes undergo leukemic progression by Meis1 independent of its binding to DNA. Leukemia 2005, 19, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Pineault, N.; Buske, C.; Feuring-Buske, M.; Abramovich, C.; Rosten, P.; Hogge, D.E.; Aplan, P.D.; Humphries, R.K. Induction of acute myeloid leukemia in mice by the human leukemia-specific fusion gene NUP98-HOXD13 in concert with Meis1. Blood 2003, 101, 4529–4538. [Google Scholar] [CrossRef]
- Calvo, K.R.; Sykes, D.B.; Pasillas, M.P.; Kamps, M.P. Nup98-HoxA9 immortalizes myeloid progenitors, enforces expression of Hoxa9, Hoxa7 and Meis1, and alters cytokine-specific responses in a manner similar to that induced by retroviral co-expression of Hoxa9 and Meis1. Oncogene 2002, 21, 4247–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salsi, V.; Ferrari, S.; Gorello, P.; Fantini, S.; Chiavolelli, F.; Mecucci, C.; Zappavigna, V. NUP98 Fusion Oncoproteins Promote Aneuploidy by Attenuating the Mitotic Spindle Checkpoint. Cancer Res. 2014, 74, 1079–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricke, R.M.; van Ree, J.H.; van Deursen, J.M. Whole chromosome instability and cancer: A complex relationship. Trends Genet. TIG 2008, 24, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salsi, V.; Fantini, S.; Zappavigna, V. NUP98 fusion oncoproteins interact with the APC/C(Cdc20) as a pseudosubstrate and prevent mitotic checkpoint complex binding. Cell Cycle 2016, 15, 2275–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taketani, T.; Taki, T.; Nakamura, T.; Kobayashi, Y.; Ito, E.; Fukuda, S.; Yamaguchi, S.; Hayashi, Y. High frequencies of simultaneous FLT3-ITD, WT1 and KIT mutations in hematological malignancies with NUP98-fusion genes. Leukemia 2010, 24, 1975–1977. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Zhou, L.; Li, Y.; Yang, E.; Liu, Y.; Lv, N.; Fu, L.; Ding, Y.; Wang, N.; Fang, N.; et al. Profiling of somatic mutations and fusion genes in acute myeloid leukemia patients with FLT3-ITD or FLT3-TKD mutation at diagnosis reveals distinct evolutionary patterns. Exp. Hematol. Oncol. 2021, 10, 27. [Google Scholar] [CrossRef]
- Fang, Y.; Han, X.; Shen, L.; Hou, J. NUP98-HOXA9 Bearing Acute Myeloid Leukemia. Blood 2022, 140 (Suppl. S1), 11614. [Google Scholar] [CrossRef]
- Chou, W.C.; Chen, C.Y.; Hou, H.A.; Lin, L.I.; Tang, J.L.; Yao, M.; Tsay, W.; Ko, B.S.; Wu, S.J.; Huang, S.Y.; et al. Acute myeloid leukemia bearing t(7;11)(p15;p15) is a distinct cytogenetic entity with poor outcome and a distinct mutation profile: Comparative analysis of 493 adult patients. Leukemia 2009, 23, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Lavallée, V.P.; Lemieux, S.; Boucher, G.; Gendron, P.; Boivin, I.; Girard, S.; Hébert, J.; Sauvageau, G. Identification of MYC mutations in acute myeloid leukemias with NUP98–NSD1 translocations. Leukemia 2016, 30, 1621–1624. [Google Scholar] [CrossRef]
- Cui, J.; Xie, J.; Qin, L.; Chen, S.; Zhao, Y.; Wu, D. A unique acute myeloid leukemia patient with cryptic NUP98-NSD1 gene and ASXL1 mutation. Leuk. Lymphoma 2016, 57, 196–198. [Google Scholar] [CrossRef]
- Kelly, L.M.; Gilliland, D.G. Genetics of myeloid leukemias. Annu. Rev. Genom. Hum. Genet. 2002, 3, 179–198. [Google Scholar] [CrossRef]
- Takahashi, S. Current findings for recurring mutations in acute myeloid leukemia. J. Hematol. Oncol. 2011, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, H.G.; Popplewell, L.; Tcheurekdjian, L.; Slovak, M.L. NUP98 gene rearrangements and the clonal evolution of chronic myelogenous leukemia. Genes Chromosomes Cancer 2001, 30, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Nakamura, Y.; Nakamura, Y.; Saito, K.; Furusawa, S. Expression of the NUP98/HOXA9 fusion transcript in the blast crisis of Philadelphia chromosome-positive chronic myelogenous leukaemia with t(7;11)(p15;p15). Br. J. Haematol. 2000, 109, 423–426. [Google Scholar] [CrossRef]
