


E–Z Photoisomerization in Proton-Modulated Photoswitchable Merocyanine Based on Benzothiazolium and o-Hydroxynaphthalene Platform

Abstract
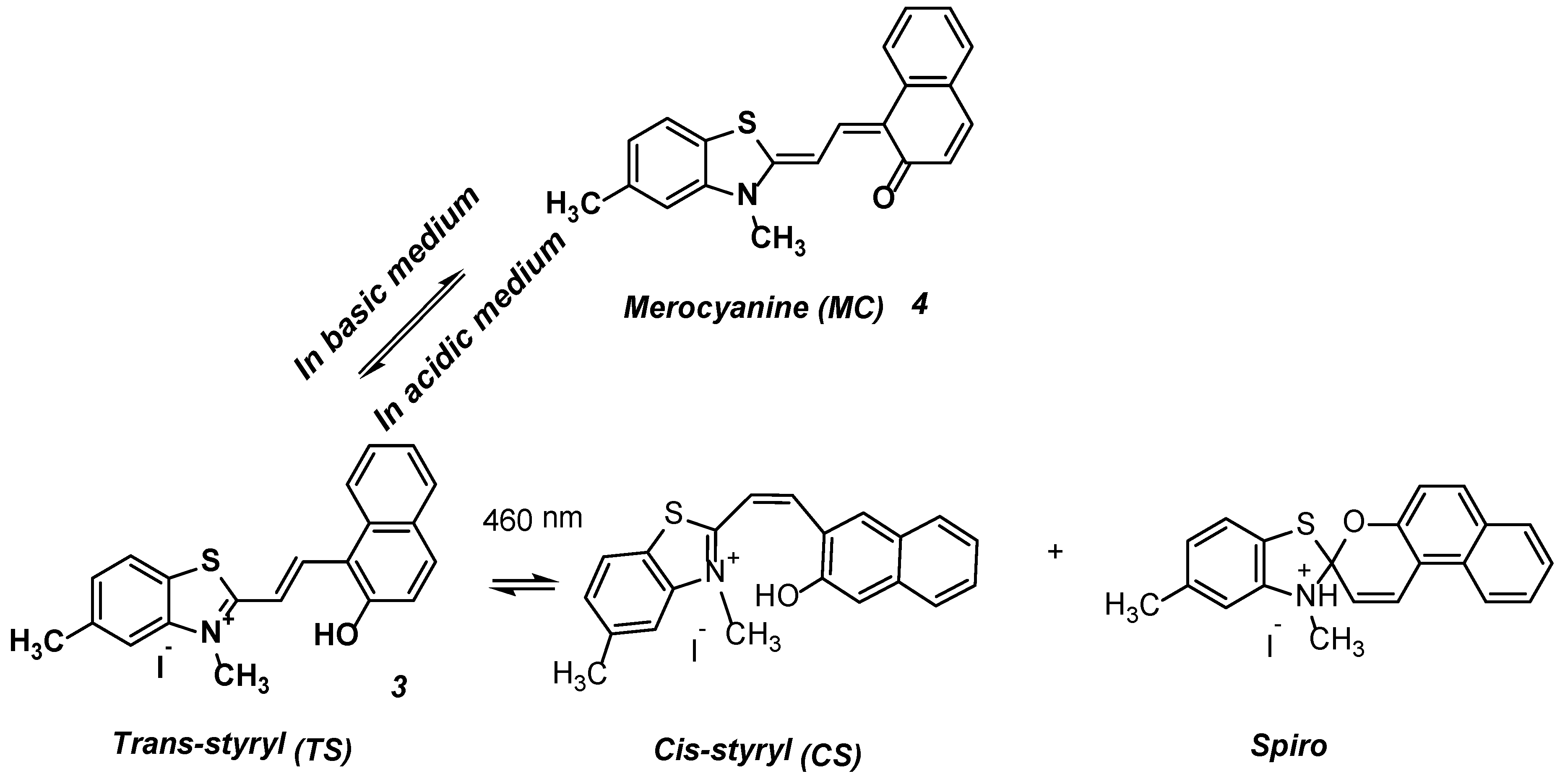
:1. Introduction
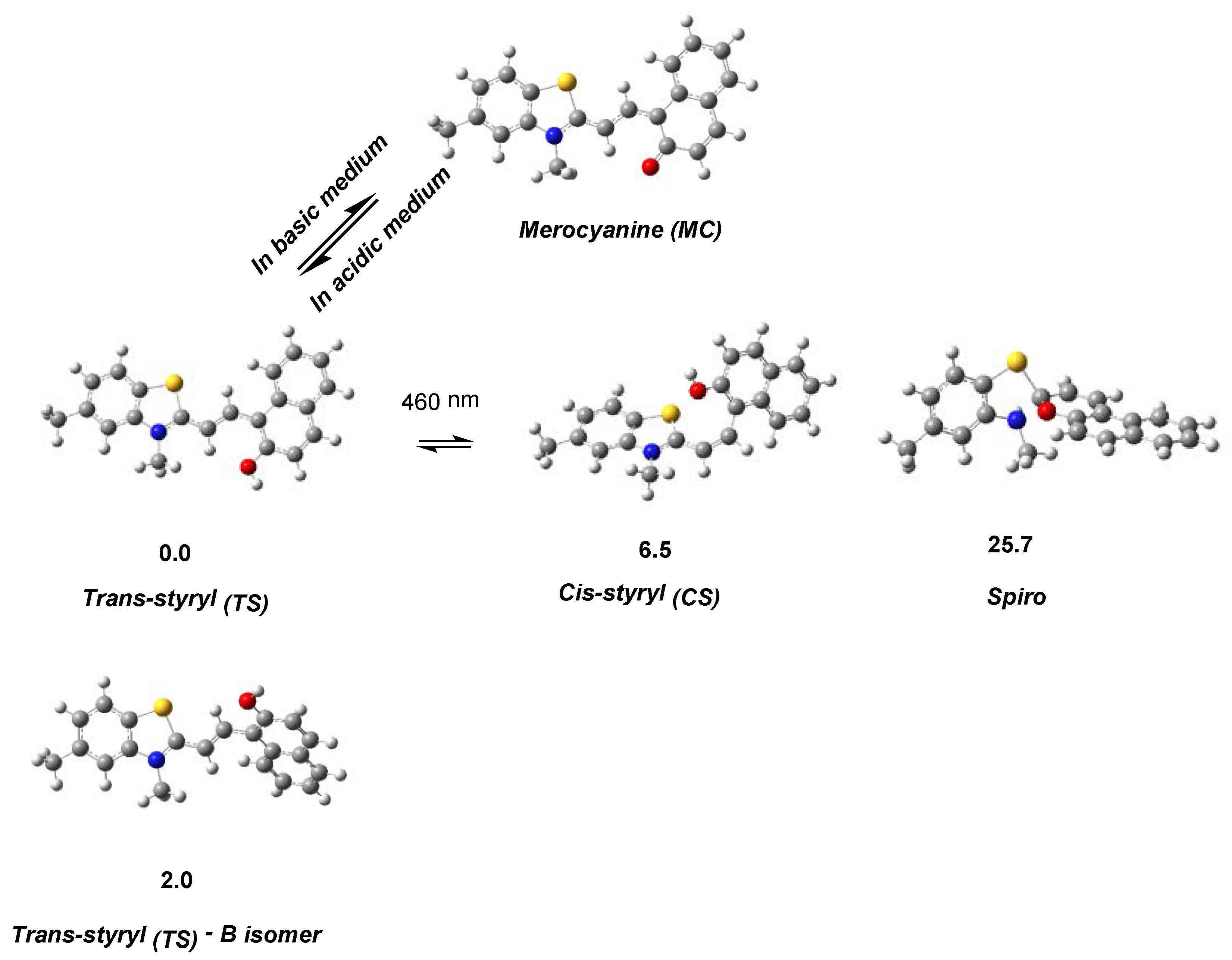
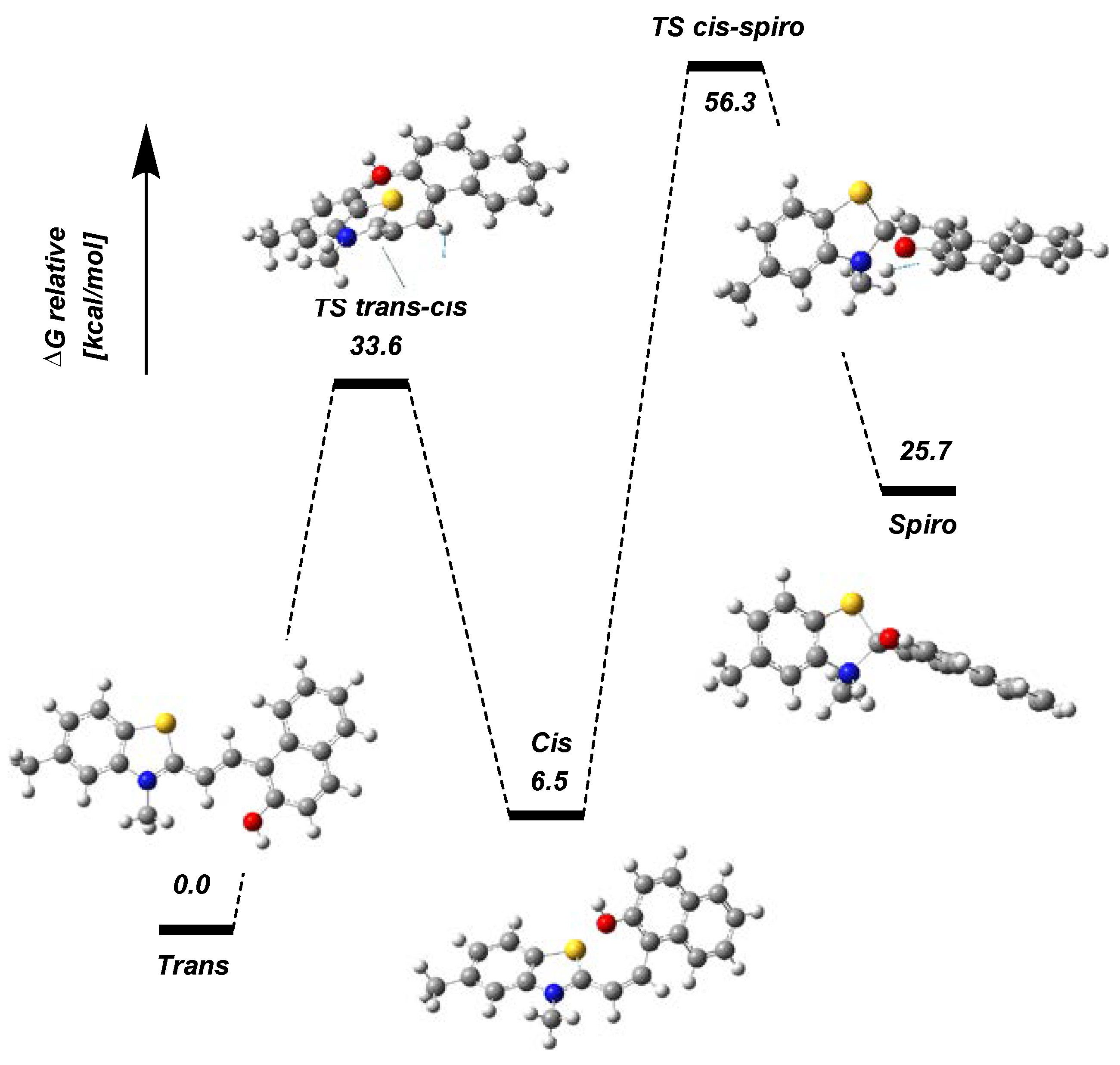
2. Results and Discussion
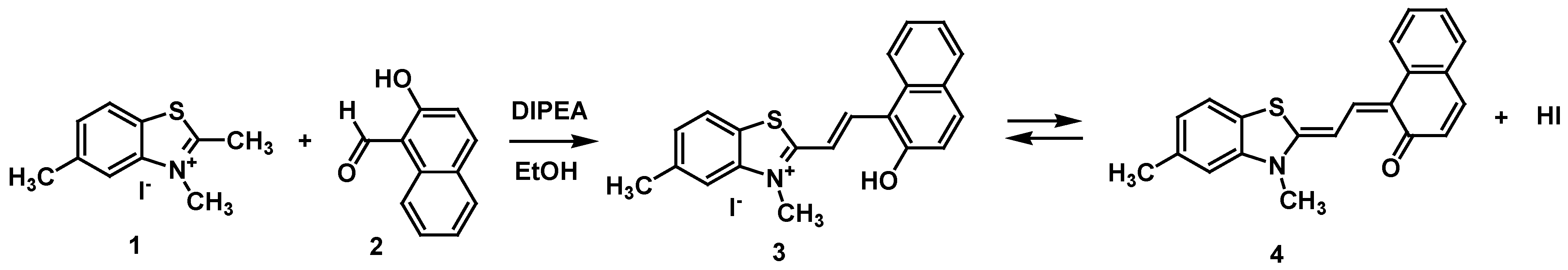
2.1. Synthesis
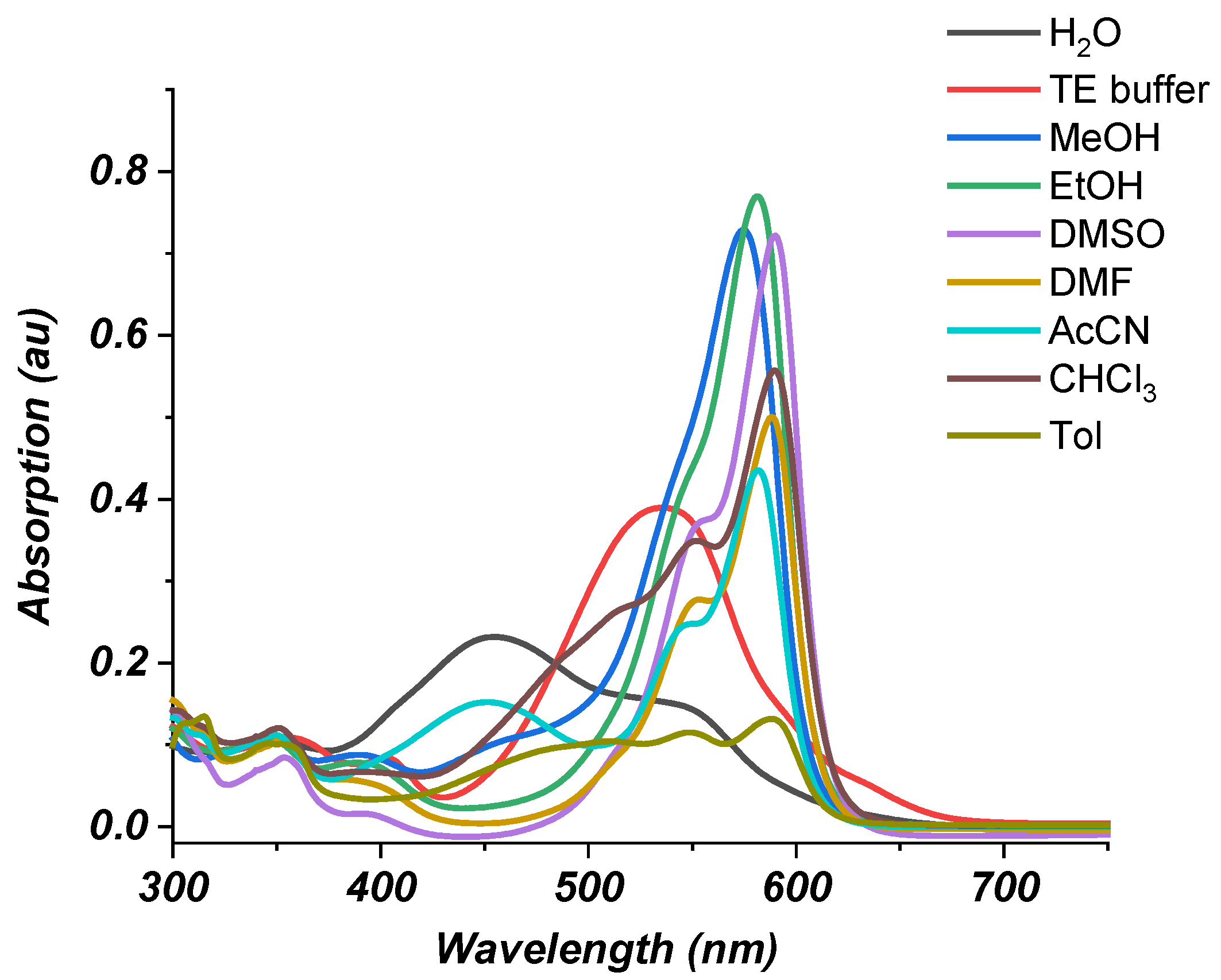
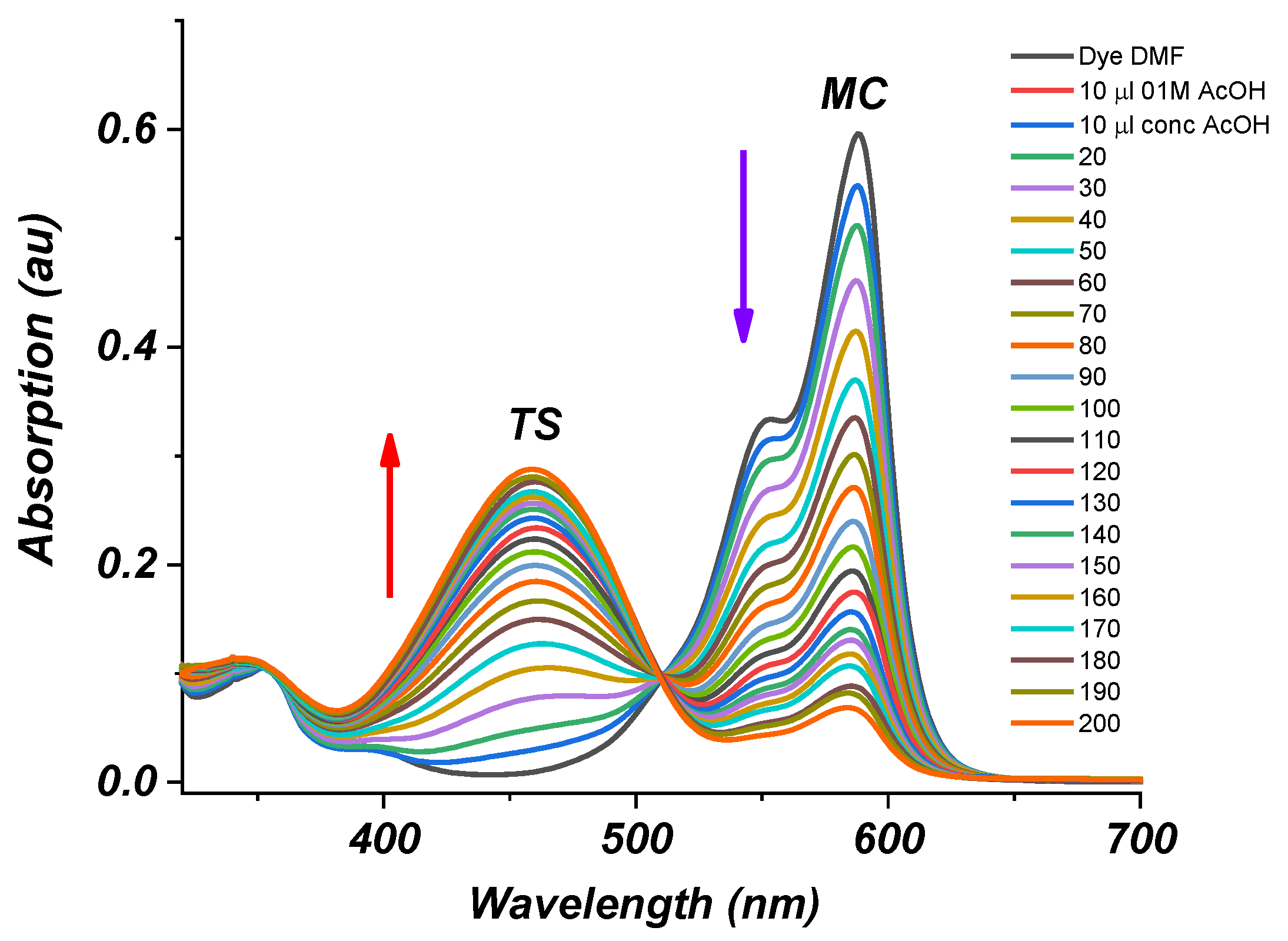
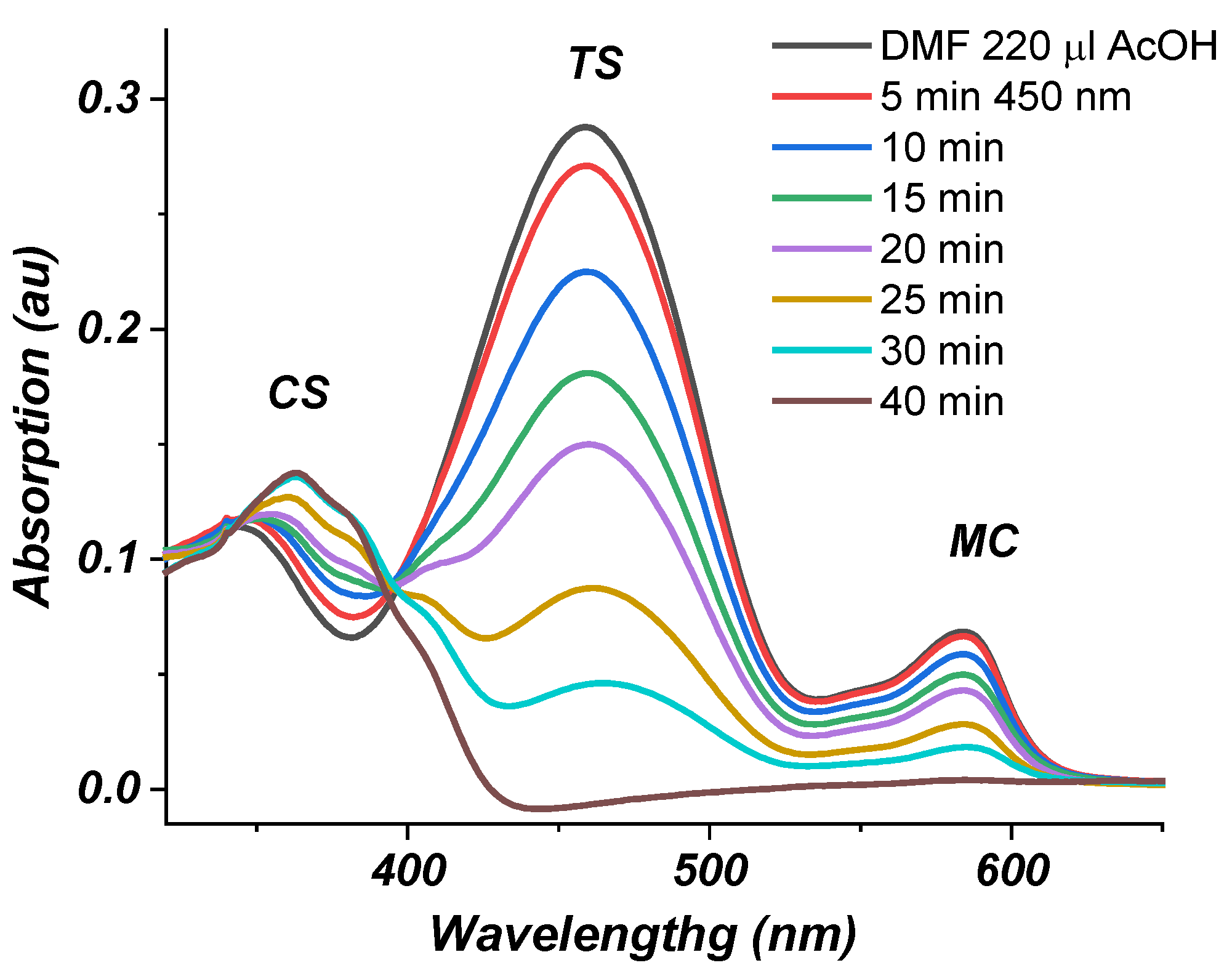
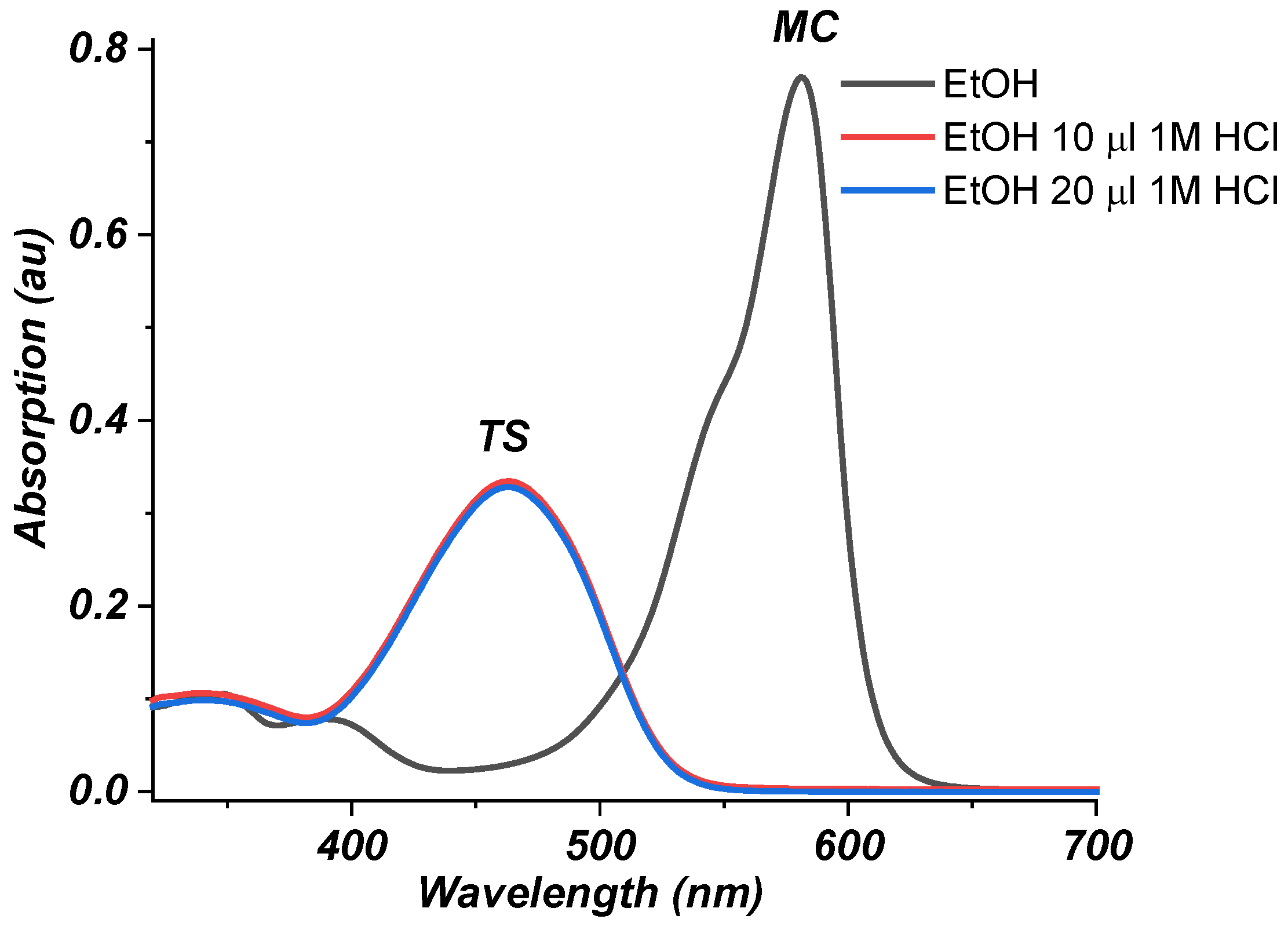
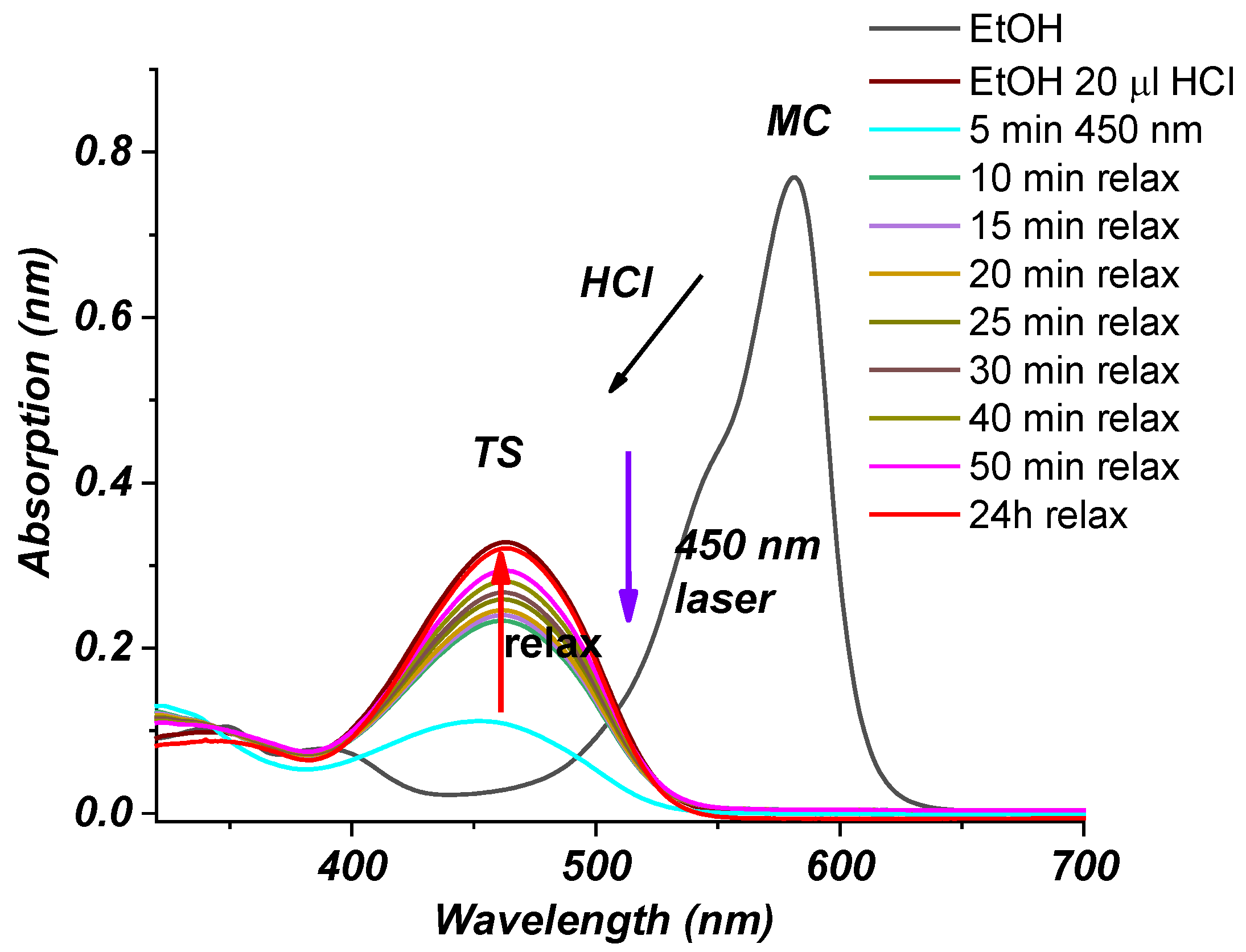
2.2. Photophysical Properties of Dye 3
Absorption Spectra
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Synthesis of (E)-2-(2-(2-Hydroxynaphthalen-1-yl)vinyl)-3,5-dimethylbenzo[d]thiazol-3-ium Iodide (3)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Volarić, J.; Szymanski, W.; Simeth, N.A.; Feringa, B.L. Molecular photoswitches in aqueous environments. Chem. Soc. Rev. 2021, 50, 12377–12449. [Google Scholar] [CrossRef] [PubMed]
- Olesińska-Mönch, M.; Deo, C. Small-molecule photoswitches for fluorescence bioimaging: Engineering and applications. Chem. Commun. 