
Survey of T1 and T2 Energies of Intramolecular Singlet Fission Chromophores


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Details
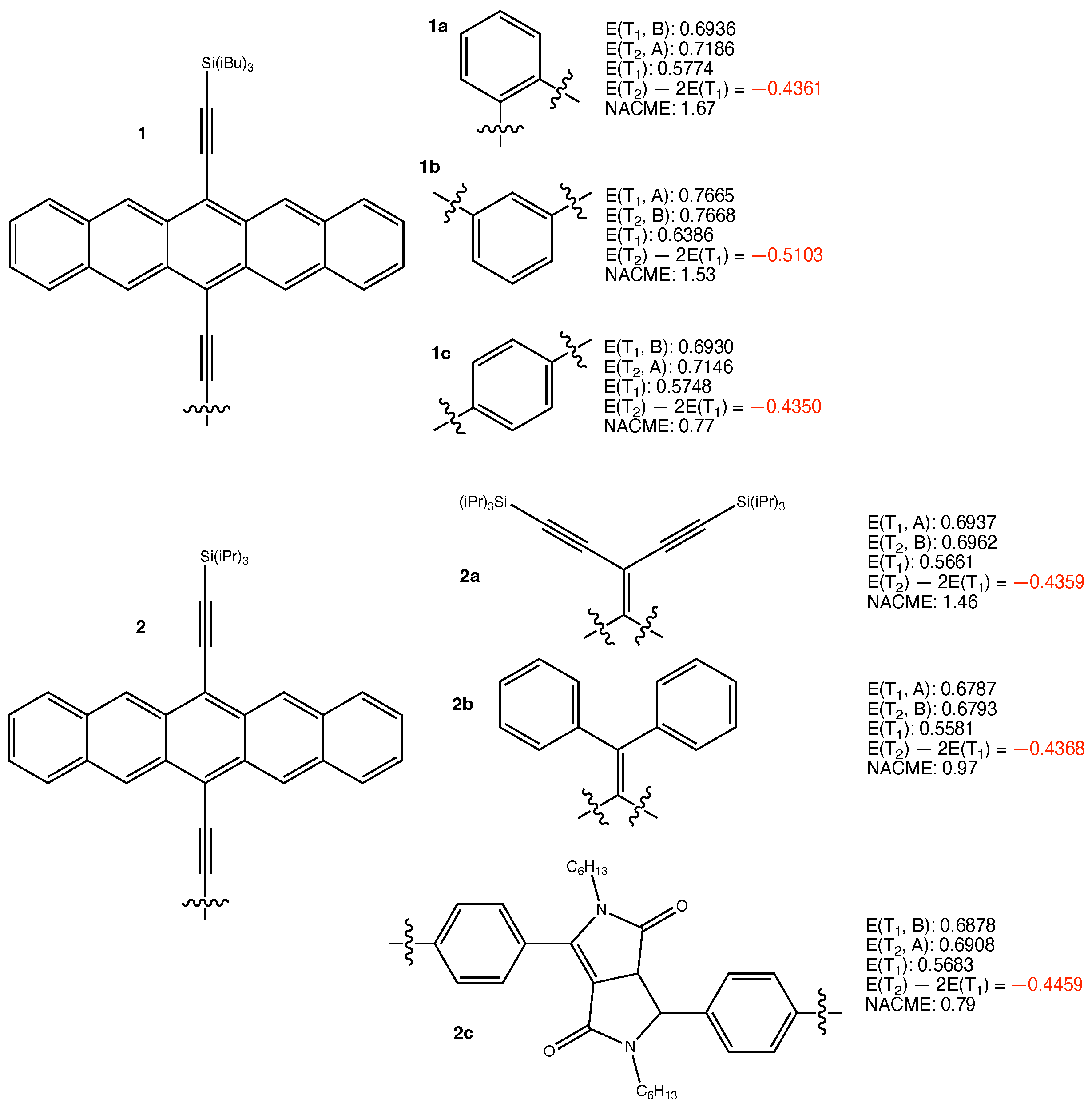
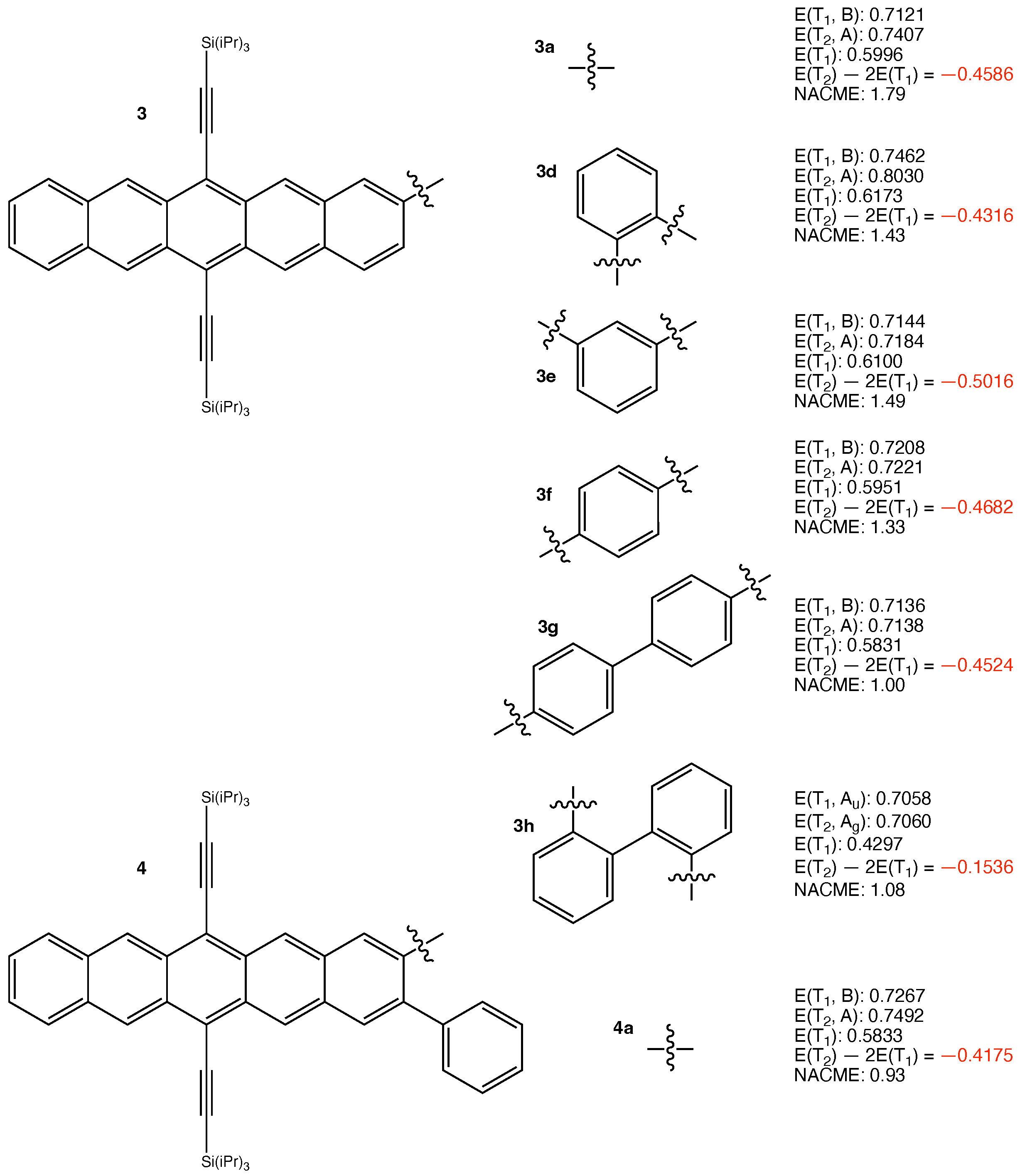
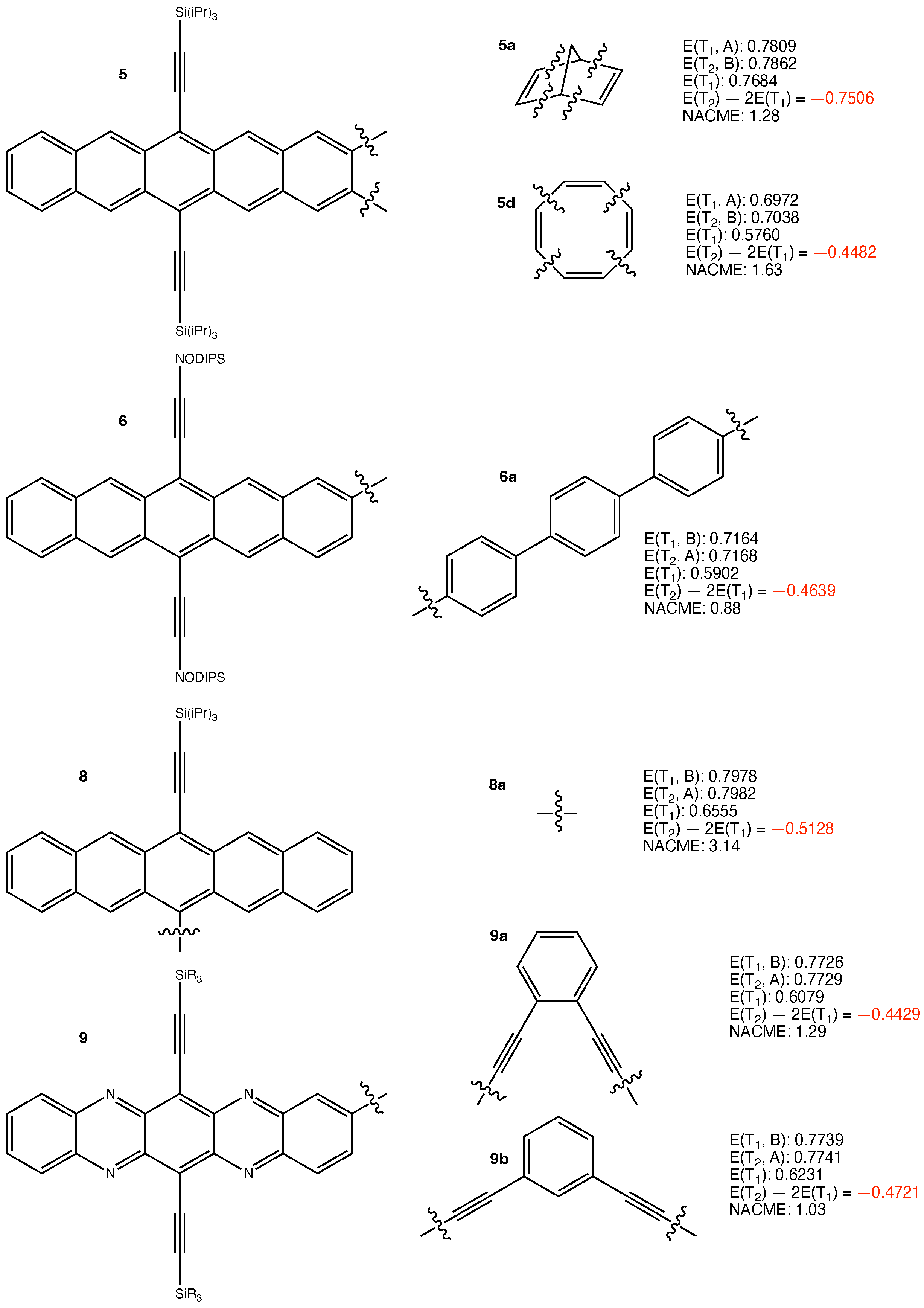
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SF | Singlet fission |
| iSF | Intramolecular singlet fission |
| TF | Triplet fusion |
| TC | Triplet-pair concentration |
| SF-TDDFT | Spin-flip time-dependent density functional theory |
References
- Smith, M.B.; Michl, J. Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B.; Michl, J. Recent Advances in Singlet Fission. Annu. Rev. Phys. Chem. 2013, 64, 361–386. [Google Scholar] [CrossRef]
- Michl, J. Unconventional Solar Energy: Singlet Fission. Mol. Front. J. 2019, 3, 84–91. [Google Scholar] [CrossRef]
- Shockley, W.; Queisser, H.J. Detailed Balance Limit of Efficiency of P-N Junction Solar Cells. J. Appl. Phys. 1961, 32, 510–519. [Google Scholar] [CrossRef]
- Hanna, M.C.; Nozik, A.J. Solar Conversion Efficiency of Photovoltaic and Photoelectrolysis Cells with Carrier Multiplication Absorbers. J. Appl. Phys. 2006, 100, 074510. [Google Scholar] [CrossRef]
- Tayebjee, M.J.Y.; McCamey, D.R.; Schmidt, T.W. Beyond Shockley–Queisser: Molecular Approaches to High-Efficiency Photovoltaics. J. Phys. Chem. Lett. 2015, 6, 2367–2378. [Google Scholar] [CrossRef]
- Singh, S.; Jones, W.J.; Siebrand, W.; Stoicheff, B.P.; Schneider, W.G. Laser generation of excitons and fluorescence in anthracene crystals. J. Chem. Phys. 1965, 42, 330–342. [Google Scholar] [CrossRef]
- Merrifield, R.E. Theory of magnetic field effects on the mutual annihilation of triplet excitons. J. Chem. Phys. 1968, 48, 4318. [Google Scholar] [CrossRef]
- Swenberg, C.E.; Stacy, W.T. Bimolecular radiationless transitions in crystalline tetracene. Chem. Phys. Lett. 1968, 2, 327–328. [Google Scholar] [CrossRef]
- Groff, R.P.; Avakian, P.; Merrifield, R.E. Coexistence of exciton fission and fusion in tetracene crystals. Phys. Rev. B 1970, 1, 815–817. [Google Scholar] [CrossRef]
