
From the Light Chain Sequence to the Tissue Microenvironment: Contribution of the Mesangial Cells to Glomerular Amyloidosis

 and
and
Abstract
:
{kind=link}
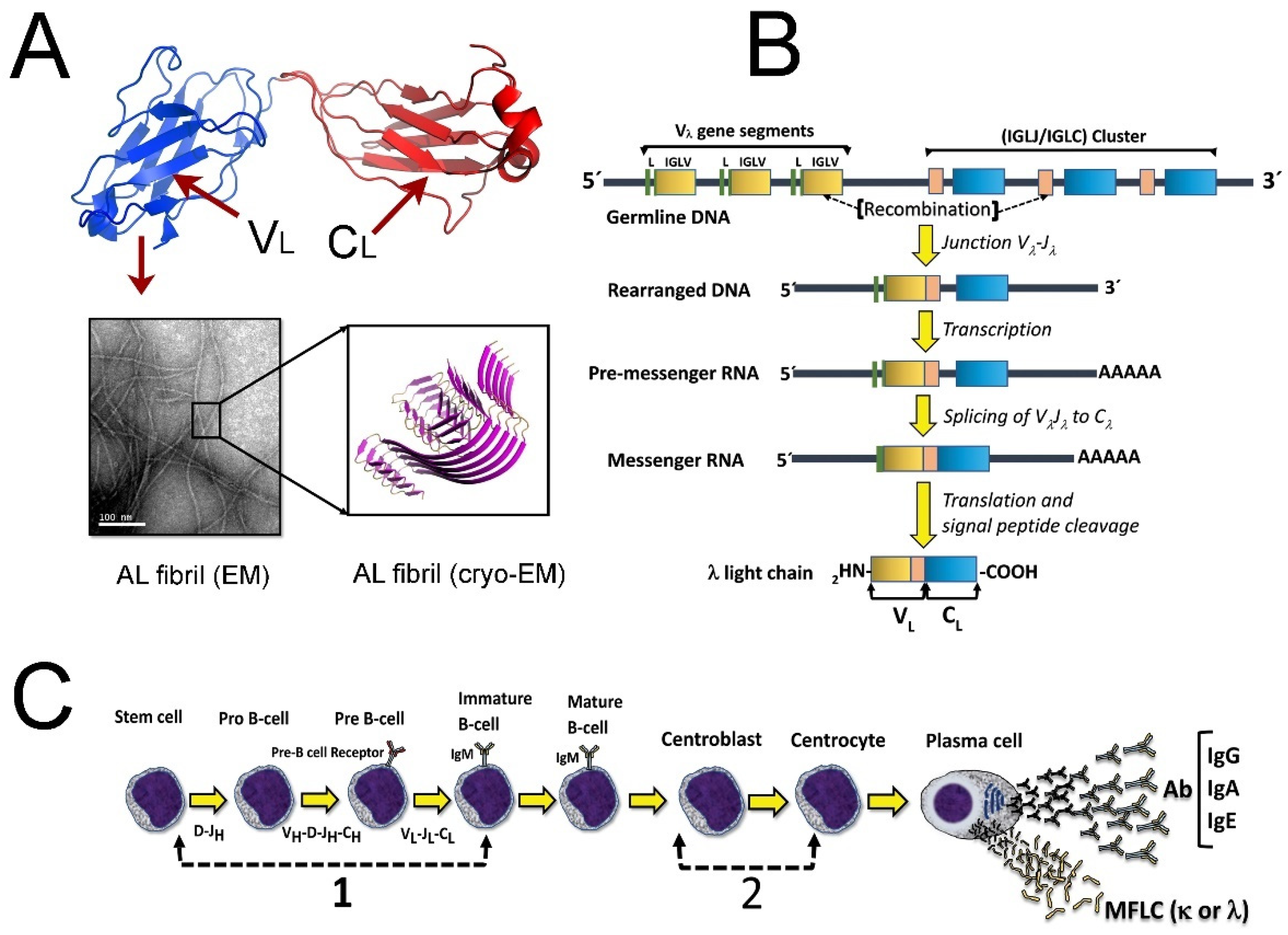{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Genetic and Structural Characteristics of the Immunoglobulin LCs
3. Structural Factors and Environmental Conditions That Drive the Amyloid Aggregation of the LCs
3.1. Pro-Fibrillogenic Sequences in LCs
3.2. The Folding Stability and Role of the Somatic Mutations
3.3. Contribution of the VL Gene Segment to the Propensity of the LCs to Form Amyloid
3.4. How Does the LC Protect Itself from Aggregation?
3.5. Intact LC as the AL Precursor and the Role of Proteolysis
3.6. Environmental Factors Modulating the Propensity of LCs to Form Amyloid
4. The Structure of AL Fibrils
5. Mechanism of Misfolding and Assembly of the LC into Amyloid Fibrils
6. Role of the Mesangial Cells in Glomerular Amyloidosis: Are They an Amyloid Factory?
7. Challenges in AL Amyloidosis Research
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Vogel, C.; Bashton, M.; Kerrison, N.D.; Chothia, C.; Teichmann, S.A. Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 2004, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Hou, J.; Chandonia, J.M.; Das, D.; Choi, I.G.; Kim, R.; Kim, S.H. Structure-based inference of molecular functions of proteins of unknown function from Berkeley Structural Genomics Center. J. Struct. Funct. Genom. 2007, 8, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Nassar, R.; Dignon, G.L.; Razban, R.M.; Dill, K.A. The Protein Folding Problem: The Role of Theory. J. Mol. Biol. 2021, 433, 167126. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inf. Ser. Workshop Genome Inf. 2000, 11, 161–171. [Google Scholar]
- Malagrino, F.; Diop, A.; Pagano, L.; Nardella, C.; Toto, A.; Gianni, S. Unveiling induced folding of intrinsically disordered proteins - Protein engineering, frustration and emerging themes. Curr. Opin. Struct. Biol. 2021, 72, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, P.P.; Orta, D.J.; Pena, I.; Flores, E.C.; Ramirez, J.U.; Beltran, H.I.; Alas, S.J. A Computational Approach to Studying Protein Folding Problems Considering the Crucial Role of the Intracellular Environment. J. Comput. Biol. 2017, 24, 995–1013. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Peng, P.H.; Hsu, K.W.; Wu, K.J. Liquid-liquid phase separation (LLPS) in cellular physiology and tumor biology. Am. J. Cancer Res. 2021, 11, 3766–3776. [Google Scholar]
- Lu, J.; Qian, J.; Xu, Z.; Yin, S.; Zhou, L.; Zheng, S.; Zhang, W. Emerging Roles of Liquid-Liquid Phase Separation in Cancer: From Protein Aggregation to Immune-Associated Signaling. Front. Cell Dev. Biol. 2021, 9, 631486. [Google Scholar] [CrossRef]
- Carey, J.L.; Guo, L. Liquid-Liquid Phase Separation of TDP-43 and FUS in Physiology and Pathology of Neurodegenerative Diseases. Front. Mol. Biosci. 2022, 9, 826719. [Google Scholar] [CrossRef]
- Temussi, P.A.; Tartaglia, G.G.; Pastore, A. The seesaw between normal function and protein aggregation: How functional interactions may increase protein solubility. Bioessays 2021, 43, e2100031. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, J.J.; Liu, S.; Zhao, J.; Jiang, Y.J.; Song, A.X.; Hu, H.Y. Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: Implication in a loss-of-function pathology. Sci. Rep. 2014, 4, 6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 2004, 279, 31374–31382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 2020, 3, 435. [Google Scholar] [CrossRef]
- Stefani, M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010, 277, 4602–4613. [Google Scholar] [CrossRef] [Green Version]
- Bemporad, F.; Ramazzotti, M. From the Evolution of Protein Sequences Able to Resist Self-Assembly to the Prediction of Aggregation Propensity. Int. Rev. Cell Mol. Biol. 2017, 329, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Monsellier, E.; Chiti, F. Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 2007, 8, 737–742. [Google Scholar] [CrossRef] [Green Version]
- Begum, T.; Ghosh, T.C. Understanding the effect of secondary structures and aggregation on human protein folding class evolution. J. Mol. Evol. 2010, 71, 60–69. [Google Scholar] [CrossRef]
- Das, M.; Gursky, O. Amyloid-Forming Properties of Human Apolipoproteins: Sequence Analyses and Structural Insights. Adv. Exp. Med. Biol. 2015, 855, 175–211. [Google Scholar] [CrossRef] [Green Version]
- Sant’Anna, R.; Braga, C.; Varejao, N.; Pimenta, K.M.; Grana-Montes, R.; Alves, A.; Cortines, J.; Cordeiro, Y.; Ventura, S.; Foguel, D. The importance of a gatekeeper residue on the aggregation of transthyretin: Implications for transthyretin-related amyloidoses. J. Biol. Chem. 2014, 289, 28324–28337. [Google Scholar] [CrossRef] [Green Version]
- De Baets, G.; Van Durme, J.; Rousseau, F.; Schymkowitz, J. A genome-wide sequence-structure analysis suggests aggregation gatekeepers constitute an evolutionary constrained functional class. J. Mol. Biol. 2014, 426, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S.; Richardson, D.C. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 2002, 99, 2754–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foy, S.G.; Wilson, B.A.; Bertram, J.; Cordes, M.H.J.; Masel, J. A Shift in Aggregation Avoidance Strategy Marks a Long-Term Direction to Protein Evolution. Genetics 2019, 211, 1345–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esperante, S.A.; Varejao, N.; Pinheiro, F.; Sant’Anna, R.; Luque-Ortega, J.R.; Alfonso, C.; Sora, V.; Papaleo, E.; Rivas, G.; Reverter, D.; et al. Disease-associated mutations impacting BC-loop flexibility trigger long-range transthyretin tetramer destabilization and aggregation. J. Biol. Chem. 2021, 297, 101039. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Hammarstrom, P.; Jiang, X.; Hurshman, A.R.; Powers, E.T.; Kelly, J.W. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. S4), 16427–16432. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.; Yerbury, J.; Poon, S.; Dabbs, R.; Wilson, M. Chapter 6: The chaperone action of Clusterin and its putative role in quality control of extracellular protein folding. Adv. Cancer Res. 2009, 104, 89–114. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Stewart, E.M.; Wyatt, A.R.; Wilson, M.R. Quality control of protein folding in extracellular space. EMBO Rep. 2005, 6, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Chevet, E.; Cameron, P.H.; Pelletier, M.F.; Thomas, D.Y.; Bergeron, J.J. The endoplasmic reticulum: Integration of protein folding, quality control, signaling and degradation. Curr. Opin. Struct. Biol. 2001, 11, 120–124. [Google Scholar] [CrossRef]
- Stefani, M. Protein aggregation diseases: Toxicity of soluble prefibrillar aggregates and their clinical significance. Methods Mol. Biol. 2010, 648, 25–41. [Google Scholar] [CrossRef]
