
Reduction Potential Predictions for Thirty-Seven 1,4-di-N-Oxide Quinoxaline-2-Carboxamide Derivatives with Anti-Tuberculosis Activity
Abstract
:1. Introduction

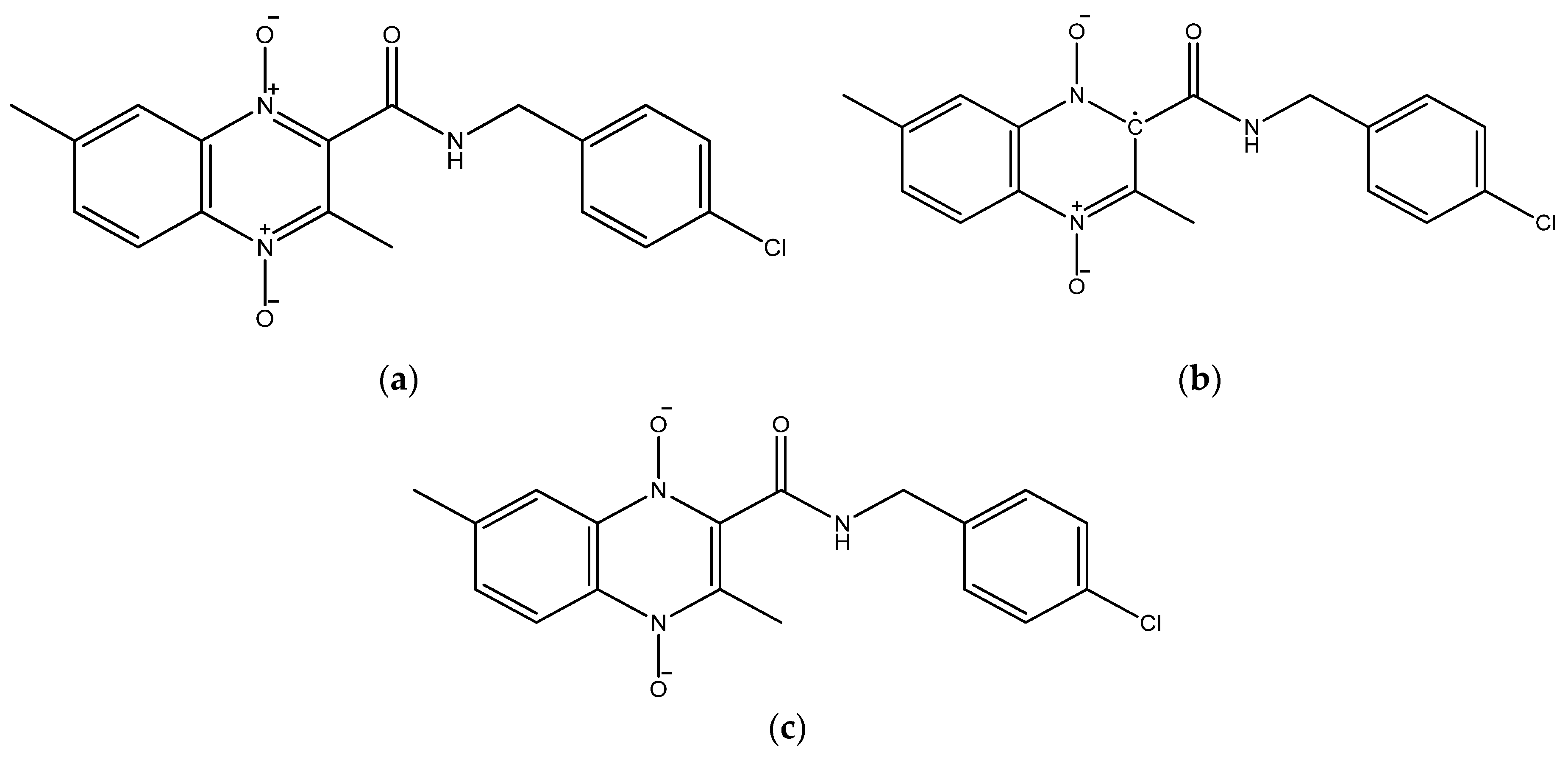{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compounds Code | R | R7/R6 |
|---|---|---|
| 1 | Benzyl | CH3/H |
| 2 | Cl/H | |
| 3 | Cl/Cl | |
| 4 | F/H | |
| 5 | CF3/H | |
| 6 | 2-Phenylethyl | H/H |
| 7 | CH3/H | |
| 8 | Cl/H | |
| 9 | Cl/Cl | |
| 10 | CF3/H | |
| 11 | F/H | |
| 12 | F/F | |
| 13 | p-Methoxybenzyl | H/H |
| 14 | CH3/H | |
| 15 | Cl/H | |
| 16 | p-Trifluoromethylbenzyl | H/H |
| 17 | CH3/H | |
| 18 | Cl/H | |
| 19 | p-Chlorobenzyl | H/H |
| 20 | CH3/H | |
| 21 | Cl/H | |
| 22 | Cl/Cl | |
| 23 | p-Bromobenzyl | H/H |
| 24 | CH3/H | |
| 24 | Cl/H | |
| 26 | Cl/Cl | |
| 27 | p-Methylbenzyl | H/H |
| 28 | CH3/H | |
| 29 | Cl/H | |
| 30 | 2,2-Diphenylethel | H/H |
| 31 | CH3/H | |
| 32 | Cl/H | |
| 33 | Cl/Cl | |
| 34 | Benzo[d] [1,3] dioxol-5-ylmethyl | H/H |
| 35 | CH3/H | |
| 36 | Cl/H | |
| 37 | Cl/Cl |
2. Materials and Methods
2.1. Drawing the Derivatives
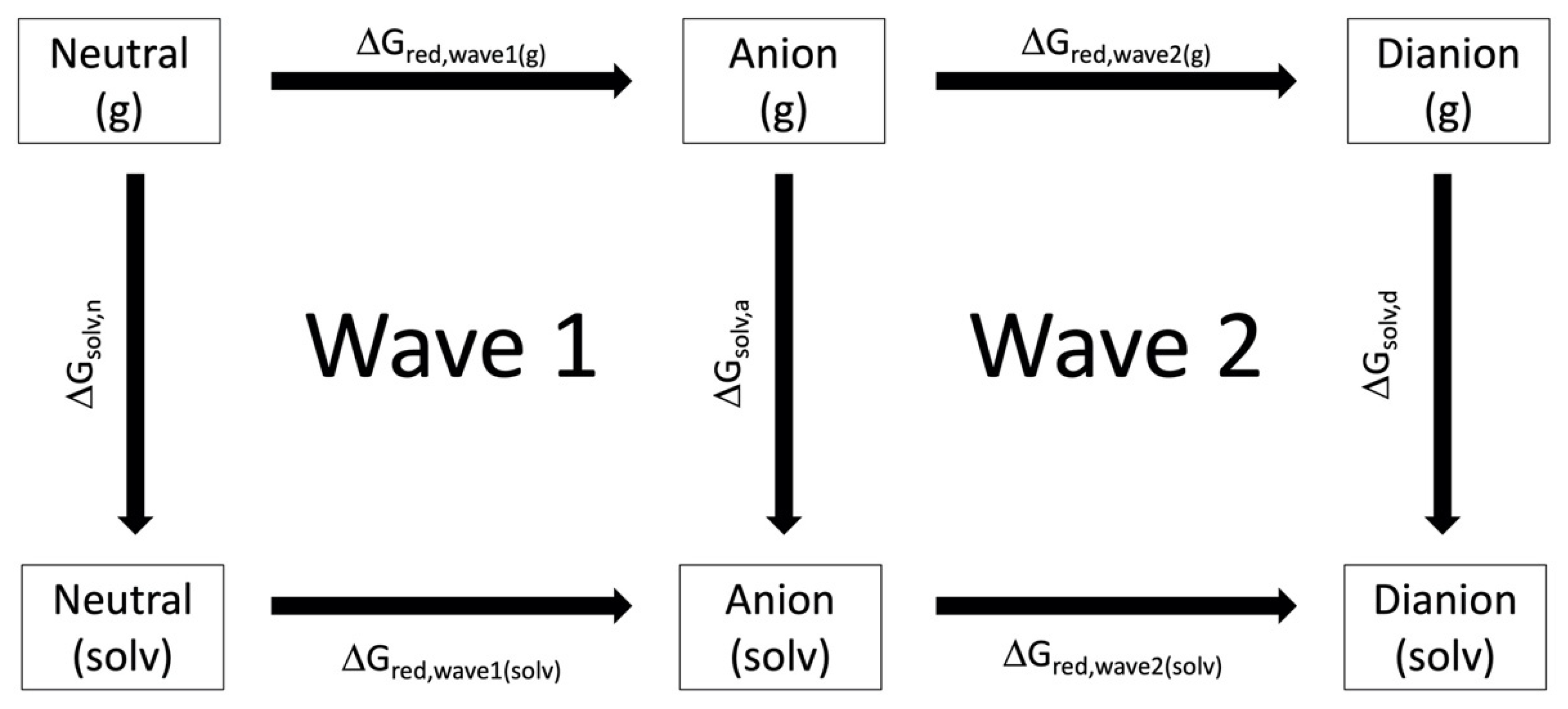
2.2. DFT Reduction Potential Predictions
3. Results and Discussion
3.1. Computational Raw Chemical Potentials
3.2. Computationally Predicted Electrochemical Cell Reaction Potentials
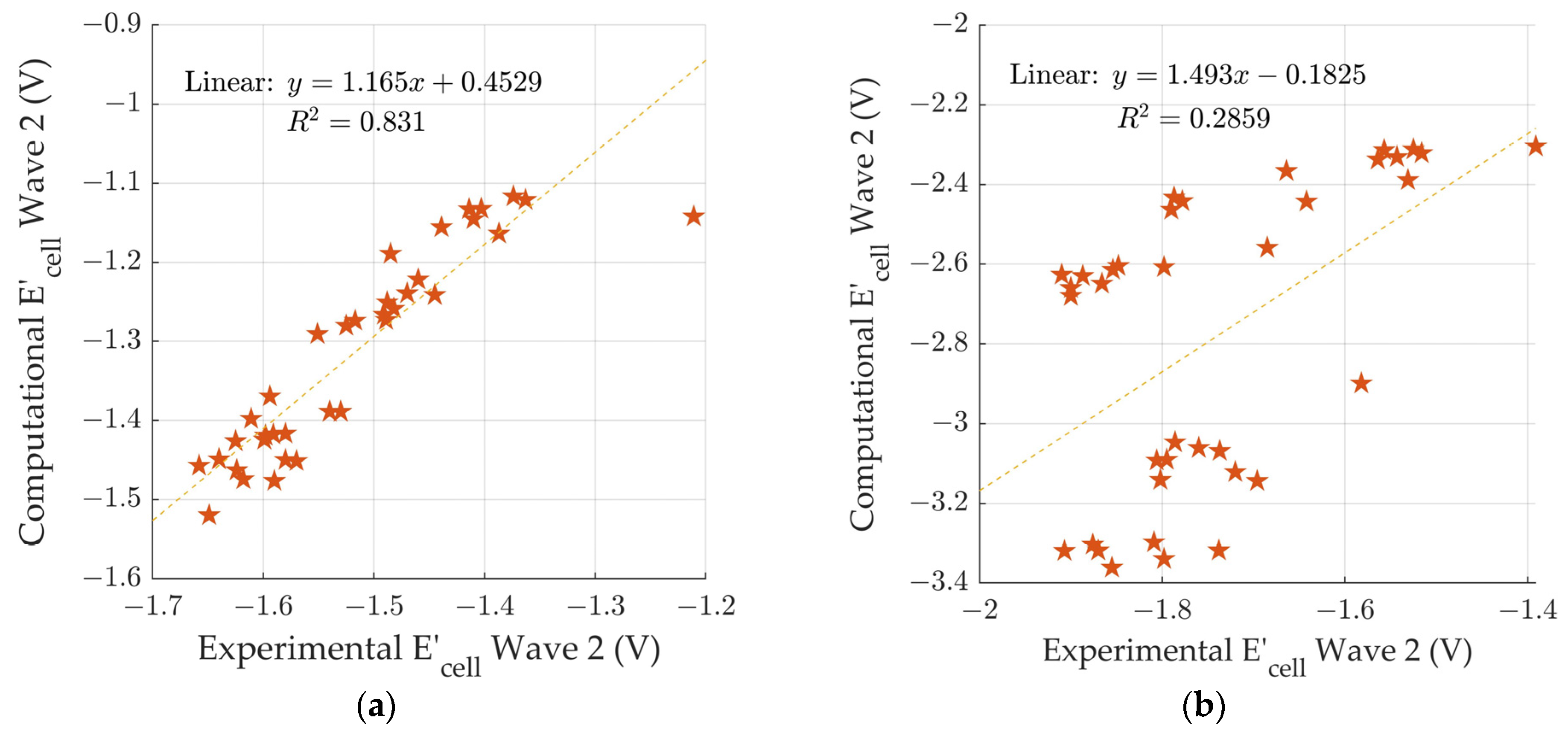
3.3. Comparison to Experimental Reduction Potentials
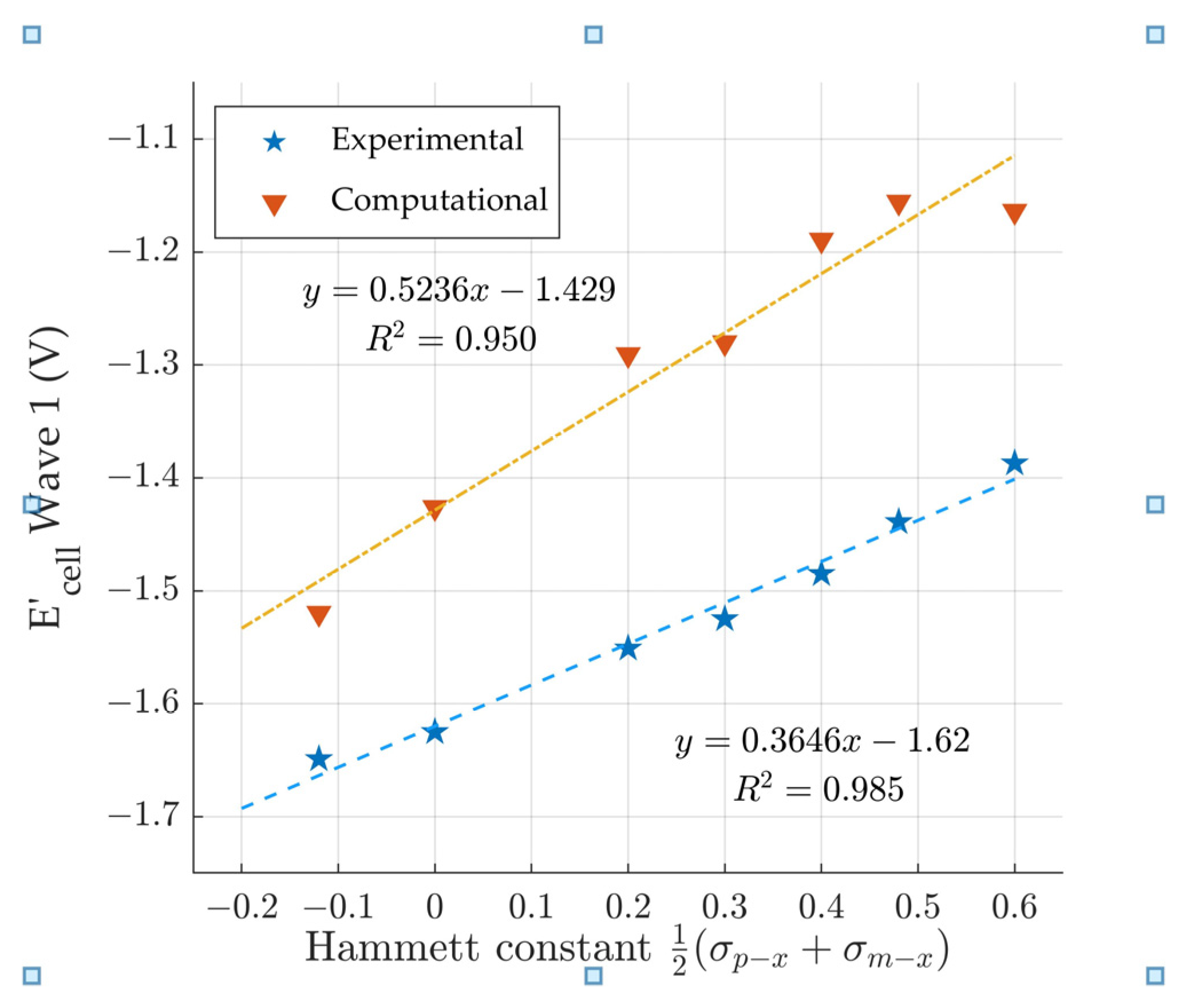
3.4. Comparison to the Modified Hammett Equation
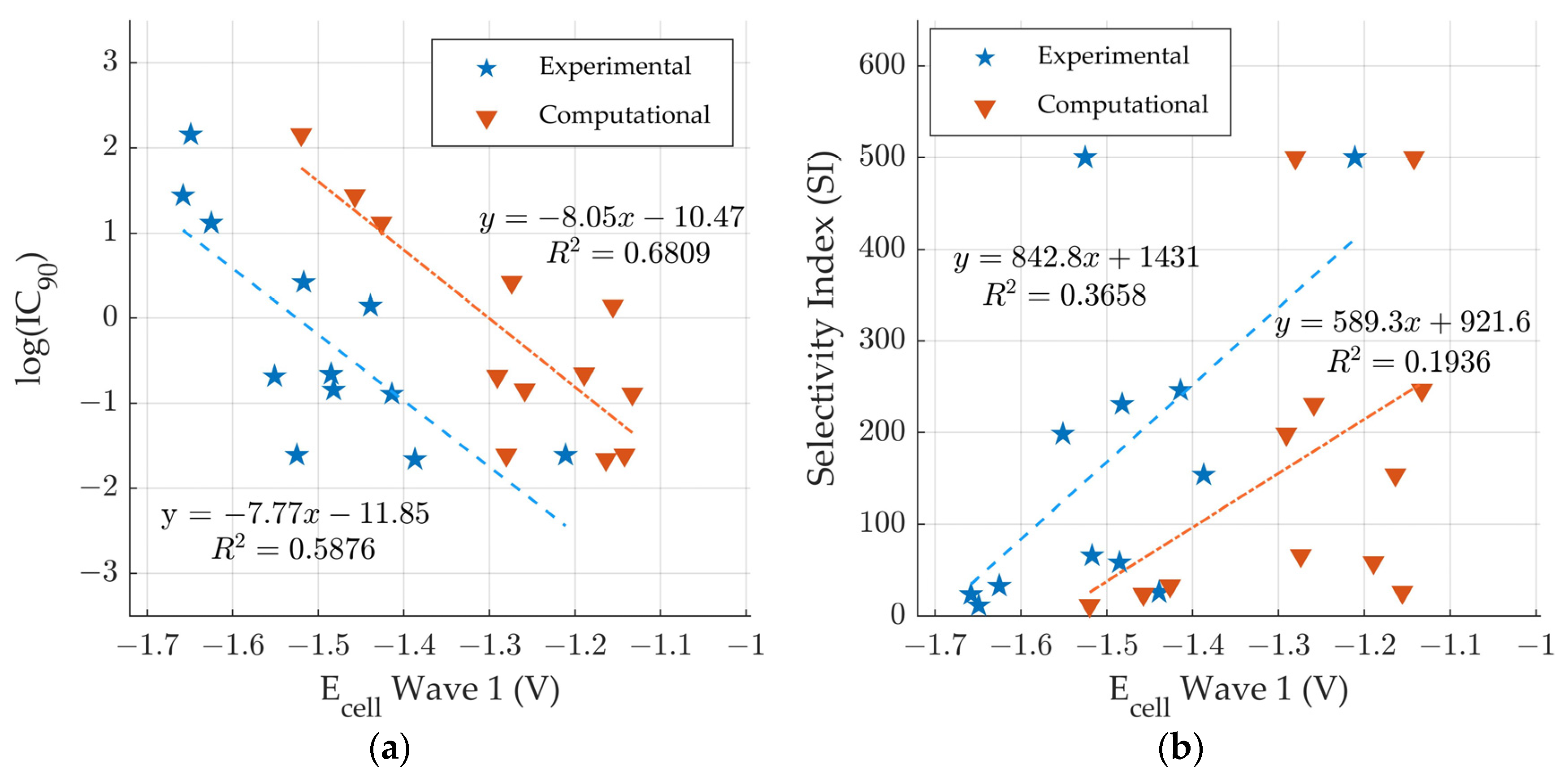
3.5. Comparison to Experimental Anti-Tuberculosis Data
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pereira, J.A.; Pessoa, A.M.; Cordeiro, M.N.; Fernandes, R.; Prudêncio, C.; Noronha, J.P.; Vieira, M. Quinoxaline, its derivatives and applications: A State of the Art review. Eur. J. Med. Chem. 2015, 97, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarranz, B.; Jaso, A.; Aldana, I.; Monge, A. Synthesis