- Dash, A.B.; Williams, I.R.; Kutok, J.L.; Tomasson, M.H.; Anastasiadou, E.; Lindahl, K.; Li, S.; Van Etten, R.A.; Borrow, J.; Housman, D.; et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proc. Natl. Acad. Sci. USA 2002, 99, 7622–7627. [Google Scholar] [CrossRef]
- Di Giacomo, D.; Pierini, V.; Barba, G.; Ceccarelli, V.; Vecchini, A.; Mecucci, C. Blast crisis Ph+ chronic myeloid leukemia with NUP98/HOXA13 up-regulating MSI2. Mol. Cytogenet. 2014, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kakihana, K.; Kurosu, T.; Murakami, N.; Miura, O. Clonal evolution with inv(11)(p15q22) and NUP98/DDX10 fusion gene in imatinib-resistant chronic myelogenous leukemia. Cancer Genet. Cytogenet. 2005, 157, 104–108. [Google Scholar] [CrossRef]
- Mohanty, S.; Heuser, M. Mouse Models of Frequently Mutated Genes in Acute Myeloid Leukemia. Cancers 2021, 13, 6192. [Google Scholar] [CrossRef]
- Greenblatt, S.; Li, L.; Slape, C.; Nguyen, B.; Novak, R.; Duffield, A.; Huso, D.; Desiderio, S.; Borowitz, M.J.; Aplan, P.; et al. Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood 2012, 119, 2883–2894. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.; Jyotsana, N.; Sharma, A.; Kloos, A.; Gabdoulline, R.; Othman, B.; Lai, C.K.; Schottmann, R.; Mandhania, M.; Schmoellerl, J.; et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia. Cancers 2020, 12, 2766. [Google Scholar] [CrossRef] [PubMed]
- Thanasopoulou, A.; Tzankov, A.; Schwaller, J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica 2014, 99, 1465–1471. [Google Scholar] [CrossRef] [Green Version]
- Matsukawa, T.; Yin, M.; Nigam, N.; Negi, V.; Li, L.; Small, D.; Zhu, Y.J.; Walker, R.L.; Meltzer, P.S.; Aplan, P.D. NUP98::Nsd1 and FLT3-ITD collaborate to generate acute myeloid leukemia. Leukemia 2023, 37, 1545–1548. [Google Scholar] [CrossRef]
- Nakamura, T. Retroviral insertional mutagenesis identifies oncogene cooperation. Cancer Sci. 2005, 96, 7–12. [Google Scholar] [CrossRef]
- Slape, C.; Hartung, H.; Lin, Y.W.; Bies, J.; Wolff, L.; Aplan, P.D. Retroviral insertional mutagenesis identifies genes that collaborate with NUP98-HOXD13 during leukemic transformation. Cancer Res. 2007, 67, 5148–5155. [Google Scholar] [CrossRef] [Green Version]
- Slape, C.; Liu, L.Y.; Beachy, S.; Aplan, P.D. Leukemic transformation in mice expressing a NUP98-HOXD13 transgene is accompanied by spontaneous mutations in Nras, Kras, and Cbl. Blood 2008, 112, 2017–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, M.; Tallman, M.S. Acute promyelocytic leukemia (APL): Remaining challenges towards a cure for all. Leuk. Lymphoma 2019, 60, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Stegmaier, K. Targeted therapy for fusion-driven high-risk acute leukemia. Blood 2018, 132, 1241–1247. [Google Scholar] [CrossRef] [Green Version]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Budida, R.; Scherr, M.; Kuchenbauer, F.; Lindner, R.; Noyan, F.; Sühs, K.W.; Stangel, M.; et al. Lipid nanoparticle-mediated siRNA delivery for safe targeting of human CML in vivo. Ann. Hematol. 2019, 98, 1905–1918. [Google Scholar] [CrossRef]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Scherr, M.; Kuchenbauer, F.; Sajti, L.; Barchanski, A.; Lindner, R.; Noyan, F.; Sühs, K.W.; et al. RNA interference efficiently targets human leukemia driven by a fusion oncogene in vivo. Leukemia 2018, 32, 224–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, H.; Swart, L.E.; Rasouli, M.; Ashtiani, M.; Nakjang, S.; Jyotsana, N.; Schuschel, K.; Heuser, M.; Blair, H.; Heidenreich, O. Nanoparticle-mediated targeting of the fusion gene RUNX1/ETO in t(8;21)-positive acute myeloid leukaemia. Leukemia 2023, 37, 820–834. [Google Scholar] [CrossRef]