- Nevone, A.; Merlini, G.; Nuvolone, M. Treating Protein Misfolding Diseases: Therapeutic Successes Against Systemic Amyloidoses. Front. Pharm. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Howie, A.J. Origins of a pervasive, erroneous idea: The "green birefringence" of Congo red-stained amyloid. Int. J. Exp. Pathol. 2019, 100, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Howie, A.J.; Brewer, D.B.; Howell, D.; Jones, A.P. Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab. Investig. 2008, 88, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, S.M.; Fine, G. Thioflavin-T for amyloid detection. Am. J. Clin. Pathol. 1967, 47, 588–593. [Google Scholar] [CrossRef]
- Rogers, D.R. Screening for Amyloid with the Thioflavin-T Fluorescent Method. Am. J. Clin. Pathol. 1965, 44, 59–61. [Google Scholar] [CrossRef] [Green Version]
- LeVine, H., 3rd. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzym. 1999, 309, 274–284. [Google Scholar] [CrossRef]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef]
- Chatani, E.; Yuzu, K.; Ohhashi, Y.; Goto, Y. Current Understanding of the Structure, Stability and Dynamic Properties of Amyloid Fibrils. Int J. Mol. Sci. 2021, 22, 4349. [Google Scholar] [CrossRef]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.I.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef]
- Vaxman, I.; Gertz, M. Recent Advances in the Diagnosis, Risk Stratification, and Management of Systemic Light-Chain Amyloidosis. Acta. Haematol. 2019, 141, 93–106. [Google Scholar] [CrossRef]
- Fotiou, D.; Dimopoulos, M.A.; Kastritis, E. Systemic AL Amyloidosis: Current Approaches to Diagnosis and Management. Hemasphere 2020, 4, e454. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Larson, D.R.; Kurtin, P.J.; Kumar, S.; Cerhan, J.R.; Therneau, T.M.; Rajkumar, S.V.; Vachon, C.M.; Dispenzieri, A. Incidence of AL Amyloidosis in Olmsted County, Minnesota, 1990 through 2015. Mayo Clin. Proc. 2019, 94, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Desport, E.; Bridoux, F.; Sirac, C.; Delbes, S.; Bender, S.; Fernandez, B.; Quellard, N.; Lacombe, C.; Goujon, J.M.; Lavergne, D.; et al. Al Amyloidosis . Orphanet. J. Rare Dis. 2012, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, N.; Mollee, P.; Augustson, B.; Brown, R.; Catley, L.; Gibson, J.; Harrison, S.; Ho, P.J.; Horvath, N.; Jaksic, W.; et al. Management of systemic AL amyloidosis: Recommendations of the Myeloma Foundation of Australia Medical and Scientific Advisory Group. Intern. Med. J. 2015, 45, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, M.; Usher, S.; Habib, M.H.; Ahmed, N.; Ali, J.; Begemann, M.; Shabbir, S.A.; Shune, L.; Al-Hilli, J.; Cossor, F.; et al. Current Updates on the Management of AL Amyloidosis. J. Hematol. 2020, 10, 147–161. [Google Scholar] [CrossRef]
- Quock, T.P.; Yan, T.; Chang, E.; Guthrie, S.; Broder, M.S. Epidemiology of AL amyloidosis: A real-world study using US claims data. Blood Adv. 2018, 2, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Bork, P.; Holm, L.; Sander, C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Eulitz, M.; Murphy, C.; Weiss, D.T.; Solomon, A. Serologic and chemical differentiation of human lambda III light chain variable regions. J. Immunol. 1991, 146, 3091–3096. [Google Scholar]
- Bentley, D.L.; Rabbitts, T.H. Evolution of immunoglobulin V genes: Evidence indicating that recently duplicated human V kappa sequences have diverged by gene conversion. Cell 1983, 32, 181–189. [Google Scholar] [CrossRef]
- Retter, I.; Althaus, H.H.; Munch, R.; Muller, W. VBASE2, an integrative V gene database. Nucleic Acids Res. 2005, 33, D671–D674. [Google Scholar] [CrossRef] [Green Version]
- Giudicelli, V.; Chaume, D.; Lefranc, M.P. IMGT/GENE-DB: A comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005, 33, D256–D261. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Stone, M.J. Dangerous small B-cell clones. Blood 2006, 108, 2520–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palladini, G.; Milani, P.; Merlini, G. Management of AL amyloidosis in 2020. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Bellotti, V.; Mangione, P.; Merlini, G. Review: Immunoglobulin light chain amyloidosis—The archetype of structural and pathogenic variability. J. Struct. Biol. 2000, 130, 280–289. [Google Scholar] [CrossRef]
- Rennella, E.; Morgan, G.J.; Kelly, J.W.; Kay, L.E. Role of domain interactions in the aggregation of full-length immunoglobulin light chains. Proc. Natl. Acad. Sci. USA 2019, 116, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Desikan, K.R.; Dhodapkar, M.V.; Hough, A.; Waldron, T.; Jagannath, S.; Siegel, D.; Barlogie, B.; Tricot, G. Incidence and impact of light chain associated (AL) amyloidosis on the prognosis of patients with multiple myeloma treated with autologous transplantation. Leuk Lymphoma 1997, 27, 315–319. [Google Scholar] [CrossRef]
- Xu, J.; Wang, M.; Shen, Y.; Yan, M.; Xie, W.; Wang, B.; Liu, H.; Cen, X. Effects of Amyloid Light-Chain Amyloidosis on Clinical Characteristics and Prognosis in Multiple Myeloma: A Single-Center Retrospective Study. Cancer. Manag. Res. 2021, 13, 1343–1356. [Google Scholar] [CrossRef]
- Del Pozo-Yauner, L.B.; Becerril, B.; Ochoa-Leyva, L.; Rodríguez-Ambriz, S.L.; Pérez Carrión, J.I.; Zavala-Padilla, G.; Sánchez-López, R.; Fernández Velasco, D.A. The structural determinants of the immunoglobulin light chain amyloid aggregation. In Physical Biology of Proteins and Peptides; Olivares-Quiroz, L.G.-L.O., Jardón-Valadez, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 1–28. [Google Scholar]
- Herrera, G.A.; Teng, J.; Turbat-Herrera, E.A.; Zeng, C.; Del Pozo-Yauner, L. Understanding Mesangial Pathobiology in AL-Amyloidosis and Monoclonal Ig Light Chain Deposition Disease. Kidney Int. Rep. 2020, 5, 1870–1893. [Google Scholar] [CrossRef]
- Davis, D.P.; Gallo, G.; Vogen, S.M.; Dul, J.L.; Sciarretta, K.L.; Kumar, A.; Raffen, R.; Stevens, F.J.; Argon, Y. Both the environment and somatic mutations govern the aggregation pathway of pathogenic immunoglobulin light chain. J. Mol. Biol. 2001, 313, 1021–1034. [Google Scholar] [CrossRef]
- Fandrich, M.; Dobson, C.M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002, 21, 5682–5690. [Google Scholar] [CrossRef] [Green Version]
- Iconomidou, V.A.; Chryssikos, G.D.; Gionis, V.; Galanis, A.S.; Cordopatis, P.; Hoenger, A.; Hamodrakas, S.J. Amyloid fibril formation propensity is inherent into the hexapeptide tandemly repeating sequence of the central domain of silkmoth chorion proteins of the A-family. J. Struct. Biol. 2006, 156, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Reches, M.; Gazit, E. Amyloidogenic hexapeptide fragment of medin: Homology to functional islet amyloid polypeptide fragments. Amyloid 2004, 11, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tenidis, K.; Waldner, M.; Bernhagen, J.; Fischle, W.; Bergmann, M.; Weber, M.; Merkle, M.L.; Voelter, W.; Brunner, H.; Kapurniotu, A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 2000, 295, 1055–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fandrich, M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell Mol. Life Sci. 2007, 64, 2066–2078. [Google Scholar] [CrossRef]