2023, 59, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Skov, A.B.; Broman, S.L.; Gertsen, A.S.; Elm, J.; Jevric, M.; Cacciarini, M.; Kadziola, A.; Mikkelsen, K.V.; Nielsen, M.B. Aromaticity-Controlled Energy Storage Capacity of the Dihydroazulene-Vinylheptafulvene Photochromic System. Chem. Eur. J. 2016, 22, 14567. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Christensen, O.; Kilde, M.D.; Abildgaard, M.; Metz, L.; Kadziola, A.; Jevric, M.; Mikkelsen, K.V.; Nielsen, M.B. Molecular Solar Thermal Energy Storage Systems with Long Discharge Times Based on the Dihydroazulene/Vinylheptafulvene Couple. Eur. J. Org. Chem. 2019, 2019, 1986–1993. [Google Scholar] [CrossRef]
- Moth-Poulsen, K.; Ćoso, D.; Börjesson, K.; Vinokurov, N.; Meier, S.K.; Majumdar, A.; Vollhardt, K.P.C.; Segalman, R.A. Molecular solar thermal (MOST) energy storage and release system. Energy Environ. Sci. 2012, 5, 8534–8537. [Google Scholar] [CrossRef] [Green Version]
- Bren’, V.A.; Dubonosov, A.D.; Minkin, V.I.; Chernoivanov, V.A. Norbornadiene–quadricyclane—An effective molecular system for the storage of solar energy. Russ. Chem. Rev. 1991, 60, 451–469. [Google Scholar] [CrossRef]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Velema, W.A.; Szymanski, W.; Feringa, B.L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Feng, Y.; Wang, L.; Feng, W. Azobenzene-based