- Johnson, R.; Merrifield, R. Effects of magnetic fields on the mutual annihilation of triplet excitons in anthracene crystals. Phys. Rev. B 1970, 1, 896–902. [Google Scholar] [CrossRef]
- Paci, I.; Johnson, J.C.; Chen, X.D.; Rana, G.; Popović, D.; David, D.E.; Nozik, A.J.; Ratner, M.A.; Michl, J. Singlet Fission for Dye-Sensitizerd Solar Cells: Can a Suitable Sensitizer be Found? J. Am. Chem. Soc. 2006, 128, 16546–16553. [Google Scholar] [CrossRef] [PubMed]
- Schwerin, A.F.; Johnson, J.C.; Smith, M.B.; Sreearunothai, P.; Popović, D.; Černý, J.; Havlas, Z.; Paci, I.; Akdag, A.; MacLeod, M.K.; et al. Toward Designed Singlet Fission: Electronic States and Photophysics of 1,3-Diphenylisobenzofuran. J. Phys. Chem. A 2010, 114, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Greyson, E.C.; Stepp, B.R.; Chen, X.; Schwerin, A.F.; Paci, I.; Smith, M.B.; Akdag, A.; Johnson, J.C.; Nozik, A.J.; Michl, J.; et al. Singlet Exciton Fission for Solar Cell Applications: Energy Aspects of Interchromophore Coupling. J. Phys. Chem. B 2010, 114, 14223–14232. [Google Scholar] [CrossRef] [PubMed]
- Zirzlmeier, J.; Lehnherr, D.; Coto, P.B.; Chernick, E.T.; Casillas, R.; Basel, B.S.; Thoss, M.; Tykwinski, R.R.; Guldi, D.M. Singlet Fission in Pentacene Dimers. Proc. Natl. Acad. Sci. USA 2015, 112, 5325–5330. [Google Scholar] [CrossRef]
- Zirzlmeier, J.; Casillas, R.; Reddy, S.R.; Coto, P.B.; Lehnherr, D.; Chernick, E.T.; Papadopoulos, I.; Thoss, M.; Tykwinski, R.R.; Guldi, D.M. Solution-Based Intramolecular Singlet Fission in Cross-Conjugated Pentacene Dimers. Nanoscale 2016, 8, 10113–10123. [Google Scholar] [CrossRef]
- Hertzer, C.; Basel, B.S.; Kopp, S.M.; Hampel, F.; White, F.J.; Clark, T.; Guldi, D.M.; Tykwinski, R.R. Chromophore Multiplication to enable exciton delocalization and triplet diffusion following singlet fission in tetrameric pentacene. Angew. Chem. Int. Ed. 2019, 58, 115263–115267. [Google Scholar]
- Chien, A.D.; Molina, A.R.; Abeyasinghe, N.; Varnavski, O.P.; Goodson, T.; Zimmerman, P.M. Structure and Dynamics fo the 1(TT) State in a Quinoidal Bithiophene: Characterizing a Promising Intramolecular Singlet Fission Candidate. J. Phys. Chem. C 2015, 119, 28258–28268. [Google Scholar] [CrossRef]