- Sabate, R.; Espargaro, A.; de Groot, N.S.; Valle-Delgado, J.J.; Fernandez-Busquets, X.; Ventura, S. The role of protein sequence and amino acid composition in amyloid formation: Scrambling and backward reading of IAPP amyloid fibrils. J. Mol. Biol. 2010, 404, 337–352. [Google Scholar] [CrossRef]
- Lopez de la Paz, M.; Serrano, L. Sequence determinants of amyloid fibril formation. Proc. Natl. Acad. Sci. USA 2004, 101, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Esteras-Chopo, A.; Serrano, L.; Lopez de la Paz, M. The amyloid stretch hypothesis: Recruiting proteins toward the dark side. Proc. Natl. Acad. Sci. USA 2005, 102, 16672–16677. [Google Scholar] [CrossRef] [Green Version]
- Ventura, S.; Zurdo, J.; Narayanan, S.; Parreno, M.; Mangues, R.; Reif, B.; Chiti, F.; Giannoni, E.; Dobson, C.M.; Aviles, F.X.; et al. Short amino acid stretches can mediate amyloid formation in globular proteins: The Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. USA 2004, 101, 7258–7263. [Google Scholar] [CrossRef] [Green Version]
- Teng, P.K.; Eisenberg, D. Short protein segments can drive a non-fibrillizing protein into the amyloid state. Protein Eng. Des. Sel. 2009, 22, 531–536. [Google Scholar] [CrossRef]
- Pastor, M.T.; Esteras-Chopo, A.; Serrano, L. Hacking the code of amyloid formation: The amyloid stretch hypothesis. Prion 2007, 1, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makin, O.S.; Atkins, E.; Sikorski, P.; Johansson, J.; Serpell, L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA 2005, 102, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiltzius, J.J.; Sievers, S.A.; Sawaya, M.R.; Cascio, D.; Popov, D.; Riekel, C.; Eisenberg, D. Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin). Protein Sci. 2008, 17, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Zamora, R.A.; Guillaume, S.; Al-Hilaly, Y.K.; Al-Garawi, Z.; Rodriguez-Alvarez, F.J.; Zavala-Padilla, G.; Perez-Carreon, J.I.; Rodriguez-Ambriz, S.L.; Herrera, G.A.; Becerril-Lujan, B.; et al. The CDR1 and Other Regions of Immunoglobulin Light Chains are Hot Spots for Amyloid Aggregation. Sci. Rep. 2019, 9, 3123. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Ladiwala, A.R.; Bhattacharya, M.; Tessier, P.M. Structure-based design of conformation- and sequence-specific antibodies against amyloid beta. Proc. Natl. Acad. Sci. USA 2012, 109, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Zhang, J.; Li, X.Y.; Yuan, L.; Pan, Y.F.; Chen, X.R.; Gao, T.M.; Qiao, J.T.; Qi, J.S. A novel antibody targeting sequence 31-35 in amyloid beta protein attenuates Alzheimer’s disease-related neuronal damage. Hippocampus 2017, 27, 122–133. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L.; Kumar, A.; Frangione, B.; Soto, C. Beta-sheet breaker peptide inhibitor of Alzheimer’s amyloidogenesis with increased blood-brain barrier permeability and resistance to proteolytic degradation in plasma. J. Neurobiol. 1999, 39, 371–382. [Google Scholar] [CrossRef]
- Thompson, M.J.; Sievers, S.A.; Karanicolas, J.; Ivanova, M.I.; Baker, D.; Eisenberg, D. The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4074–4078. [Google Scholar] [CrossRef] [Green Version]
- Brumshtein, B.; Esswein, S.R.; Sawaya, M.R.; Rosenberg, G.; Ly, A.T.; Landau, M.; Eisenberg, D.S. Identification of two principal amyloid-driving segments in variable domains of Ig light chains in systemic light-chain amyloidosis. J. Biol. Chem. 2018, 293, 19659–19671. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Annamalai, K.; Schmidt, M.; Grigorieff, N.; Fandrich, M. Cryo-EM reveals the steric zipper structure of a light chain-derived amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, 6200–6205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Pozo Yauner, L.; Ortiz, E.; Sanchez, R.; Sanchez-Lopez, R.; Guereca, L.; Murphy, C.L.; Allen, A.; Wall, J.S.; Fernandez-Velasco, D.A.; Solomon, A.; et al. Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human lambda 6 light chains. Proteins 2008, 72, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Belli, M.; Ramazzotti, M.; Chiti, F. Prediction of amyloid aggregation in vivo. EMBO Rep. 2011, 12, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Cui, W.; He, Z.; Shi, X.; Feng, K.; Ma, B.; Cai, Y.D. Cooperativity among short amyloid stretches in long amyloidogenic sequences. PLoS ONE 2012, 7, e39369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecoq, L.; Wiegand, T.; Rodriguez-Alvarez, F.J.; Cadalbert, R.; Herrera, G.A.; Del Pozo-Yauner, L.; Meier, B.H.; Bockmann, A. A Substantial Structural Conversion of the Native Monomer Leads to in-Register Parallel Amyloid Fibril Formation in Light-Chain Amyloidosis. Chembiochem 2019, 20, 1027–1031. [Google Scholar] [CrossRef]
- Hurle, M.R.; Helms, L.R.; Li, L.; Chan, W.; Wetzel, R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc. Natl. Acad. Sci. USA 1994, 91, 5446–5450. [Google Scholar] [CrossRef] [Green Version]
- Helms, L.R.; Wetzel, R. Specificity of abnormal assembly in immunoglobulin light chain deposition disease and amyloidosis. J. Mol. Biol. 1996, 257, 77–86. [Google Scholar] [CrossRef]
- Wetzel, R. Domain stability in immunoglobulin light chain deposition disorders. Adv. Protein Chem. 1997, 50, 183–242. [Google Scholar] [CrossRef]
- Kim, Y.S.; Cape, S.P.; Chi, E.; Raffen, R.; Wilkins-Stevens, P.; Stevens, F.J.; Manning, M.C.; Randolph, T.W.; Solomon, A.; Carpenter, J.F. Counteracting effects of renal solutes on amyloid fibril formation by immunoglobulin light chains. J. Biol. Chem. 2001, 276, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Blancas-Mejia, L.M.; Hammernik, J.; Marin-Argany, M.; Ramirez-Alvarado, M. Differential effects on light chain amyloid formation depend on mutations and type of glycosaminoglycans. J. Biol. Chem. 2015, 290, 4953–4965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blancas-Mejia, L.M.; Tischer, A.; Thompson, J.R.; Tai, J.; Wang, L.; Auton, M.; Ramirez-Alvarado, M. Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations. J. Mol. Biol. 2014, 426, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Pozo-Yauner, L.; Wall, J.S.; Gonzalez Andrade, M.; Sanchez-Lopez, R.; Rodriguez-Ambriz, S.L.; Perez Carreon, J.I.; Ochoa-Leyva, A.; Fernandez-Velasco, D.A. The N-terminal strand modulates immunoglobulin light chain fibrillogenesis. Biochem. Biophys. Res. Commun. 2014, 443, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Andrade, M.; Becerril-Lujan, B.; Sanchez-Lopez, R.; Cecena-Alvarez, H.; Perez-Carreon, J.I.; Ortiz, E.; Fernandez-Velasco, D.A.; del Pozo-Yauner, L. Mutational and genetic determinants of lambda6 light chain amyloidogenesis. FEBS J. 2013, 280, 6173–6183. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wall, J.S.; Meyer, J.; Murphy, C.; Randolph, T.W.; Manning, M.C.; Solomon, A.; Carpenter, J.F. Thermodynamic modulation of light chain amyloid fibril formation. J. Biol. Chem. 2000, 275, 1570–1574. [Google Scholar] [CrossRef] [Green Version]
