
Indolent T- and NK-Cell Lymphoproliferative Disorders of the Gastrointestinal Tract: Current Understanding and Outstanding Questions


Abstract
:1. Introduction
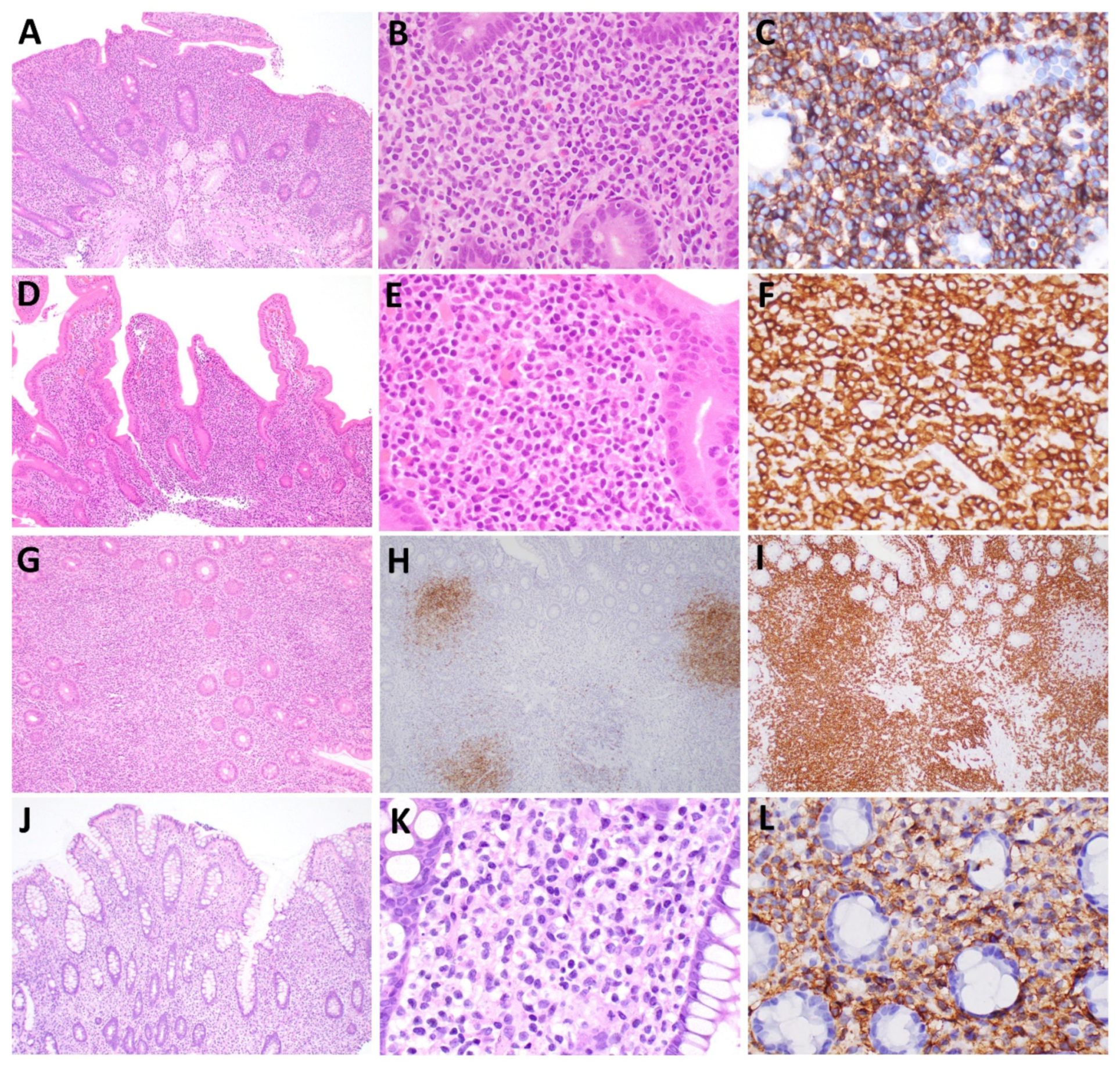
2. Indolent T-Cell Lymphoproliferative Disorder of the Gastrointestinal Tract (ITLPD-GI)
2.1. Immunophenotype

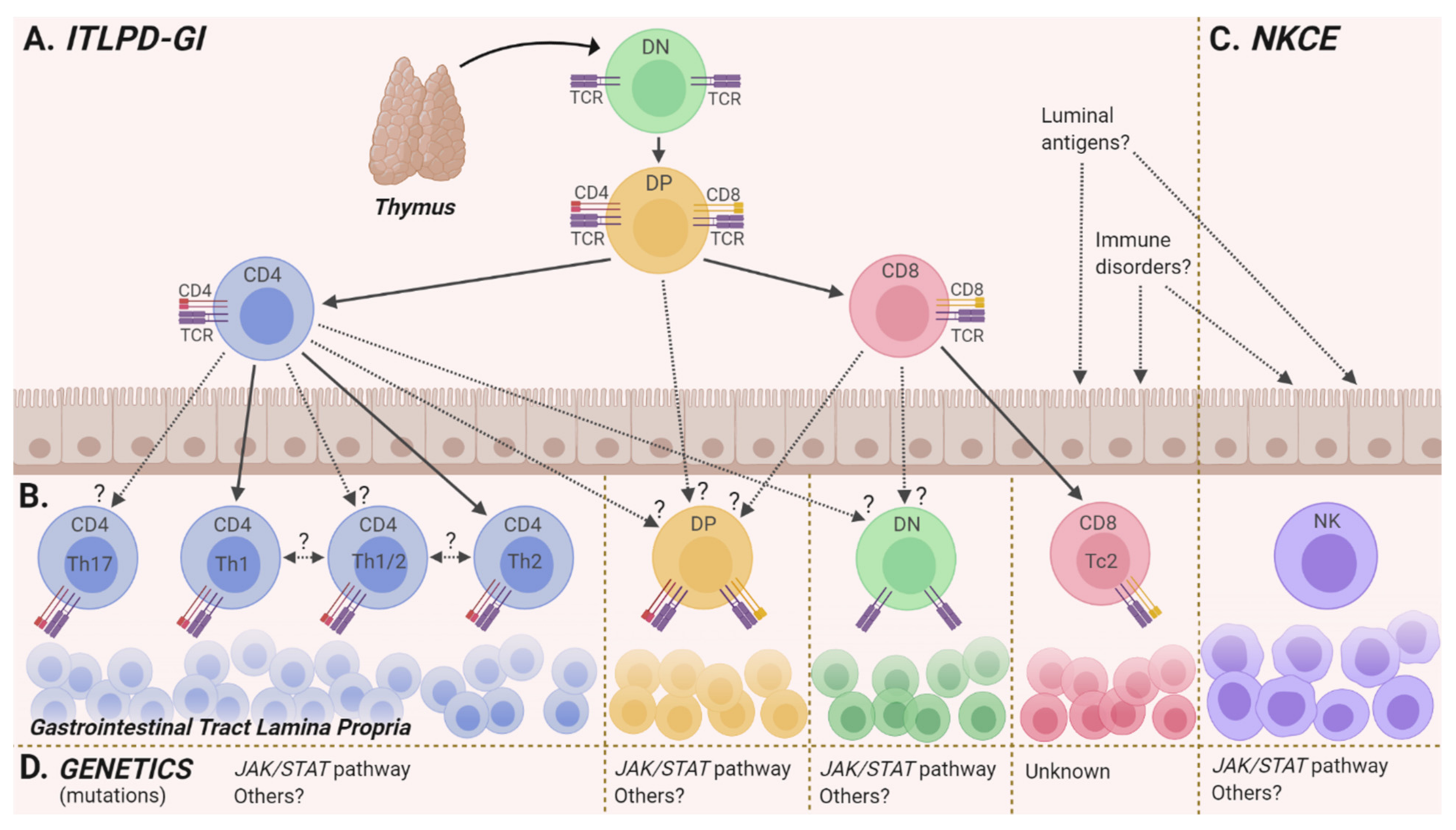
2.2. Cell(s) of Origin
2.3. Genetics
2.3.1. Clonality, Karyotyping, and Chromosome Microarray Analyses
2.3.2. Next-Generation Sequencing (NGS)
JAK/STAT Pathway Alterations
IL2 Structural Alterations
Other Altered Genes and Pathways
2.3.3. Genetic Stability and Evolution
2.4. Environmental and Immunologic Factors

{kind=link}
{kind=link}
| ITLPD-GI | NKCE | ||
|---|---|---|---|
| Site of involvement | GI tract: Small intestine (84%, 53/63), colon (48%, 30/63), stomach (38%, 24/63), oral cavity (5 cases), esophagus (2 cases) Abdominal lymph nodes: enlarged (47%, 22/47), biopsy confirmed involvement (12 cases) Other: Bone marrow (9 cases), blood, liver, peripheral lymph nodes | GI tract: Stomach (73%, 35/48), small intestine (31%, 15/48), colon (27%, 13/48) Other: Gallbladder, cystic duct lymph node, esophagus, vagina * | |
| Cytomorphology | Small size, round, oval or mildly irregular nuclei, condensed chromatin, inconspicuous nucleoli, scant/moderate cytoplasm | Medium/large size, ovoid or irregular nuclei, fine chromatin, inconspicuous nucleoli, moderate pale cytoplasm with occasional azurophilic granules | |
| Immunophenotype | CD4+: 48% (34/71), CD8+: 41% (29/71), DN: 7% (5/71), DP: 4% (3/71) Typical: CD2+, CD3+, CD5+, CD7+, TCRαβ+, CD103−, CD10−, BCL6−, PD1−, CXCL13−, FOXP3−, CD30−, MUM1−, MATK− Variant: CD5− (14%, 6/42), CD7− (24%, 9/38), CD103+ (5 cases), CD20+ (5 cases), CD56+ (1 case), PD1+ (2 cases), CXCL13+ and CD10+ (1 case) Cytotoxic markers: CD8: TIA1+ (96%), GrzB+ (30%) CD4: TIA+ (0%), GrzB+ (0%) Ki-67 index: <10% | Typical: sCD3−, cCD3+, CD5−, CD2+, CD7+, CD56+, CD4−, CD8−, TIA1+, GrzB+ Variant: CD2− (30%, 7/23), CD7− (1 case), CD8+ (11%, 5/46) Ki-67 index: <40% | |