- Basel, B.S.; Young, R.M.; Krzyaniak, M.D.; Papadopoulos, L.; Hetzer, C.; Gao, Y.; La Porte, N.T.; Phelan, B.T.; Clark, T.; Tykwinski, R.R.; et al. Influence of the Heavy-Atom Effect on Singlet Fission: A Study of Platinum-Bridged Pentacene Dimers. Chem. Sci. 2019, 10, 11130–11140. [Google Scholar] [CrossRef]
- Huang, Z.; Fujihashi, Y.; Zhao, Y. Effects of Off-Diagonal Exciton-Phonon Coupling on Intramolecular Singlet Fission. J. Phys. Chem. Lett. 2017, 8, 3306–3312. [Google Scholar] [CrossRef]
- Pradhan, E.; Zeng, T. Triplet Separation after the Fastest Intramolecular Singlet Fission in the Smallest Chromophore. J. Chem. Theory Comput. 2023, 19, 2092–2101. [Google Scholar] [CrossRef]
- Pradhan, E.; Zeng, T. Design of the Smallest Intramolecular Singlet Fission Chromophore with the Fastest Singlet Fission. J. Phys. Chem. Lett. 2022, 13, 11076–11085. [Google Scholar] [CrossRef]
- Zeng, T.; Goel, P. Design of Small Intramolecular Singlet Fission Chromophores: An Azaborine Candidate and General Small Size Effects. J. Phys. Chem. Lett. 2016, 7, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Chen, J.; Zhang, G.; Yao, J.; Zhang, D.; Fu, H. Intramolecular Singlet Fission in an Antiaromatic Polycyclic Hydrocarbon. Angew. Chem. Int. Ed. 2017, 56, 9400–9404. [Google Scholar] [CrossRef] [PubMed]
- Varnavski, O.; Abeyasinghe, N.; Aragó, J.; Serrano-Pérez, J.J.; Ortí, E.; López Navarrete, J.T.; Takimiya, K.; Casanova, D.; Casado, J.; Goodson, T. High Yield Ultrafast Intramolecular Singlet Exciton Fission in a Quinoidal Bithiiophene. J. Phys. Chem. Lett. 2015, 6, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T. Through-Linker Intramolecular Singlet Fission: General Mechanism and Designing Small Chromophores. J. Phys. Chem. Lett. 2016, 7, 4405–4412. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.R.; Coto, P.B.; Thoss, M. Intramolecular Singlet Fission: Insights from Quantum Dynamical Simulations. J. Phys. Chem. Lett. 2018, 9, 5979–5986. [Google Scholar] [CrossRef]
- Sanders, S.N.; Kumarasamy, E.; Pun, A.B.; Appavoo, K.; Steigerwald, M.L.; Campos, L.M.; Sfeir, M.Y. Exciton Correlations in Intramolecular Singlet Fission. J. Am. Chem. Soc. 2016, 138, 7289–7297. [Google Scholar] [CrossRef]