- Marin-Argany, M.; Guell-Bosch, J.; Blancas-Mejia, L.M.; Villegas, S.; Ramirez-Alvarado, M. Mutations can cause light chains to be too stable or too unstable to form amyloid fibrils. Protein Sci. 2015, 24, 1829–1840. [Google Scholar] [CrossRef] [Green Version]
- Poshusta, T.L.; Sikkink, L.A.; Leung, N.; Clark, R.J.; Dispenzieri, A.; Ramirez-Alvarado, M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS ONE 2009, 4, e5169. [Google Scholar] [CrossRef]
- Sikkink, L.A.; Ramirez-Alvarado, M. Biochemical and aggregation analysis of Bence Jones proteins from different light chain diseases. Amyloid 2008, 15, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Wall, J.S.; Gupta, V.; Wilkerson, M.; Schell, M.; Loris, R.; Adams, P.; Solomon, A.; Stevens, F.; Dealwis, C. Structural basis of light chain amyloidogenicity: Comparison of the thermodynamic properties, fibrillogenic potential and tertiary structural features of four Vlambda6 proteins. J. Mol. Recognit. 2004, 17, 323–331. [Google Scholar] [CrossRef]
- Blancas-Mejia, L.M.; Tellez, L.A.; del Pozo-Yauner, L.; Becerril, B.; Sanchez-Ruiz, J.M.; Fernandez-Velasco, D.A. Thermodynamic and kinetic characterization of a germ line human lambda6 light-chain protein: The relation between unfolding and fibrillogenesis. J. Mol. Biol. 2009, 386, 1153–1166. [Google Scholar] [CrossRef]
- Wall, J.; Schell, M.; Murphy, C.; Hrncic, R.; Stevens, F.J.; Solomon, A. Thermodynamic instability of human lambda 6 light chains: Correlation with fibrillogenicity. Biochemistry 1999, 38, 14101–14108. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Alvarado, M.; Merkel, J.S.; Regan, L. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 8979–8984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Z.; Hu, D.; Zhu, M.; Fink, A.L. Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation. Biochemistry 2007, 46, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Misra, P.; Ramirez-Alvarado, M. Early events in light chain aggregation at physiological pH reveal new insights on assembly, stability, and aggregate dissociation. Amyloid 2021, 28, 113–124. [Google Scholar] [CrossRef]
- Rottenaicher, G.J.; Weber, B.; Ruhrnossl, F.; Kazman, P.; Absmeier, R.M.; Hitzenberger, M.; Zacharias, M.; Buchner, J. Molecular mechanism of amyloidogenic mutations in hypervariable regions of antibody light chains. J. Biol. Chem. 2021, 296, 100334. [Google Scholar] [CrossRef]
- Garay Sanchez, S.A.; Rodriguez Alvarez, F.J.; Zavala-Padilla, G.; Mejia-Cristobal, L.M.; Cruz-Rangel, A.; Costas, M.; Fernandez Velasco, D.A.; Melendez-Zajgla, J.; Del Pozo-Yauner, L. Stability and aggregation propensity do not fully account for the association of various germline variable domain gene segments with light chain amyloidosis. Biol. Chem. 2017, 398, 477–489. [Google Scholar] [CrossRef]
- Baden, E.M.; Owen, B.A.; Peterson, F.C.; Volkman, B.F.; Ramirez-Alvarado, M.; Thompson, J.R. Altered dimer interface decreases stability in an amyloidogenic protein. J. Biol. Chem. 2008, 283, 15853–15860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baden, E.M.; Randles, E.G.; Aboagye, A.K.; Thompson, J.R.; Ramirez-Alvarado, M. Structural insights into the role of mutations in amyloidogenesis. J. Biol. Chem. 2008, 283, 30950–30956. [Google Scholar] [CrossRef] [Green Version]
- Peterson, F.C.; Baden, E.M.; Owen, B.A.; Volkman, B.F.; Ramirez-Alvarado, M. A single mutation promotes amyloidogenicity through a highly promiscuous dimer interface. Structure 2010, 18, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Nawata, M.; Tsutsumi, H.; Kobayashi, Y.; Unzai, S.; Mine, S.; Nakamura, T.; Uegaki, K.; Kamikubo, H.; Kataoka, M.; Hamada, D. Heat-induced native dimerization prevents amyloid formation by variable domain from immunoglobulin light-chain REI. FEBS J. 2017, 284, 3114–3127. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Pondaven, S.P.; Hand, K.; Madine, J.; Jaroniec, C.P. Effect of amino acid mutations on the conformational dynamics of amyloidogenic immunoglobulin light-chains: A combined NMR and in silico study. Sci. Rep. 2017, 7, 10339. [Google Scholar] [CrossRef] [Green Version]
- Peterle, D.; Klimtchuk, E.S.; Wales, T.E.; Georgescauld, F.; Connors, L.H.; Engen, J.R.; Gursky, O. A Conservative Point Mutation in a Dynamic Antigen-binding Loop of Human Immunoglobulin lambda6 Light Chain Promotes Pathologic Amyloid Formation. J. Mol. Biol. 2021, 433, 167310. [Google Scholar] [CrossRef] [PubMed]
- Kazman, P.; Vielberg, M.T.; Pulido Cendales, M.D.; Hunziger, L.; Weber, B.; Hegenbart, U.; Zacharias, M.; Kohler, R.; Schonland, S.; Groll, M.; et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. eLife 2020, 9, e52300. [Google Scholar] [CrossRef] [PubMed]
- Bodi, K.; Prokaeva, T.; Spencer, B.; Eberhard, M.; Connors, L.H.; Seldin, D.C. AL-Base: A visual platform analysis tool for the study of amyloidogenic immunoglobulin light chain sequences. Amyloid 2009, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Gertz, M.A. Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Semin. Hematol. 1995, 32, 45–59. [Google Scholar]
- Solomon, A.; Frangione, B.; Franklin, E.C. Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J. Clin. Investig. 1982, 70, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Abe, M.; Wolfenbarger, D.; Weiss, D.T.; Solomon, A. Preferential expression of human lambda-light-chain variable-region subgroups in multiple myeloma, AL amyloidosis, and Waldenstrom’s macroglobulinemia. Clin. Immunol. Immunopathol. 1994, 71, 183–189. [Google Scholar] [CrossRef]
- Abraham, R.S.; Geyer, S.M.; Price-Troska, T.L.; Allmer, C.; Kyle, R.A.; Gertz, M.A.; Fonseca, R. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood 2003, 101, 3801–3808. [Google Scholar] [CrossRef] [PubMed]