solar thermal fuels: Design, properties, and applications. Chem. Soc. Rev. 2018, 47, 7339–7368. [Google Scholar] [CrossRef]
- Shi, Y.; Gerkman, M.A.; Qiu, Q.; Zhang, S.; Han, G.G.D. Sunlight-activated phase change materials for controlled heat storage and triggered release. J. Mater. Chem. A 2021, 9, 9798–9808. [Google Scholar] [CrossRef]
- Qiu, Q.; Shi, Y.; Han, G.G.D. Solar energy conversion and storage by photoswitchable organic materials in solution, liquid, solid, and changing phases. J. Mater. Chem. C 2021, 9, 11444–11463. [Google Scholar] [CrossRef]
- Volarić, J.; Thallmair, S.; Feringa, B.L.; Szymanski, W. Photoswitchable, Water-Soluble Bisazobenzene Cross-Linkers with Enhanced Properties for Biological Applications. ChemPhotoChem 2022, 6, e202200170. [Google Scholar] [CrossRef]
- Matsuda, K.; Irie, M. Diarylethene as a photoswitching unit. J. Photochem. Photobiol. C 2004, 5, 169–182. [Google Scholar] [CrossRef]
- Kudernac, T.; van der Molen, S.J.; van Wees, B.J.; Feringa, B.L. Uni- and bi-directional light-induced switching of diarylethenes on gold nanoparticles. Chem. Commun. 2006, 34, 3597–3599. [Google Scholar] [CrossRef]