- Schmoellerl, J.; Barbosa, I.A.M.; Eder, T.; Brandstoetter, T.; Schmidt, L.; Maurer, B.; Troester, S.; Pham, H.T.T.; Sagarajit, M.; Ebner, J.; et al. CDK6 is an essential direct target of NUP98 fusion proteins in acute myeloid leukemia. Blood 2020, 136, 387–400. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673.e11. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rücker, F.G.; Döhner, K.; McGeehan, G.M.; et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Pommert, L.; Tarlock, K. The evolution of targeted therapy in pediatric AML: Gemtuzumab ozogamicin, FLT3/IDH/BCL2 inhibitors, and other therapies. Hematology 2022, 2022, 603–610. [Google Scholar] [CrossRef]
- Heikamp, E.B.; Henrich, J.A.; Perner, F.; Wong, E.M.; Hatton, C.; Wen, Y.; Barwe, S.P.; Gopalakrishnapillai, A.; Xu, H.; Uckelmann, H.J.; et al. The menin-MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood 2022, 139, 894–906. [Google Scholar] [CrossRef]
- Issa, G.C.; Ravandi, F.; DiNardo, C.D.; Jabbour, E.; Kantarjian, H.M.; Andreeff, M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia 2021, 35, 2482–2495. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 2023, 615, 920–924. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Mura, S.; Yamada, K.; Sangel, P.; Hirata, S.; Maehara, K.; Kawakami, K.; Tachibana, T.; Ohkawa, Y.; Kimura, H.; et al. Chromatin-prebound Crm1 recruits Nup98-HoxA9 fusion to induce aberrant expression of Hox cluster genes. eLife 2016, 5, e09540. [Google Scholar] [CrossRef]
- Ren, Z.; Kim, A.; Huang, Y.T.; Pi, W.C.; Gong, W.; Yu, X.; Qi, J.; Jin, J.; Cai, L.; Roeder, R.G.; et al. A PRC2-Kdm5b axis sustains tumorigenicity of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2022, 119, e2122940119. [Google Scholar] [CrossRef]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- Straining, R.; Eighmy, W. Tazemetostat: EZH2 Inhibitor. J. Adv. Pract. Oncol. 2022, 13, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Kivioja, J.L.; Thanasopoulou, A.; Kumar, A.; Kontro, M.; Yadav, B.; Majumder, M.M.; Javarappa, K.K.; Eldfors, S.; Schwaller, J.; Porkka, K.; et al. Dasatinib and navitoclax act synergistically to target NUP98-NSD1(+)/FLT3-ITD(+) acute myeloid leukemia. Leukemia 2019, 33, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Doki, N.; Aoki, J.; Mori, J.; Machida, S.; Masuko, M.; Uchida, N.; Najima, Y.; Fukuda, T.; Kanamori, H.; et al. Outcomes after allogeneic hematopoietic stem cell transplantation in patients with acute myeloid leukemia harboring t(7;11)(p15;p15). Haematologica 2018, 103, e69–e72. [Google Scholar] [CrossRef] [Green Version]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, T.; Song, X.; Liu, B.; Wei, J. Gene fusion neoantigens: Emerging targets for cancer immunotherapy. Cancer Lett. 2021, 506, 45–54. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Imamura, T.; Tanaka, S.; Urata, T.; Yoshida, H.; Shiba, N.; Iehara, T. The Nup98::Nsd1 fusion gene induces CD123 expression in 32D cells. Int. J. Hematol. 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Thiollier, C.; Lopez, C.K.; Gerby, B.; Ignacimouttou, C.; Poglio, S.; Duffourd, Y.; Guégan, J.; Rivera-Munoz, P.; Bluteau, O.; Mabialah, V.; et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J. Exp. Med. 2012, 209, 2017–2031. [Google Scholar] [CrossRef]
- Mohanty, S.; Jyotsana, N.; Sharma, A.; Othman, B.; Kloos, A.; Mandhania, M.; Schottmann, R.; Ramsay, E.; Vornlocher, H.-P.; Ganser, A.; et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in AML. Blood 2019, 134, 2545. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of general functions of important NUP98 protein motifs. GLEBS motif of NUP98 aids nuclear export of mRNAs. GLFG repeats of NUP98 protein are required for multiple functions. It binds to IMPORTIN-B family members for nuclear import, and it binds to CRM1/XPO1 for nuclear export of proteins. By interacting with HDAC and CBP/p300, it drives gene expression. The figure was created with BioRender.com.