- Comenzo, R.L.; Wally, J.; Kica, G.; Murray, J.; Ericsson, T.; Skinner, M.; Zhang, Y. Clonal immunoglobulin light chain variable region germline gene use in AL amyloidosis: Association with dominant amyloid-related organ involvement and survival after stem cell transplantation. Br. J. Haematol. 1999, 106, 744–751. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Perfetti, V.; Casarini, S.; Palladini, G.; Vignarelli, M.C.; Klersy, C.; Diegoli, M.; Ascari, E.; Merlini, G. Analysis of V(lambda)-J(lambda) expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (lambdaIII) as a new amyloid-associated germline gene segment. Blood 2002, 100, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Pokkuluri, P.R.; Solomon, A.; Weiss, D.T.; Stevens, F.J.; Schiffer, M. Tertiary structure of human lambda 6 light chains. Amyloid 1999, 6, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Dasari, S.; Kourelis, T.V.; Dispenzieri, A.; Murray, D.L.; King, R.L.; McPhail, E.D.; Ramirez-Alvarado, M.; Kumar, S.K.; Gertz, M.A. IGVL gene region usage correlates with distinct clinical presentation in IgM vs non-IgM light chain amyloidosis. Blood Adv. 2021, 5, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Comenzo, R.L.; Zhang, Y.; Martinez, C.; Osman, K.; Herrera, G.A. The tropism of organ involvement in primary systemic amyloidosis: Contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001, 98, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, J.S.; Getzoff, E.D.; Richardson, D.C. The beta bulge: A common small unit of nonrepetitive protein structure. Proc. Natl. Acad. Sci. USA 1978, 75, 2574–2578. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Santoyo, A.; del Pozo Yauner, L.; Fuentes-Silva, D.; Ortiz, E.; Rudino-Pinera, E.; Sanchez-Lopez, R.; Horjales, E.; Becerril, B.; Rodriguez-Romero, A. A single mutation at the sheet switch region results in conformational changes favoring lambda6 light-chain fibrillogenesis. J. Mol. Biol. 2010, 396, 280–292. [Google Scholar] [CrossRef]
- Radamaker, L.; Karimi-Farsijani, S.; Andreotti, G.; Baur, J.; Neumann, M.; Schreiner, S.; Berghaus, N.; Motika, R.; Haupt, C.; Walther, P.; et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat. Commun. 2021, 12, 6434. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Sutton, L.; Hayden, M.R. Small changes, big impact: Posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist 2011, 17, 475–492. [Google Scholar] [CrossRef]
- Lu, Y.; Jiang, Y.; Prokaeva, T.; Connors, L.H.; Costello, C.E. Oxidative Post-Translational Modifications of an Amyloidogenic Immunoglobulin Light Chain Protein. Int. J. Mass Spectrom. 2017, 416, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Zottig, X.; Laporte Wolwertz, M.; Golizeh, M.; Ohlund, L.; Sleno, L.; Bourgault, S. Effects of oxidative post-translational modifications on structural stability and self-assembly of lambda6 immunoglobulin light chain. Biophys. Chem. 2016, 219, 59–68. [Google Scholar] [CrossRef]
- Souillac, P.O.; Uversky, V.N.; Millett, I.S.; Khurana, R.; Doniach, S.; Fink, A.L. Elucidation of the molecular mechanism during the early events in immunoglobulin light chain amyloid fibrillation. Evidence for an off-pathway oligomer at acidic pH. J. Biol. Chem. 2002, 277, 12666–12679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Lopez, I.; Valdes-Garcia, G.; Romero Romero, S.; Maya Martinez, R.; Leal-Cervantes, A.I.; Costas, M.; Sanchez-Lopez, R.; Amero, C.; Pastor, N.; Fernandez Velasco, D.A. Localized conformational changes trigger the pH-induced fibrillogenesis of an amyloidogenic lambda light chain protein. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Epstein, W.V.; Tan, M.; Wood, I.S. Formation of "amyloid" fibrils in vitro by action of human kidney lysosomal enzymes on Bence Jones proteins. J. Lab. Clin. Med. 1974, 84, 107–110. [Google Scholar]
- Mukherjee, S.; Pondaven, S.P.; Jaroniec, C.P. Conformational flexibility of a human immunoglobulin light chain variable domain by relaxation dispersion nuclear magnetic resonance spectroscopy: Implications for protein misfolding and amyloid assembly. Biochemistry 2011, 50, 5845–5857. [Google Scholar] [CrossRef] [PubMed]
- Tzotzos, S.; Doig, A.J. Amyloidogenic sequences in native protein structures. Protein Sci. 2010, 19, 327–348. [Google Scholar] [CrossRef] [Green Version]
- Del Pozo Yauner, L.; Ortiz, E.; Becerril, B. The CDR1 of the human lambdaVI light chains adopts a new canonical structure. Proteins 2006, 62, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Weiss, D.T.; Williams, T.K. Experimental model of human light-chain-associated disease. Curr. Top. Microbiol. Immunol. 1992, 182, 261–267. [Google Scholar] [CrossRef]
- Stevens, P.W.; Raffen, R.; Hanson, D.K.; Deng, Y.L.; Berrios-Hammond, M.; Westholm, F.A.; Murphy, C.; Eulitz, M.; Wetzel, R.; Solomon, A.; et al. Recombinant immunoglobulin variable domains generated from synthetic genes provide a system for in vitro characterization of light-chain amyloid proteins. Protein Sci. 1995, 4, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Maya-Martinez, R.; French-Pacheco, L.; Valdes-Garcia, G.; Pastor, N.; Amero, C. Different Dynamics in 6aJL2 Proteins Associated with AL Amyloidosis, a Conformational Disease. Int. J. Mol. Sci. 2019, 20, 3078. [Google Scholar] [CrossRef] [Green Version]
- Maya-Martinez, R.; Gil-Rodriguez, P.; Amero, C. Solution structure of 6aJL2 and 6aJL2-R24G amyloidogenics light chain proteins. Biochem. Biophys. Res. Commun. 2015, 456, 695–699. [Google Scholar] [CrossRef]
- Pelaez-Aguilar, A.E.; Valdes-Garcia, G.; French-Pacheco, L.; Pastor, N.; Amero, C.; Rivillas-Acevedo, L. Site-Specific Interactions with Copper Promote Amyloid Fibril Formation for lambda6aJL2-R24G. ACS Omega 2020, 5, 7085–7095. [Google Scholar] [CrossRef]
- Valdes-Garcia, G.; Millan-Pacheco, C.; Pastor, N. Convergent mechanisms favor fast amyloid formation in two lambda 6a Ig light chain mutants. Biopolymers 2017, 107, e23027. [Google Scholar] [CrossRef] [PubMed]
- Khurana, R.; Gillespie, J.R.; Talapatra, A.; Minert, L.J.; Ionescu-Zanetti, C.; Millett, I.; Fink, A.L. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry 2001, 40, 3525–3535. [Google Scholar] [CrossRef] [PubMed]
- Klimtchuk, E.S.; Gursky, O.; Patel, R.S.; Laporte, K.L.; Connors, L.H.; Skinner, M.; Seldin, D.C. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry 2010, 49, 9848–9857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, G.J.; Kelly, J.W. The Kinetic Stability of a Full-Length Antibody Light Chain Dimer Determines whether Endoproteolysis Can Release Amyloidogenic Variable Domains. J. Mol. Biol. 2016, 428, 4280–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blancas-Mejia, L.M.; Martin, E.B.; Williams, A.; Wall, J.S.; Ramirez-Alvarado, M. Kinetic stability and sequence/structure studies of urine-derived Bence-Jones proteins from multiple myeloma and light chain amyloidosis patients. Biophys. Chem. 2017, 230, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Ein, D.; Eanes, E.D.; Bladen, H.A.; Terry, W.; Page, D.L. Creation of "amyloid" fibrils from Bence Jones proteins in vitro. Science 1971, 174, 712–714. [Google Scholar] [CrossRef]
- Linke, R.P.; Tischendorf, F.W.; Zucker-Franklin, D.; Franklin, E.C. The formation of amyloid-like fibrils in vitro from Bence Jones Proteins of the VlambdaI subclass. J. Immunol. 1973, 111, 24–26. [Google Scholar]
- Rocken, C.; Hegenbarth, V.; Schmitz, M.; Stix, B.; Schade, G.; Mohnert, A.; Roessner, A. Plasmacytoma of the tonsil with AL amyloidosis: Evidence of post-fibrillogenic proteolysis of the fibril protein. Virchows Arch. 2000, 436, 336–344. [Google Scholar] [CrossRef]