- Broichhagen, J.; Frank, J.A.; Trauner, D. A Roadmap to Success in Photopharmacology. Acc. Chem. Res. 2015, 48, 1947–1960. [Google Scholar] [CrossRef]
- Lerch, M.M.; Hansen, M.J.; van Dam, G.M.; Szymanski, W.; Feringa, B.L. Emerging Targets in Photopharmacology. Angew. Chem. Int. Ed. 2016, 55, 10978–10999. [Google Scholar] [CrossRef]
- Fischer, E.; Hirshberg, Y. Formation of Coloured Forms of Spirans by Low-Temperature Irradiation. J. Chem. Soc. 1952, 4522–4524. [Google Scholar] [CrossRef]
- Klajn, R. Spiropyran-based dynamic materials. Chem. Soc. Rev. 2014, 43, 148–184. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.H.; Tian, Z.Y.; Wu, W.W.; Wan, W.; Li, A.D.Q. Single-Molecule Photoswitching Enables High-Resolution Optical Imaging. Microsc. Microanal. 2009, 15, 840–841. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.Y.; Li, A.D.Q.; Hu, D.H. Super-resolution fluorescence nanoscopy applied to imaging core–shell photoswitching nanoparticles and their self-assemblies. Chem. Commun. 2011, 47, 1258–1260. [Google Scholar] [CrossRef]
- Montagnoli, G.; Pieroni, O.; Suzuki, S. Control of peptide chain conformation by photoisomerising chromophores: Enzymes and model compounds. Polym. Photochem. 1983, 3, 279–294. [Google Scholar] [CrossRef]