- Sanders, S.N.; Kumarasamy, E.; Pun, A.B.; Trinh, M.T.; Choi, B.; Xia, J.; Taffet, E.J.; Low, J.Z.; Miller, J.R.; Roy, X.; et al. Quantitative Intramolecular Singlet Fission in Bipentacene. J. Am. Chem. Soc. 2015, 137, 8965–8972. [Google Scholar] [CrossRef]
- Pun, A.B.; Asadpoordarvish, A.; Kumarasamy, E.; Tayebjee, M.J.Y.; Niesner, D.; McCamey, D.R.; Sanders, S.N.; Campos, L.M.; Sfeir, M.Y. Ultra-fast intramolecular singlet fission to persistent multiexcitons by molecular design. Nat. Chem. 2019, 11, 821–828. [Google Scholar] [CrossRef]
- Ito, S.; Nagami, T.; Nakano, M. Design Principles of Electronic Couplings for Intramolecular Singlet Fission in Covalently Linked Systems. J. Phys. Chem. A 2016, 120, 6236–6241. [Google Scholar] [CrossRef] [PubMed]
- Momenti, M.R. Intramolecular Singlet Fission in Quinoidal Bi- and Tetrathiophenes: A Comparative Study of Low-Lying Excited Electronic States and Potential Energy Surfaces. J. Chem. Theory Comput. 2016, 12, 5067–5075. [Google Scholar] [CrossRef]
- Margulies, E.A.; Miller, C.E.; Wu, Y.; Ma, L.; Schatz, G.C.; Young, R.M.; Wasielewski, M.R. Enabling Singlet Fission by Controlling Intramolecular Charge Transfer in π-Stacked Covalent Terrylenediimide Dimers. Nat. Chem. 2016, 8, 1120–1125. [Google Scholar] [CrossRef]
- Basel, B.S.; Zirzlmeier, J.; Hetzer, C.; Phelan, B.T.; Krzyaniak, M.D.; Reddy, S.R.; Coto, P.B.; Horwitz, N.E.; Young, R.M.; White, F.J.; et al. Unified Model for Singlet Fission within a Non-Conjugated Covalent Pentacene Dimer. Nat. Commun. 2017, 8, 15171. [Google Scholar] [CrossRef] [PubMed]
- Krishnapriya, K.C.; Musser, A.J.; Patil, S. Molecular Design Strategies for Efficient Intramolecular Singlet Exciton Fission. ACS Energy Lett. 2019, 4, 192–202. [Google Scholar] [CrossRef]
- Hasobe, T.; Nakamura, S.; Tkachenko, N.V.; Kobori, Y. Molecular design strategy for high-yield and long-lived individual doubled triplet excitons through intramolecular singlet fission. ACS Energy Lett. 2022, 7, 390–400. [Google Scholar] [CrossRef]