and anticancer activity evaluation of new 2-alkylcarbonyl and 2-benzoyl-3-trifluoromethyl-quinoxaline 1,4-di-N-oxide derivatives. Bioorganic Med. Chem. 2004, 12, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; Ismail, M.M.F.; Noaman, E.; Soliman, D.H.; Ammar, Y.A. New quinoxaline 1,4-di-N-oxides. Part 1: Hypoxia-selective cytotoxins and anticancer agents derived from quinoxaline 1,4-di-N-oxides. Bioorganic Med. Chem. 2006, 14, 6917–6923. [Google Scholar] [CrossRef] [PubMed]
- Estevez, Y.; Quiliano, M.; Burguete, A.; Cabanillas, B.; Zimic, M.; Málaga, E.; Verástegui, M.; Pérez-Silanes, S.; Aldana, I.; Monge, A.; et al. Trypanocidal properties, structure–activity relationship and computational studies of quinoxaline 1,4-di-N-oxide derivatives. Exp. Parasitol. 2011, 127, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ancizu, S.; Moreno, E.; Torres, E.; Burguete, A.; Pérez-Silanes, S.; Benítez, D.; Villar, R.; Solano, B.; Marín, A.; Aldana, I.; et al. Heterocyclic-2-carboxylic Acid (3-Cyano-1,4-di-N-oxidequinoxalin-2-yl)amide Derivatives as Hits for the Development of Neglected Disease Drugs. Molecules 2009, 14, 2256–2272. [Google Scholar] [CrossRef] [Green Version]
- Torres, E.; Moreno-Viguri, E.; Galiano, S.; Devarapally, G.; Crawford, P.W.; Azqueta, A.; Arbillaga, L.; Varela, J.; Birriel, E.; Di Maio, R.; et al. Novel quinoxaline 1,4-di-N-oxide derivatives as new potential antichagasic agents. Eur. J. Med. Chem. 2013, 66, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Carta, A.; Paglietti, G.; Rahbar Nikookar, M.E.; Sanna, P.; Sechi, L.; Zanetti, S. Novel substituted quinoxaline 1,4-dioxides with in vitro antimycobacterial and anticandida activity. Eur. J. Med. Chem. 2002, 37, 355–366. [Google Scholar] [CrossRef]