- Ciardelli, F.; Fabbri, D.; Pieroni, O.; Fissi, A. Photomodulation of polypeptide conformation by sunlight in spiropyran-containing poly(L-glutamic acid). J. Am. Chem. Soc. 1989, 111, 3470–3472. [Google Scholar] [CrossRef]
- Sakata, T.; Yan, Y.L.; Marriott, G. Optical switching of dipolar interactions on proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 4759–4764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fissi, A.; Pieroni, O.; Angelini, N.; Lenci, F. Photoresponsive Polypeptides. Photochromic and Conformational Behavior of Spiropyran-Containing Poly-l-Glutamate)s under Acid Conditions. Macromolecules 1999, 32, 7116–7121. [Google Scholar] [CrossRef]
- Wojtyk, J.T.C.; Wasey, A.; Xiao, N.N.; Kazmaier, P.M.; Hoz, S.; Yu, C.; Lemieux, R.P.; Buncel, E. Elucidating the Mechanisms of Acidochromic Spiropyran-Merocyanine Interconversion. J. Phys. Chem. A 2007, 111, 2511–2516. [Google Scholar] [CrossRef]
- Remon, P.; Li, S.M.; Grotli, M.; Pischel, U.; Andreasson, J. An Acido- and Photochromic Molecular Device That Mimics Triode Action. Chem. Commun. 2016, 52, 4659–4662. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.B.; Kempe, F.; Brügner, O.; Walter, M.; Sommer, M. Alkyl-Substituted Spiropyrans: Electronic Effects, Model Compounds and Synthesis of Aliphatic Main-Chain Copolymers. Polym. Chem. 2017, 8, 5407–5414. [Google Scholar] [CrossRef]
- Roxburgh, C.J.; Sammes, P.G. On the Acid Catalysed Isomerisation of Some Substituted Spirobenzopyrans. Dyes Pigments 1995, 27, 63–69. [Google Scholar] [CrossRef]
- Shiozaki, H. Molecular Orbital Calculations for Acid Induced Ring Opening Reaction of Spiropyran. Dyes Pigments 1997, 33, 229–237. [Google Scholar] [CrossRef]
- Kortekaas, L.; Chen, J.; Jacquemin, D.; Browne, W.R. Proton-Stabilized Photochemically Reversible E/Z Isomerization of Spiropyrans. J. Phys. Chem. B 2018, 122, 6423–6430. [Google Scholar] [CrossRef]
- Vasilev, A.; Dimitrova, R.; Kandinska, M.; Landfester, K. Baluschev. S. Accumulation of the photonic energy of the deep-red part of the terrestrial sun irradiation by rare-earth metal-free E–Z photoisomerization. J. Mater. Chem. C 2021, 9, 7119–7126. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Labanowski, J.K.; Andzelm, J.W. (Eds.) Density Functional Methods in Chemistry; Springer: New York, NY, USA, 1991. [Google Scholar]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. The accuracy of the