- Lin, H.H.; Kue, K.Y.; Claudio, G.C.; Hsu, C.P. First Principle Prediction of Intramolecular Singlet Fission and Triplet Triplet Annihilation Rates. J. Chem. Theory Comput. 2019, 15, 2246–2253. [Google Scholar] [CrossRef]
- Fuemmeler, E.G.; Sanders, S.N.; Pun, A.B.; Kumarasamy, E.; Zeng, T.; Miyata, K.; Steigerwald, M.L.; Zhu, X.Y.; Sfeir, M.Y.; Campos, L.M.; et al. A Direct Mechanism of Ultrafast Intramolecular Singlet Fission in Pentacene Dimers. ACS Cent. Sci. 2016, 2, 316–324. [Google Scholar] [CrossRef]
- Busby, E.; Xia, J.; Wu, Q.; Low, J.Z.; Song, R.; Miller, J.R.; Zhu, X.Y.; Campos, L.M.; Sfeir, M.Y. A Design Strategy for Intramolecular Singlet Fission Mediated by Charge-Transfer States in Donor–Acceptor Organic Materials. Nat. Mater. 2015, 14, 426–433. [Google Scholar] [CrossRef]
- Kefer, O.; Ahrens, L.; Han, J.; Wollscheid, N.; Misselwitz, E.; Rominger, F.; Freudenberg, J.; Dreuw, A.; Bunz, U.H.F.; Buckup, T. Efficient Intramolecular Singlet Fission in Spiro-Linked Heterodimers. J. Am. Chem. Soc. 2023, 145, 17965–17974. [Google Scholar] [CrossRef]
- James, D.; Pradhan, E.; Zeng, T. Design of Singlet Fission Chromophores by the Introduction of N-Oxyl Fragments. J. Chem. Phys. 2022, 156, 034303. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, I.; Reddy, S.R.; Coto, P.B.; Lehnherr, D.; Thiel, D.; Thoss, M.; Tykwinski, R.R.; Guldi, D.M. Parallel versus Twisted Pentacenes: Conformational Impact on Singlet Fission. J. Phys. Chem. Lett. 2022, 13, 5094–5100. [Google Scholar] [CrossRef] [PubMed]
- Tayebjee, M.J.Y.; Sanders, S.N.; Kumarasamy, E.; Campos, L.M.; Sfeir, M.Y.; McCamey, D.R. Quintet Multiexciton Dynamics in Singlet Fission. Nat. Phys. 2017, 13, 182–188. [Google Scholar] [CrossRef]
- Scholes, G.D. Correlated Pair States Formed by Singlet Fission and Exciton–Exciton Annihilation. J. Phys. Chem. A 2015, 119, 12699–12705. [Google Scholar] [CrossRef]
- Minami, T.; Ito, S.; Nakano, M. Theoretical Study of Singlet Fission in Oligorylenes. J. Phys. Chem. Lett. 2012, 3, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Minami, T.; Nakano, M. Diradical Character Based Design for Singlet Fission of Condensed-Ring Systems with 4nπ Electrons. J. Phys. Chem. C 2012, 116, 19729–19736. [Google Scholar] [CrossRef]