- Vicente, E.; Pérez-Silanes, S.; Lima, L.M.; Ancizu, S.; Burguete, A.; Solano, B.; Villar, R.; Aldana, I.; Monge, A. Selective activity against Mycobacterium tuberculosis of new quinoxaline 1,4-di-N-oxides. Bioorganic Med. Chem. 2009, 17, 385–389. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, Q.; Zhang, J.; Qu, W.; Xie, S.; Huang, L.; Yuan, Z.; Pan, Y. Discovery of novel nitrogenous heterocyclic-containing quinoxaline-1,4-di-N-oxides as potent activator of autophagy in M.tb-infected macrophages. Eur. J. Med. Chem. 2021, 223, 113657. [Google Scholar] [CrossRef]
- Agrawal, N.; Bhardwaj, A. An appraisal on synthetic and pharmaceutical perspectives of quinoxaline 1,4-di-N-oxide scaffold. Chem. Biol. Drug Des. 2022, 100, 346–363. [Google Scholar] [CrossRef]
- Anderson, R.F.; Yadav, P.; Shinde, S.S.; Hong, C.R.; Pullen, S.M.; Reynisson, J.; Wilson, W.R.; Hay, M.P. Radical Chemistry and Cytotoxicity of Bioreductive 3-Substituted Quinoxaline Di-N-Oxides. Chem. Res. Toxicol. 2016, 29, 1310–1324. [Google Scholar] [CrossRef]
- Divya, K.M.; Savitha, D.P.; Krishna, G.A.; Dhanya, T.M.; Mohanan, P.V. Crystal structure, DFT studies, Hirshfeld surface and energy framework analysis of 4-(5-nitro-thiophen-2-yl)-pyrrolo [1, 2-a] quinoxaline: A potential SARS-CoV-2 main protease inhibitor. J. Mol. Struct. 2022, 1251, 131932. [Google Scholar] [CrossRef]
- Kucuk, C.; Yurdakul, S.; Celik, S.; Erdem, B. Experimental and DFT studies of 2-methyl-quinoxaline and its silver (I) complex: Non-covalent interaction analysis, antimicrobial activity and molecular docking study. Inorg. Chem. Commun. 2022, 145, 109935. [Google Scholar] [CrossRef]
- Bouanane, Z.; Bounekhel, M.; Elkolli, M.; Abrigach, F.; Khoutoul, M.; Bouyala, R.; Touzani, R.; Hellal, A. Synthesis, structural, catecholase, tyrosinase and DFT studies of pyrazoloquinoxaline derivatives. J. Mol. Struct. 2017, 1139, 238–246. [Google Scholar] [CrossRef]
- Maltsev, D.V.; Skripka, M.O.; Spasov, A.A.; Vassiliev, P.M.; Perfiliev, M.A.; Divaeva, L.N.; Zubenko, A.A.; Morkovnik, A.S.; Klimenko, A.I.; Miroshnikov, M.V.; et al. Design, Synthesis and Pharmacological Evaluation of Novel C2,C3-Quinoxaline Derivatives as Promising Anxiolytic Agents. Int. J. Mol. Sci. 2022, 23, 14401. [Google Scholar] [CrossRef]