pseudopotential approximation. II. A comparison of various core sizes for indium pseudopotentials in calculations for spectroscopic constants of InH, InF, and InCl. J. Chem. Phys. 1996, 105, 1052–1059. [Google Scholar] [CrossRef]
- Haas, J.; Bissmire, S.; Wirth, T. Iodine Monochloride–Amine Complexes: An Experimental and Computational Approach to New Chiral Electrophiles. Chem. Eur. J. 2005, 11, 5777–5785. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision 16.A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zonjić, I.; Radić Stojković, M.; Crnolatac, I.; Tomašić Paić, A.; Pšeničnik, S.; Vasilev, A.; Kandinska, M.; Mondeshki, M.; Baluschev, S.; Landfester, K.; et al. Styryl dyes with N-Methylpiperazine and N-Phenylpiperazine Functionality: AT-DNA and G-quadruplex binding ligands and theranostic agents. Bioorg. Chem. 2022, 127, 105999. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Polarity Index | Dielectric Permittivity ε | λmax1 (nm) Styryl | ε1 (L × mol−1 × cm−1) | λmax2 (nm) MC | ε2 (L × mol−1 × cm−1) |
|---|---|---|---|---|---|---|
| Toluene | 2.4 | 2.3741 | 481 | 9 490 | 589 | 13,160 |
| CHCl3 | 4.1 | 4.71 | ----- | ----- | 590 | 55,700 |
| MeOH | 5.1 | 32.613 | 459 | 10 510 | 574 | 72,860 |