- Enqvist, S.; Sletten, K.; Westermark, P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J. Pathol. 2009, 219, 473–480. [Google Scholar] [CrossRef]
- Lavatelli, F.; Mazzini, G.; Ricagno, S.; Iavarone, F.; Rognoni, P.; Milani, P.; Nuvolone, M.; Swuec, P.; Caminito, S.; Tasaki, M.; et al. Mass spectrometry characterization of light chain fragmentation sites in cardiac AL amyloidosis: Insights into the timing of proteolysis. J. Biol. Chem. 2020, 295, 16572–16584. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, G.; Ricagno, S.; Caminito, S.; Rognoni, P.; Milani, P.; Nuvolone, M.; Basset, M.; Foli, A.; Russo, R.; Merlini, G.; et al. Protease-sensitive regions in amyloid light chains: What a common pattern of fragmentation across organs suggests about aggregation. FEBS J. 2021, 289, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Stevens, F.J.; Kisilevsky, R. Immunoglobulin light chains, glycosaminoglycans, and amyloid. Cell Mol. Life Sci. 2000, 57, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.; Shafton, A.; Altobell, L.J., 3rd; Tripuraneni, S.; Rogel, J.K.; Wentworth, A.D.; Lerner, R.A.; Wentworth, P., Jr. Lipid-derived aldehydes accelerate light chain amyloid and amorphous aggregation. Biochemistry 2008, 47, 7695–7705. [Google Scholar] [CrossRef]
- Herrera, G.A.; Del Pozo-Yauner, L.; Teng, J.; Zeng, C.; Shen, X.; Moriyama, T.; Ramirez Alcantara, V.; Liu, B.; Turbat-Herrera, E.A. Glomerulopathic Light Chain-Mesangial Cell Interactions: Sortilin-Related Receptor (SORL1) and Signaling. Kidney Int. Rep. 2021, 6, 1379–1396. [Google Scholar] [CrossRef]
- McLaughlin, R.W.; De Stigter, J.K.; Sikkink, L.A.; Baden, E.M.; Ramirez-Alvarado, M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein Sci. 2006, 15, 1710–1722. [Google Scholar] [CrossRef] [Green Version]
- Ren, R.; Hong, Z.; Gong, H.; Laporte, K.; Skinner, M.; Seldin, D.C.; Costello, C.E.; Connors, L.H.; Trinkaus-Randall, V. Role of glycosaminoglycan sulfation in the formation of immunoglobulin light chain amyloid oligomers and fibrils. J. Biol. Chem. 2010, 285, 37672–37682. [Google Scholar] [CrossRef] [Green Version]
- Murphy, R.M. Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim. Biophys. Acta 2007, 1768, 1923–1934. [Google Scholar] [CrossRef] [Green Version]
- Marin-Argany, M.; Lin, Y.; Misra, P.; Williams, A.; Wall, J.S.; Howell, K.G.; Elsbernd, L.R.; McClure, M.; Ramirez-Alvarado, M. Cell Damage in Light Chain Amyloidosis: Fibril internalization, toxicity and cell-mediated seeding. J. Biol Chem. 2016, 291, 19813–19825. [Google Scholar] [CrossRef] [Green Version]
- Makin, O.S.; Serpell, L.C. Examining the structure of the mature amyloid fibril. Biochem. Soc. Trans. 2002, 30, 521–525. [Google Scholar] [CrossRef]
- Eanes, E.D.; Glenner, G.G. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 1968, 16, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Muller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-beta amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schormann, N.; Murrell, J.R.; Liepnieks, J.J.; Benson, M.D. Tertiary structure of an amyloid immunoglobulin light chain protein: A proposed model for amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1995, 92, 9490–9494. [Google Scholar] [CrossRef] [Green Version]
- Serpell, L.C.; Sunde, M.; Benson, M.D.; Tennent, G.A.; Pepys, M.B.; Fraser, P.E. The protofilament substructure of amyloid fibrils. J. Mol. Biol. 2000, 300, 1033–1039. [Google Scholar] [CrossRef]
- Khurana, R.; Ionescu-Zanetti, C.; Pope, M.; Li, J.; Nielson, L.; Ramirez-Alvarado, M.; Regan, L.; Fink, A.L.; Carter, S.A. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys. J. 2003, 85, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Radamaker, L.; Lin, Y.H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schonland, S.O.; Fritz, G.; Schmidt, M.; Fandrich, M. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef] [Green Version]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schonland, S.; Bansal, A.; Schmidt, M.; Fandrich, M. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef]
- Piehl, D.W.; Blancas-Mejia, L.M.; Ramirez-Alvarado, M.; Rienstra, C.M. Solid-state NMR chemical shift assignments for AL-09 VL immunoglobulin light chain fibrils. Biomol. NMR Assign. 2017, 11, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, T.; Annamalai, K.; Sarkar, R.; Hegenbart, U.; Schonland, S.; Fandrich, M.; Reif, B. Solid state NMR assignments of a human lambda-III immunoglobulin light chain amyloid fibril. Biomol. NMR Assign. 2021, 15, 9–16. [Google Scholar] [CrossRef]
- Pradhan, T.; Annamalai, K.; Sarkar, R.; Huhn, S.; Hegenbart, U.; Schonland, S.; Fandrich, M.; Reif, B. Seeded fibrils of the germline variant of human lambda-III immunoglobulin light chain FOR005 have a similar core as patient fibrils with reduced stability. J. Biol. Chem. 2020, 295, 18474–18484. [Google Scholar] [CrossRef] [PubMed]
- Hora, M.; Sarkar, R.; Morris, V.; Xue, K.; Prade, E.; Harding, E.; Buchner, J.; Reif, B. MAK33 antibody light chain amyloid fibrils are similar to oligomeric precursors. PLoS ONE 2017, 12, e0181799. [Google Scholar] [CrossRef] [PubMed]
- Piehl, D.W.; Blancas-Mejia, L.M.; Wall, J.S.; Kennel, S.J.; Ramirez-Alvarado, M.; Rienstra, C.M. Immunoglobulin Light Chains Form an Extensive and Highly Ordered Fibril Involving the N- and C-Termini. ACS Omega 2017, 2, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Gonzalez, L.H.; Muresanu, L.; del Pozo-Yauner, L.; Sanchez, R.; Guereca, L.; Becerril, B.; Lucke, C. (1)H, (13)C and (15)N resonance assignment of 6aJL2(R25G), a highly fibrillogenic lambdaVI light chain variable domain. Biomol. NMR Assign. 2007, 1, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Rudino-Pinera, E.; Pelaez-Aguilar, A.E.; Amero, C.; Diaz-Vilchis, A. Crystal structure of 6aJL2-R24G light chain variable domain: Does crystal packing explain amyloid fibril formation? Biochem. Biophys. Rep. 2019, 20, 100682. [Google Scholar] [CrossRef]
- Honda, R.; Kuwata, K. Evidence for a central role of PrP helix 2 in the nucleation of amyloid fibrils. FASEB J. 2018, 32, 3641–3652. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.; Hughes, E.; Hussain, R.; Siligardi, G.; Baldock, S.; Madine, J.; Middleton, D.A. Heparin and Methionine Oxidation Promote the Formation of Apolipoprotein A-I Amyloid Comprising alpha-Helical and beta-Sheet Structures. Biochemistry 2017, 56, 1632–1644. [Google Scholar] [CrossRef]
- Chen, Y.C. Impact of a discordant helix on beta-amyloid structure, aggregation ability and toxicity. Eur. Biophys. J. 2017, 46, 681–687. [Google Scholar] [CrossRef]
- Lo, C.J.; Wang, C.C.; Huang, H.B.; Chang, C.F.; Shiao, M.S.; Chen, Y.C.; Lin, T.H. The Arctic mutation accelerates Abeta aggregation in SDS through reducing the helical propensity of residues 15-25. Amyloid 2015, 22, 8–18. [Google Scholar] [CrossRef]