- Fan, D.; Liu, P.; Jiang, Y.; He, X.; Zhang, L.; Wang, L.; Yang, T. Discovery and SAR Study of Quinoxaline–Arylfuran Derivatives as a New Class of Antitumor Agents. Pharmaceutics 2022, 14, 2420. [Google Scholar] [CrossRef]
- Crawford, P.W.; Scamehorn, R.G.; Hollstein, U.; Ryan, M.D.; Kovacic, P. Cyclic voltammetry of phenazines and quinoxalines including mono- and di-N-oxides. Relation to structure and antimicrobial activity. Chem.-Biol. Interact. 1986, 60, 67–84. [Google Scholar] [CrossRef]
- Moreno, E.; Pérez-Silanes, S.; Gouravaram, S.; Macharam, A.; Ancizu, S.; Torres, E.; Aldana, I.; Monge, A.; Crawford, P.W. 1,4-Di-N-oxide quinoxaline-2-carboxamide: Cyclic voltammetry and relationship between electrochemical behavior, structure and anti-tuberculosis activity. Electrochim. Acta 2011, 56, 3270–3275. [Google Scholar] [CrossRef]
- Pérez-Silanes, S.; Devarapally, G.; Torres, E.; Moreno-Viguri, E.; Aldana, I.; Monge, A.; Crawford, P.W. Cyclic Voltammetric Study of Some Anti-Chagas-Active 1,4-Dioxidoquinoxalin-2-yl Ketone Derivatives. Helv. Chim. Acta 2013, 96, 217–227. [Google Scholar] [CrossRef]
- Verbitskiy, E.V.; le Poul, P.; Bureš, F.; Achelle, S.; Barsella, A.; Kvashnin, Y.A.; Rusinov, G.L.; Charushin, V.N. Push-Pull Derivatives Based on 2,4′-Biphenylene Linker with Quinoxaline, [1,2,5]Oxadiazolo[3,4-B]Pyrazine and [1,2,5]Thiadiazolo[3,4-B]Pyrazine Electron Withdrawing Parts. Molecules 2022, 27, 4250. [Google Scholar] [CrossRef]
- Rupar, J.; Aleksić, M.M.; Nikolić, K.; Popović-Nikolić, M.R. Comparative electrochemical studies of kinetic and thermodynamic parameters of Quinoxaline and Brimonidine redox process. Electrochim. Acta 2018, 271, 220–231. [Google Scholar] [CrossRef]