- Minami, T.; Nakano, M. Diradical Character View of Singlet Fission. J. Phys. Chem. Lett. 2012, 3, 145–150. [Google Scholar] [CrossRef]
- Minami, T.; Ito, S.; Nakano, M. Fundamental of Diradical-Character-Based Molecular Design for Singlet Fission. J. Phys. Chem. Lett. 2013, 4, 2133–2137. [Google Scholar] [CrossRef]
- Zeng, T.; Hoffmann, R.; Ananth, N. The Low-Lying Electronic States of Pentacene and Their Roles in Singlet Fission. J. Am. Chem. Soc. 2014, 136, 5755–5764. [Google Scholar] [CrossRef]
- Zeng, T.; Ananth, N.; Hoffmann, R. Seeking Small Molecules for Singlet Fission: A Heteroatom Substitution Strategy. J. Am. Chem. Soc. 2014, 136, 12638–12647. [Google Scholar] [CrossRef]
- Japahuge, A.; Zeng, T. Theoretical Studies of Singlet Fission: Searching for Materials and Exploring Mechanisms. ChemPlusChem 2018, 83, 146–182. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Mellerup, S.K.; Yang, D.; Wang, X.; Wang, S.; Stamplecoskie, K. Identifying (BN)2-pyrenes as a New Class of Singlet Fission Chromophores: Significance of Azaborine Substitution. J. Phys. Chem. Lett. 2018, 9, 2919–2927. [Google Scholar] [CrossRef] [PubMed]
- Japahuge, A.; Lee, S.; Choi, C.H.; Zeng, T. Design of Singlet Fission Chromophores with Cyclic (Alkyl)(Amino) Carbene Building Blocks. J. Chem. Phys. 2019, 150, 234306. [Google Scholar] [CrossRef]
- Pradhan, E.; Bentley, J.N.; Caputo, C.B.; Zeng, T. Designs of Singlet Fission Chromophores with a Diazadiborinine Framework. ChemPhotoChem 2020, 4, 5279–5287. [Google Scholar] [CrossRef]
- Pradhan, E.; Lee, S.; Choi, C.H.; Zeng, T. Diboron- and Diaza-Doped Anthracenes and Phenanthrenes: Their Electronic Structures for Being Singlet Fission Chromophores. J. Phys. Chem. A 2020, 124, 8159–8172. [Google Scholar] [CrossRef]
- James, D.; Pradhan, E.; Lee, S.; Choi, C.H.; Zeng, T. Dicarbonyl Anthracenes and Phenanthrenes as Singlet Fission Chromophores. Can. J. Chem. 2022, 100, 520–529. [Google Scholar] [CrossRef]
- Shaik, S.; Hiberty, P.C. A Chemist’s Guide to Valence Bond Theory; Wiley-Interscience: Hoboken, NJ, USA, 2008. [Google Scholar]