- Berhanu, W.M.; Alred, E.J.; Hansmann, U.H. Stability of Osaka Mutant and Wild-Type Fibril Models. J. Phys. Chem. B 2015, 119, 13063–13070. [Google Scholar] [CrossRef]
- Grasso, G.; Leanza, L.; Morbiducci, U.; Danani, A.; Deriu, M.A. Aminoacid substitutions in the glycine zipper affect the conformational stability of amyloid beta fibrils. J. Biomol. Struct. Dyn. 2020, 38, 3908–3915. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Shivaprasad, S.; Kheterpal, I.; Wetzel, R. Thermodynamics of A beta(1-40) amyloid fibril elongation. Biochemistry 2005, 44, 12709–12718. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Shibata, H.; Oyama, K.; Ueda, T. Effect of O-glycosylation on amyloid fibril formation of the variable domain in the Vlambda6 light chain mutant Wil. Int J. Biol. Macromol. 2021, 166, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Omtvedt, L.A.; Bailey, D.; Renouf, D.V.; Davies, M.J.; Paramonov, N.A.; Haavik, S.; Husby, G.; Sletten, K.; Hounsell, E.F. Glycosylation of immunoglobulin light chains associated with amyloidosis. Amyloid 2000, 7, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Murray, D.; Dasari, S.; Milani, P.; Barnidge, D.; Madden, B.; Kourelis, T.; Arendt, B.; Merlini, G.; Ramirez-Alvarado, M.; et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: A potential path to earlier AL diagnosis for a subset of patients. Leukemia 2019, 33, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.L.; Maar, K.; Redhage, K.R.; Misra, P.; Blancas-Mejia, L.M.; Dick, C.J.; Wall, J.S.; Williams, A.; Dietz, A.B.; van Wijnen, A.J.; et al. Light chain amyloidosis induced inflammatory changes in cardiomyocytes and adipose-derived mesenchymal stromal cells. Leukemia 2020, 34, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- McWilliams-Koeppen, H.P.; Foster, J.S.; Hackenbrack, N.; Ramirez-Alvarado, M.; Donohoe, D.; Williams, A.; Macy, S.; Wooliver, C.; Wortham, D.; Morrell-Falvey, J.; et al. Light Chain Amyloid Fibrils Cause Metabolic Dysfunction in Human Cardiomyocytes. PLoS ONE 2015, 10, e0137716. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; Del Monte, F.; Ward, J.E.; Connors, L.H.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.A.; Jain, M.; Pimentel, D.R.; Wang, B.; Connors, L.H.; Skinner, M.; Apstein, C.S.; Liao, R. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ. Res. 2004, 94, 1008–1010. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.J.; Ramirez-Alvarado, M. Glycosaminoglycans promote fibril formation by amyloidogenic immunoglobulin light chains through a transient interaction. Biophys. Chem. 2011, 158, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Alexandrescu, A.T. Amyloid accomplices and enforcers. Protein Sci. 2005, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Martinez, O.D.; Hernandez-Santoyo, A.; Villalba-Velazquez, M.I.; Sanchez-Alcala, R.; Fernandez-Velasco, D.A.; Becerril, B. Stabilizing an amyloidogenic lambda6 light chain variable domain. FEBS J. 2017, 284, 3702–3717. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Abe, Y.; Ohkuri, T.; Mishima, T.; Monji, A.; Kanba, S.; Ueda, T. Mechanism for retardation of amyloid fibril formation by sugars in Vlambda6 protein. Protein Sci. 2013, 22, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishima, T.; Ohkuri, T.; Monji, A.; Kanemaru, T.; Abe, Y.; Ueda, T. Residual structures in the acid-unfolded states of Vlambda6 proteins affect amyloid fibrillation. J. Mol. Biol. 2009, 392, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Kazman, P.; Absmeier, R.M.; Engelhardt, H.; Buchner, J. Dissection of the amyloid formation pathway in AL amyloidosis. Nat. Commun. 2021, 12, 6516. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Esswein, S.R.; Salwinski, L.; Phillips, M.L.; Ly, A.T.; Cascio, D.; Sawaya, M.R.; Eisenberg, D.S. Inhibition by small-molecule ligands of formation of amyloid fibrils of an immunoglobulin light chain variable domain. eLife 2015, 4, e10935. [Google Scholar] [CrossRef]
- Wolwertz, M.L.; Nguyen, P.T.; Quittot, N.; Bourgault, S. Probing the role of lambda6 immunoglobulin light chain dimerization in amyloid formation. Biochim. Biophys. Acta 2016, 1864, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Owczarz, M.; Muller-Spath, T.; Rognoni, P.; Beeg, M.; Wu, H.; Salmona, M.; Morbidelli, M. In vitro aggregation behavior of a non-amyloidogenic lambda light chain dimer deriving from U266 multiple myeloma cells. PLoS ONE 2012, 7, e33372. [Google Scholar] [CrossRef] [Green Version]
- Myatt, E.A.; Westholm, F.A.; Weiss, D.T.; Solomon, A.; Schiffer, M.; Stevens, F.J. Pathogenic potential of human monoclonal immunoglobulin light chains: Relationship of in vitro aggregation to in vivo organ deposition. Proc. Natl. Acad. Sci. USA 1994, 91, 3034–3038. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.J.; Barrow, C.J. Protein conformational misfolding and amyloid formation: Characteristics of a new class of disorders that include Alzheimer’s and Prion diseases. Curr. Med. Chem. 2002, 9, 1751–1762. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.R. Techniques to study amyloid fibril formation in vitro. Methods 2004, 34, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Webster, P.; Taddei, N.; Clark, A.; Stefani, M.; Ramponi, G.; Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA 1999, 96, 3590–3594. [Google Scholar] [CrossRef] [Green Version]
- Bonifacio, M.J.; Sakaki, Y.; Saraiva, M.J. ‘In vitro’ amyloid fibril formation from transthyretin: The influence of ions and the amyloidogenicity of TTR variants. Biochim. Biophys. Acta 1996, 1316, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Janciauskiene, S.; Carlemalm, E.; Eriksson, S. In vitro amyloid fibril formation from alpha 1-antitrypsin. Biol. Chem. Hoppe Seyler 1995, 376, 103–109. [Google Scholar] [CrossRef]
- Klafki, H.W.; Pick, A.I.; Pardowitz, I.; Cole, T.; Awni, L.A.; Barnikol, H.U.; Mayer, F.; Kratzin, H.D.; Hilschmann, N. Reduction of disulfide bonds in an amyloidogenic Bence Jones protein leads to formation of “amyloid-like” fibrils in vitro. Biol. Chem. Hoppe Seyler 1993, 374, 1117–1122. [Google Scholar] [CrossRef]
- Colon, W.; Kelly, J.W. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar] [CrossRef]
- Connors, L.H.; Shirahama, T.; Skinner, M.; Fenves, A.; Cohen, A.S. In vitro formation of amyloid fibrils from intact beta 2-microglobulin. Biochem. Biophys. Res. Commun. 1985, 131, 1063–1068. [Google Scholar] [CrossRef]
- Bucciantini, M.; Rigacci, S.; Stefani, M. Amyloid Aggregation: Role of Biological Membranes and the Aggregate-Membrane System. J. Phys. Chem. Lett. 2014, 5, 517–527. [Google Scholar] [CrossRef]
- Husby, G.; Stenstad, T.; Magnus, J.H.; Sletten, K.; Nordvag, B.Y.; Marhaug, G. Interaction between circulating amyloid fibril protein precursors and extracellular tissue matrix components in the pathogenesis of systemic amyloidosis. Clin. Immunol. Immunopathol. 1994, 70, 2–9. [Google Scholar] [CrossRef]