| Other histopathologic features | Typical: Diffuse or nodular infiltrate largely confined to the lamina propria Other: Small clusters of lymphocytes infiltrating crypt or villous epithelium, scattered B-cell follicles, and occasionally granulomas | Well-circumscribed infiltrate within the lamina propria, often surrounded by a rim of polymorphous inflammatory cells Absence of angioinvasion/angiodestruction | |
| Chromosome/ genomic structural alterations | CD4 Translocations: STAT3-JAK2 (45%, 5/11) IL2-TNFRSF17 Gains/Losses: Gains: Chr: 1p, 1q, 8q, 13q, 15q, 17q, 19q, X Losses: Chr: 1p, 3q, 4q, 7q, 9p, 10p, 15q, 16p, 19p, 19q, 20q, X | CD8 Structural alterations: IL2 3′UTR-RHOH IL2 3′UTR del/IL2-TNIP3 † | None reported |
| Mutations | CD4 JAK/STAT pathway: STAT3, SOCS1 del Epigenetics: TET2, KMT2D, EZH2 Other: TNFAIP3, DIS3 | CD8 JAK/STAT pathway: None reported Other: MCM5 | JAK/STAT pathway: JAK3 (27%, 3/11) Other: PTPRS, AURKB, AXL, ERBB4, IGF1R, PIK3CB, CUL3, CHEK2, RUNX1T1, CIC, SMARCB1, SETD5 |
3. NK-Cell Enteropathy (NKCE)
3.1. Immunophenotype
3.2. Cell of Origin
3.3. Genetics
3.4. Environmental and Immunologic Factors
4. Disease Monitoring of ITLPD-GI and NKCE

5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Carbonnel, F.; Lavergne, A.; Messing, B.; Tsapis, A.; Berger, R.; Galian, A.; Nemeth, J.; Brouet, J.C.; Rambaud, J.C. Extensive small intestinal T-cell lymphoma of low-grade malignancy associated with a new chromosomal translocation. Cancer 1994, 73, 1286–1291. [Google Scholar] [CrossRef]