Figure 1.
Schematic representation of general functions of important NUP98 protein motifs. GLEBS motif of NUP98 aids nuclear export of mRNAs. GLFG repeats of NUP98 protein are required for multiple functions. It binds to IMPORTIN-B family members for nuclear import, and it binds to CRM1/XPO1 for nuclear export of proteins. By interacting with HDAC and CBP/p300, it drives gene expression. The figure was created with BioRender.com.

Figure 2.
Fusion partners of NUP98 in AML. NUP98 fusion partners can be divided into three groups. In the first group, NUP98 has a transcription factor as a fusion partner. In the second group, NUP98 has an epigenetic regulator as a fusion partner. In the second group, the star marked LEDGF is a transcriptional coactivator. The last group includes fusion partners of NUP98 that have neither transcription factor nor epigenetic regulation properties.
Figure 2.
Fusion partners of NUP98 in AML. NUP98 fusion partners can be divided into three groups. In the first group, NUP98 has a transcription factor as a fusion partner. In the second group, NUP98 has an epigenetic regulator as a fusion partner. In the second group, the star marked LEDGF is a transcriptional coactivator. The last group includes fusion partners of NUP98 that have neither transcription factor nor epigenetic regulation properties.

Figure 3.
Mechanism of leukemic transformation by NUP98 fusions. Epigenetic and transcriptional changes that occur when hematopoietic stem and progenitor cells (HSPCs) are transformed using an NUP98 fusion oncogene. The figure was created with BioRender.com.
Figure 3.
Mechanism of leukemic transformation by NUP98 fusions. Epigenetic and transcriptional changes that occur when hematopoietic stem and progenitor cells (HSPCs) are transformed using an NUP98 fusion oncogene. The figure was created with BioRender.com.

Figure 4.
Generalized model of AML development by NUP98 fusions in cooperation with other oncogenic abnormalities. The BCR::ABL oncogene causes a CML-like phenotype and undergoes a CML blast crisis (AML phenotype) when it acquires a NUP98 fusion oncogene. The NUP98 fusion oncogene shows the MDS or MPN phenotype and transforms into AML when it acquires mutations in signaling genes. The figure was created with BioRender.com.
Figure 4.
Generalized model of AML development by NUP98 fusions in cooperation with other oncogenic abnormalities. The BCR::ABL oncogene causes a CML-like phenotype and undergoes a CML blast crisis (AML phenotype) when it acquires a NUP98 fusion oncogene. The NUP98 fusion oncogene shows the MDS or MPN phenotype and transforms into AML when it acquires mutations in signaling genes. The figure was created with BioRender.com.

Figure 5.
Potential therapeutic options for leukemia with NUP98 rearrangements. NUP98 fusions can be directly targeted using siRNA-LNP formulations. The downstream targets of NUP98 fusion like CDK6 can be inhibited by a small molecule inhibitor. Further CRM1/XPO1, MEN1, or PRC2 inhibitors can be used to eradicate NUP98 fusion driven leukemia. A small-molecule inhibitor can be used to inhibit the FLT3-ITD mutation that is commonly associated with NUP98 rearrangements. The figure was created with BioRender.com.
Figure 5.
Potential therapeutic options for leukemia with NUP98 rearrangements. NUP98 fusions can be directly targeted using siRNA-LNP formulations. The downstream targets of NUP98 fusion like CDK6 can be inhibited by a small molecule inhibitor. Further CRM1/XPO1, MEN1, or PRC2 inhibitors can be used to eradicate NUP98 fusion driven leukemia. A small-molecule inhibitor can be used to inhibit the FLT3-ITD mutation that is commonly associated with NUP98 rearrangements. The figure was created with BioRender.com.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mohanty, S. NUP98 Rearrangements in AML: Molecular Mechanisms and Clinical Implications. Onco 2023, 3, 147-164. https://doi.org/10.3390/onco3030011
AMA Style
Mohanty S. NUP98 Rearrangements in AML: Molecular Mechanisms and Clinical Implications. Onco. 2023; 3(3):147-164. https://doi.org/10.3390/onco3030011
Chicago/Turabian StyleMohanty, Sagarajit. 2023. "NUP98 Rearrangements in AML: Molecular Mechanisms and Clinical Implications" Onco 3, no. 3: 147-164. https://doi.org/10.3390/onco3030011