- Keeling, J.; Teng, J.; Herrera, G.A. AL-amyloidosis and light-chain deposition disease light chains induce divergent phenotypic transformations of human mesangial cells. Lab. Investig. 2004, 84, 1322–1338. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; do Amaral, J.B.; Guimaraes, A.; Saraiva, M.J. Up-regulation of the extracellular matrix remodeling genes, biglycan, neutrophil gelatinase-associated lipocalin, and matrix metalloproteinase-9 in familial amyloid polyneuropathy. FASEB J. 2005, 19, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Vora, M.; Kevil, C.G.; Herrera, G.A. Contribution of human smooth muscle cells to amyloid angiopathy in AL (light-chain) amyloidosis. Ultrastruct Pathol. 2017, 41, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, J.; Wang, S. Serum Amyloid A Induces a Vascular Smooth Muscle Cell Phenotype Switch through the p38 MAPK Signaling Pathway. Biomed. Res. Int. 2017, 2017, 4941379. [Google Scholar] [CrossRef] [PubMed]
- Khvotchev, M.; Sudhof, T.C. Proteolytic processing of amyloid-beta precursor protein by secretases does not require cell surface transport. J. Biol. Chem. 2004, 279, 47101–47108. [Google Scholar] [CrossRef] [Green Version]
- Fisher, Y.; Nemirovsky, A.; Baron, R.; Monsonego, A. Dendritic cells regulate amyloid-beta-specific T-cell entry into the brain: The role of perivascular amyloid-beta. J. Alzheimers Dis. 2011, 27, 99–111. [Google Scholar] [CrossRef]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Shirahama, T.; Cohen, A.S. Intralysosomal formation of amyloid fibrils. Am. J. Pathol. 1975, 81, 101–116. [Google Scholar]
- Shirahama, T.; Cohen, A.S. An analysis of the close relationship of lysosomes to early deposits of amyloid. Ultrastructural evidence in experimental mouse amyloidosis. Am. J. Pathol. 1973, 73, 97–114. [Google Scholar]
- Shirahama, T.; Cohen, A.S. Lysosomal breakdown of amyloid fibrils by macrophages. Am. J. Pathol. 1971, 63, 463–486. [Google Scholar]
- Kluve-Beckerman, B.; Manaloor, J.J.; Liepnieks, J.J. A pulse-chase study tracking the conversion of macrophage-endocytosed serum amyloid A into extracellular amyloid. Arthritis Rheum. 2002, 46, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.J.; Cardelli, J.; Harris, E.; Baier, R.J.; Herrera, G.A. Monoclonal light chain-mesangial cell interactions: Early signaling events and subsequent pathologic effects. Lab. Investig. 2001, 81, 689–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, A.; Weiss, D.T.; Schell, M.; Hrncic, R.; Murphy, C.L.; Wall, J.; McGavin, M.D.; Pan, H.J.; Kabalka, G.W.; Paulus, M.J. Transgenic mouse model of AA amyloidosis. Am. J. Pathol. 1999, 154, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Tagouri, Y.M.; Sanders, P.W.; Picken, M.M.; Siegal, G.P.; Kerby, J.D.; Herrera, G.A. In vitro AL-amyloid formation by rat and human mesangial cells. Lab Investig. 1996, 74, 290–302. [Google Scholar]
- Christensen, E.I.; Birn, H. Megalin and cubilin: Synergistic endocytic receptors in renal proximal tubule. Am. J. Physiol. Ren. Physiol. 2001, 280, F562–F573. [Google Scholar] [CrossRef]
- Verroust, P.J.; Christensen, E.I. Megalin and cubilin—The story of two multipurpose receptors unfolds. Nephrol Dial. Transpl. 2002, 17, 1867–1871. [Google Scholar] [CrossRef] [Green Version]
- Herrera, G.A. Plasticity of mesangial cells: A basis for understanding pathological alterations. Ultrastruct Pathol. 2006, 30, 471–479. [Google Scholar] [CrossRef]
- Keeling, J.; Herrera, G.A. Matrix metalloproteinases and mesangial remodeling in light chain-related glomerular damage. Kidney Int. 2005, 68, 1590–1603. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.; Turbat-Herrera, E.A.; Herrera, G.A. Extrusion of amyloid fibrils to the extracellular space in experimental mesangial AL-amyloidosis: Transmission and scanning electron microscopy studies and correlation with renal biopsy observations. Ultrastruct Pathol. 2014, 38, 104–115. [Google Scholar] [CrossRef]
- Teng, J.; Russell, W.J.; Gu, X.; Cardelli, J.; Jones, M.L.; Herrera, G.A. Different types of glomerulopathic light chains interact with mesangial cells using a common receptor but exhibit different intracellular trafficking patterns. Lab. Investig. 2004, 84, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.; Turbat-Herrera, E.A.; Herrera, G.A. Role of translational research advancing the understanding of the pathogenesis of light chain-mediated glomerulopathies. Pathol Int. 2007, 57, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Isaac, J.; Kerby, J.D.; Russell, W.J.; Dempsey, S.C.; Sanders, P.W.; Herrera, G.A. In vitro modulation of AL-amyloid formation by human mesangial cells exposed to amyloidogenic light chains. Amyloid 1998, 5, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, G.A.; Teng, J.; Zeng, C.; Xu, H.; Liang, M.; Alexander, J.S.; Liu, B.; Boyer, C.; Turbat-Herrera, E.A. Phenotypic plasticity of mesenchymal stem cells is crucial for mesangial repair in a model of immunoglobulin light chain-associated mesangial damage. Ultrastruct Pathol. 2018, 42, 262–288. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145. [Google Scholar] [CrossRef]
- Wong, C.Y.; Cheong, S.K.; Mok, P.L.; Leong, C.F. Differentiation of human mesenchymal stem cells into mesangial cells in post-glomerular injury murine model. Pathology 2008, 40, 52–57. [Google Scholar] [CrossRef]
- Wong, C.Y.; Tan, E.L.; Cheong, S.K. In vitro differentiation of mesenchymal stem cells into mesangial cells when co-cultured with injured mesangial cells. Cell Biol. Int. 2014, 38, 497–501. [Google Scholar] [CrossRef]
- Kisilevsky, R. Amyloids: Tombstones or triggers? Nat. Med. 2000, 6, 633–634. [Google Scholar] [CrossRef]
- Absmeier, R.M.; Rottenaicher, G.J.; Svilenov, H.L.; Kazman, P.; Buchner, J. Antibodies gone bad - the molecular mechanism of light chain amyloidosis. FEBS J. 2022. [Google Scholar] [CrossRef]
- Otzen, D.E. Driving forces in amyloidosis: How does a light chain make a heavy heart? J. Biol. Chem. 2021, 296, 100785. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Pozo-Yauner, L.; Turbat-Herrera, E.A.; Pérez-Carreón, J.I.; Herrera, G.A. From the Light Chain Sequence to the Tissue Microenvironment: Contribution of the Mesangial Cells to Glomerular Amyloidosis. Hemato 2022, 3, 232-267. https://doi.org/10.3390/hemato3010019
Del Pozo-Yauner L, Turbat-Herrera EA, Pérez-Carreón JI, Herrera GA. From the Light Chain Sequence to the Tissue Microenvironment: Contribution of the Mesangial Cells to Glomerular Amyloidosis. Hemato. 2022; 3(1):232-267. https://doi.org/10.3390/hemato3010019
Chicago/Turabian StyleDel Pozo-Yauner, Luis, Elba A. Turbat-Herrera, Julio I. Pérez-Carreón, and Guillermo A. Herrera. 2022. "From the Light Chain Sequence to the Tissue Microenvironment: Contribution of the Mesangial Cells to Glomerular Amyloidosis" Hemato 3, no. 1: 232-267. https://doi.org/10.3390/hemato3010019