- Aguilar-Martínez, M.; Cuevas, G.; Jiménez-Estrada, M.; González, I.; Lotina-Hennsen, B.; Macías-Ruvalcaba, N. An Experimental and Theoretical Study of the Substituent Effects on the Redox Properties of 2-[(R-phenyl)amine]-1,4-naphthalenediones in Acetonitrile. J. Org. Chem. 1999, 64, 3684–3694. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.L.; Namazian, M.; Bottle, S.E.; Coote, M.L. One-Electron Oxidation and Reduction Potentials of Nitroxide Antioxidants: A Theoretical Study. J. Phys. Chem. A 2007, 111, 13595–13605. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro da Silva, M.D.M.C.; Gomes, J.R.B.; Gonçalves, J.M.; Sousa, E.A.; Pandey, S.; Acree, W.E. Thermodynamic Properties of Quinoxaline-1,4-Dioxide Derivatives: A Combined Experimental and Computational Study. J. Org. Chem. 2004, 69, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Dhilshath Raihana, H.; Karthick, K.; Shankar, T.; Kamalesu, S.; Anish Babu, A.; Swarnalatha, K. A new tetradentate Schiff base of N,N′-bis(3,5-diiodosalicylidene)-1,2-phenylenediamine: Spectral aspects, Hirshfield surfaces, DFT computations and molecular docking. J. Mol. Struct. 2022, 1264, 133217. [Google Scholar] [CrossRef]
- Kucuk, C.; Yurdakul, S.; Erdem, B. Spectroscopic characterization, DFT calculations, and microbiological activity of 5-iodoindole. J. Mol. Struct. 2022, 1252, 132125. [Google Scholar] [CrossRef]
- Mishra, A.; Verma, C.; Srivastava, V.; Lgaz, H.; Quraishi, M.A.; Ebenso, E.E.; Chung, I.-M. Chemical, Electrochemical and Computational Studies of Newly Synthesized Novel and Environmental Friendly Heterocyclic Compounds as Corrosion Inhibitors for Mild Steel in Acidic Medium. J. Bio-Tribo-Corros. 2018, 4, 32. [Google Scholar] [CrossRef]
- Lauria, A.; Almerico, A.M.; Barone, G. The influence of substitution in the quinoxaline nucleus on 1,3-dipolar cycloaddition reactions: A DFT study. Comput. Theor. Chem. 2013, 1013, 116–122. [Google Scholar] [CrossRef]