- Padula, D.; Omar, Ö.H.; Nematiaram, T.; Troisi, A. Singlet Fission Molecules among Known Compounds: Finding a Few Needles in a Hay Stack. Energy Environ. Sci. 2019, 12, 2412–2416. [Google Scholar] [CrossRef]
- Korovina, N.V.; Pompetti, N.F.; Johnson, J.C. Lessons from Intramolecular Singlet Fission with Covalently Bound Chromophores. J. Chem. Phys. 2020, 152, 040904. [Google Scholar] [CrossRef]
- Bersuker, I.B. The Jahn-Teller Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Brown, J.; Lang, R.A.; Zeng, T. Unified Hamiltonian formalism of Jahn-Teller and pseudo-Jahn-Teller problems in axial symmetries. J. Chem. Theory Comput. 2021, 17, 4392–4402. [Google Scholar] [CrossRef]
- Shao, Y.; Head-Gordon, M.; Krylov, A.I. The Spin–Flip Approach within Time-Dependent density Functional Theory: Theory and Applications to Diradicals. J. Chem. Phys. 2023, 118, 4807–4818. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. 1. The Effect of the Exchange-Only Gradient Correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIRES Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Pradhan, E.; Zeng, T. The Lack of Triplet Fusion for an Intramolecular Singlet Fission Chromophore: The Expected, the Unexpected, and a Reconciliation. J. Phys. Chem. Lett. 2024, 15, 43–50. [Google Scholar] [CrossRef]
- Pyykkö, P. Relativistic Effects in Chemistry: More Common Than You Thought. Annu. Rev. Phys. Chem. 2012, 63, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P. Relativistic effects in structural chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Marian, C.M. Spin-Orbit Coupling in Molecules. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; WILEY-VCH: New York, NY, USA, 2001; Volume 17, pp. 99–204. [Google Scholar]
- Penfold, T.J.; Gindensperger, E.; Daniel, C.; Marian, C.M. Spin-Vibronic Mechanism for Intersystem Crossing. Chem. Rev. 2018, 118, 6975–7025. [Google Scholar] [CrossRef]
- Zeng, T.; Fedorov, D.G.; Klobukowski, M. Model core potentials for studies of scalar relativistic effects and spin–orbit coupling at Douglas-Kroll level. I. Theory and applications to Pb and Bi. J. Chem. Phys. 2009, 131, 124109. [Google Scholar] [CrossRef]