| EtOH | 5.2 | 24.85 | ----- | ----- | 581 | 76,980 |
| Acetonitrile (AcCN) | 5.8 | 35.688 | 451 | 15 220 | 582 | 43,540 |
| DMF | 6.4 | 37.219 | ----- | ----- | 588 | 50,050 |
| DMSO | 7.2 | 46.826 | ----- | ----- | 590 | 72,190 |
| H2O | 10.2 | 78.355 | 454 | 23 180 | 543 | 14,930 |
| TE buffer pH = 7 | ----- | 407 | 6 460 | 535 | 38,960 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilev, A.A.; Baluschev, S.; Ilieva, S.; Cheshmedzhieva, D. E–Z Photoisomerization in Proton-Modulated Photoswitchable Merocyanine Based on Benzothiazolium and o-Hydroxynaphthalene Platform. Photochem 2023, 3, 301-312. https://doi.org/10.3390/photochem3020018
Vasilev AA, Baluschev S, Ilieva S, Cheshmedzhieva D. E–Z Photoisomerization in Proton-Modulated Photoswitchable Merocyanine Based on Benzothiazolium and o-Hydroxynaphthalene Platform. Photochem. 2023; 3(2):301-312. https://doi.org/10.3390/photochem3020018
Chicago/Turabian StyleVasilev, Aleksey A., Stanislav Baluschev, Sonia Ilieva, and Diana Cheshmedzhieva. 2023. "E–Z Photoisomerization in Proton-Modulated Photoswitchable Merocyanine Based on Benzothiazolium and o-Hydroxynaphthalene Platform" Photochem 3, no. 2: 301-312. https://doi.org/10.3390/photochem3020018