- Miller, E.M.; Brazel, C.J.; Brillos-Monia, K.A.; Crawford, P.W.; Hufford, H.C.; Loncaric, M.R.; Mruzik, M.N.; Nenninger, A.W.; Ragain, C.M. Reduction Potential Predictions for Some 3-Aryl-Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives with Known Anti-Tumor Properties. Computation 2019, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.E.; Xia, Q.; Cella, E.M.; Nenninger, W.A.; Mruzik, N.M.; Brillos-Monia, A.K.; Hu, Z.Y.; Sheng, R.; Ragain, M.C.; Crawford, W.P. Voltammetric Study of Some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide Derivatives with Anti-Tumor Activities. Molecules 2017, 22, 1442. [Google Scholar] [CrossRef]
- Bhattarai, S.; Mareta, P.; Crawford, P.W.; Kessler, J.M.; Ragain, C.M. Improved Computational Prediction of the Electrochemical Reduction Potential of Twenty 3-Aryl-Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives. Computation 2023, 11, 9. [Google Scholar] [CrossRef]
- Schaefer, H.F. (Ed.) Applications of Electronic Structure Theory; Plenum Press: New York, NY, USA, 1977; Volume 4. [Google Scholar]
- ChemDraw. Available online: https://perkinelmerinformatics.com/products/research/chemdraw (accessed on 30 November 2022).
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertuš, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Pascual-ahuir, J.L.; Silla, E.; Tuñon, I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Zuman, P. Substituent Effects in Organic Polarography; Plenum Press: New York, NY, USA, 1967. [Google Scholar]
- Strier, M.P.; Cavagnol, J.C. The Polarography of Quinoxaline. II. 6-Substituted Derivatives. J. Am. Chem. Soc. 1958, 80, 1565–1568. [Google Scholar] [CrossRef]






| Compounds | Wave 1 Half-Cell Potentials (V) | Wave 2 Half-Cell Potentials (V) |
|---|---|---|
| 1 | 3.34 | 2.17 |
| 2 | 3.54 | 1.75 * |
| 3 | 3.65 | 2.49 |
| 4 | 3.52 | 2.35 |
| 5 | 3.66 | 2.47 |
| 6 | 3.37 | 2.19 |
| 7 | 3.27 | 1.47 |
| 8 | 3.51 | 1.67 * |
| 9 | 3.63 | 2.48 |
| 10 | 3.64 | 2.46 |
| 11 | 3.50 | 1.65 |
| 12 | 3.60 | 2.43 |
| 13 | 3.37 | 2.15 |
| 14 | 3.32 | 1.49 |
| 15 | 3.52 | 2.33 * |