- Zeng, T.; Fedorov, D.G.; Klobukowski, M. Performance of Dynamically Weighted Multiconfiguration Self-Consistent Field and Spin-Orbit Coupling Calculations of Diatomic Molecules of Group 14 Elements. J. Chem. Phys. 2011, 134, 024108. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Fedorov, D.G.; Schmidt, M.W.; Klobukowski, M. Two-component natural spinors from two-step spin–orbit coupled wave functions. J. Chem. Phys. 2011, 134, 214107. [Google Scholar] [CrossRef]
- Zeng, T.; Fedorov, D.G.; Schmidt, M.W.; Klobukowski, M. Effects of spin–orbit coupling on covalent bonding and the Jahn-Teller e ect are revealed with the natural language of spinors. J. Chem. Theory Comput. 2011, 7, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Fedorov, D.G.; Schmidt, M.W.; Klobukowski, M. Nautral spinors reveal how the spin–orbit coupling affects the Jahn-Teller distortions in the hexafluorotungstate(V) anion. J. Chem. Theory Comput. 2012, 8, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Lower, S.K.; El-Sayed, M.A. The Triplet State and Molecular Electronic Processes in Organic Molecules. Chem. Rev. 1966, 66, 199–241. [Google Scholar] [CrossRef]
- El-Sayed, M.A. Triplet state. Its radiative and nonradiative properties. Acc. Chem. Res. 1968, 1, 8–16. [Google Scholar] [CrossRef]
- Pokhilko, P.; Krylov, A.I. Quantitative El-Sayed Rules for Many-Body Wave Functions from Spinless Transition Density Matrices. J. Phys. Chem. Lett. 2019, 10, 4857–4862. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, G.; Yang, Z.; Zeng, T. Survey of T1 and T2 Energies of Intramolecular Singlet Fission Chromophores. Photochem 2024, 4, 14-23. https://doi.org/10.3390/photochem4010002
Yao G, Yang Z, Zeng T. Survey of T1 and T2 Energies of Intramolecular Singlet Fission Chromophores. Photochem. 2024; 4(1):14-23. https://doi.org/10.3390/photochem4010002
Chicago/Turabian StyleYao, Guoying, Zhenyu Yang, and Tao Zeng. 2024. "Survey of T1 and T2 Energies of Intramolecular Singlet Fission Chromophores" Photochem 4, no. 1: 14-23. https://doi.org/10.3390/photochem4010002