| 16 | 3.42 | 2.13 |
| 17 | 3.37 | 2.11 |
| 18 | 3.57 | 2.35 |
| 19 | 3.40 | 1.48 * |
| 20 | 3.34 | 1.45 * |
| 21 | 3.55 | 1.73 * |
| 22 | 3.68 | 1.89 |
| 23 | 3.40 | 1.70 * |
| 24 | 3.34 | 1.70 * |
| 25 | 3.55 | 2.24 * |
| 26 | 3.67 | 2.46 |
| 27 | 3.38 | 2.19 |
| 28 | 3.32 | 1.50 |
| 29 | 3.52 | 2.36 * |
| 30 | 3.38 | 1.43 |
| 31 | 3.34 | 1.48 |
| 32 | 3.53 | 1.65 * |
| 33 | 3.65 | 2.40 |
| 34 | 3.40 | 2.18 |
| 35 | 3.33 | 2.16 |
| 36 | 3.54 | 1.73 * |
| 37 | 3.66 | 2.48 |
| Compounds | DFT Predicted | Experimental [18] | Anti-Tuberculosis Activity [18] | |||
|---|---|---|---|---|---|---|
| (V) | (V) | (V) | (V) | IC90 (μg/mL) | SI | |
| 1 | −1.46 | −2.63 | −1.658 | −1.91 | 4.21 | >23.77 |
| 2 | −1.26 | −3.05 * | −1.482 | −1.786 | 0.43 | >230.94 |
| 3 | −1.14 | −2.30 | −1.211 | −1.391 | <0.2 | >500 |
| 4 | −1.27 | −2.44 | −1.517 | −1.642 | 1.52 | >65.7 |
| 5 | −1.13 | −2.32 | −1.414 | −1.516 | 0.41 | >246.3 |
| 6 | −1.43 | −2.60 | −1.625 | −1.848 | 3.05 | >32.81 |
| 7 | −1.52 | −3.32 | −1.649 | −1.907 | 8.62 | >11.6 |
| 8 | −1.28 | −3.12 * | −1.525 | −1.72 | <0.2 | >500 |
| 9 | −1.16 | −2.31 | −1.387 | −1.557 | <0.19 | >153.84 |
| 10 | −1.16 | −2.33 | −1.439 | −1.543 | 1.153 | >26.02 |
| 11 | −1.29 | −3.14 | −1.551 | −1.696 | 0.504 | >198.41 |
| 12 | −1.19 | −2.37 | −1.485 | −1.664 | 0.517 | >58.03 |
| 13 | −1.42 | −2.65 | −1.598 | −1.866 | 2.85 | >10.54 |
| 14 | −1.47 | −3.30 | −1.618 | −1.876 | 8.9 | >3.37 |
| 15 | −1.27 | −2.46 * | −1.489 | −1.79 | ---- | ---- |
| 16 | −1.37 | −2.66 | −1.594 | −1.9 | 16.81 | ---- |
| 17 | −1.42 | −2.68 | −1.599 | −1.9 | >100 | ---- |
| 18 | −1.22 | −2.44 | −1.46 | −1.778 | 6.13 | ---- |
| 19 | −1.39 | −3.32 * | −1.53 | −1.738 | 11.04 | >6.52 |
| 20 | −1.45 | −3.34 * | −1.57 | −1.798 | 14.56 | ---- |
| 21 | −1.24 | −3.07 * | −1.47 | −1.737 | 29.68 | ---- |
| 22 | −1.12 | −2.90 | −1.374 | −1.582 | 51.86 | ---- |
| 23 | −1.39 | −3.09 * | −1.54 | −1.795 | 15.61 | ---- |
| 24 | −1.45 | −3.09 * | −1.58 | −1.806 | 78.22 | ---- |
| 25 | −1.24 | −2.56 * | −1.445 | −1.685 | 5.33 | >7.5 |
| 26 | −1.12 | −2.34 | −1.363 | −1.564 | 6.92 | >5.78 |
| 27 | −1.42 | −2.61 | −1.58 | −1.798 | 6.76 | >5.92 |
| 28 | −1.48 | −3.30 | −1.59 | −1.809 | 99.91 | ---- |
| 29 | −1.27 | −2.43 | −1.489 | −1.787 | 32.04 | ---- |
| 30 | −1.42 | −3.36 | −1.591 | −1.855 | 15.99 | ---- |
| 31 | −1.45 | −3.32 | −1.64 | −1.87 | 16.79 | ---- |
| 32 | −1.27 | −3.14 * | −1.491 | −1.802 | 60.43 | ---- |
| 33 | −1.15 | −2.39 | −1.41 | −1.531 | 66.54 | ---- |
| 34 | −1.40 | −2.61 | −1.611 | −1.854 | 22.75 | ---- |
| 35 | −1.46 | −2.63 | −1.624 | −1.887 | 13.22 | ---- |
| 36 | −1.25 | −3.06 * | −1.488 | −1.76 | 6.99 | >5.72 |
| 37 | −1.13 | −2.31 | −1.403 | −1.525 | 34.92 | ---- |
| Compound | R7/R6 | |||
|---|---|---|---|---|
| 6 | H/H | 0 | 0 | 0 |
| 7 | CH3/H | −0.17 | −0.07 | −0.12 |
| 8 | Cl/H | 0.23 | 0.37 | 0.30 |
| 9 | Cl/Cl | 0.46 | 0.74 | 0.60 |
| 10 | CF3/H | 0.55 | 0.41 | 0.48 |
| 11 | F/H | 0.06 | 0.34 | 0.20 |
| 12 | F/F | 0.12 | 0.68 | 0.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pooladian, F.; Crawford, P.W.; Kessler, J.M.; Casey, G.R.; Ragain, C.M. Reduction Potential Predictions for Thirty-Seven 1,4-di-N-Oxide Quinoxaline-2-Carboxamide Derivatives with Anti-Tuberculosis Activity. Compounds 2023, 3, 83-95. https://doi.org/10.3390/compounds3010007
Pooladian F, Crawford PW, Kessler JM, Casey GR, Ragain CM. Reduction Potential Predictions for Thirty-Seven 1,4-di-N-Oxide Quinoxaline-2-Carboxamide Derivatives with Anti-Tuberculosis Activity. Compounds. 2023; 3(1):83-95. https://doi.org/10.3390/compounds3010007
Chicago/Turabian StylePooladian, Faranak, Philip W. Crawford, Jonathan M. Kessler, Garrett R. Casey, and Christina M. Ragain. 2023. "Reduction Potential Predictions for Thirty-Seven 1,4-di-N-Oxide Quinoxaline-2-Carboxamide Derivatives with Anti-Tuberculosis Activity" Compounds 3, no. 1: 83-95. https://doi.org/10.3390/compounds3010007