- Carbonnel, F.; D’Almagne, H.; Lavergne, A.; Matuchansky, C.; Brouet, J.C.; Sigaux, F.; Beaugerie, L.; Nemeth, J.; Coffin, B.; Cosnes, J.; et al. The clinicopathological features of extensive small intestinal CD4 T cell infiltration. Gut 1999, 45, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Fukayama, M.; Kawaguchi, K.; Hishima, T.; Hayashi, Y.; Funata, N.; Ibuka, T.; Koike, M.; Miyashita, H.; Tajima, T. Relapsing oral and colonic ulcers with monoclonal T-cell infiltration. A low grade mucosal T-lymphoproliferative disease of the digestive tract. Cancer 1995, 75, 1728–1733. [Google Scholar] [CrossRef]
- Leventaki, V.; Manning, J.T.; Luthra, R.; Mehta, P.; Oki, Y.; Romaguera, J.E.; Medeiros, L.J.; Vega, F. Indolent peripheral T-cell lymphoma involving the gastrointestinal tract. Hum. Pathol. 2014, 45, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Oishi, N.; Boddicker, R.L.; Hu, G.; Benson, H.K.; Ketterling, R.P.; Greipp, P.T.; Knutson, D.L.; Kloft-Nelson, S.M.; He, R.; et al. Recurrent STAT3-JAK2 fusions in indolent T-cell lymphoproliferative disorder of the gastrointestinal tract. Blood 2018, 131, 2262. [Google Scholar] [CrossRef] [Green Version]
- Edison, N.; Belhanes-Peled, H.; Eitan, Y.; Guthmann, Y.; Yeremenko, Y.; Raffeld, M.; Elmalah, I.; Trougouboff, P. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract after treatment with adalimumab in resistant Crohn’s colitis. Hum. Pathol. 2016, 57, 45–50. [Google Scholar] [CrossRef]
- Wang, X.; Ng, C.-S.; Chen, C.; Yu, G.; Yin, W. An unusual case report of indolent T-cell lymphoproliferative disorder with aberrant CD20 expression involving the gastrointestinal tract and bone marrow. Diagn. Pathol. 2018, 13, 82. [Google Scholar] [CrossRef]
- Zanelli, M.; Zizzo, M.; Sanguedolce, F.; Martino, G.; Soriano, A.; Ricci, S.; Ruiz, C.C.; Annessi, V.; Ascani, S. Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract: A tricky diagnosis of a gastric case. BMC Gastroenterol. 2020, 20, 336. [Google Scholar] [CrossRef]
- Guo, L.; Wen, Z.; Su, X.; Xiao, S.; Wang, Y. Indolent T-cell lymphoproliferative disease with synchronous diffuse large B-cell lymphoma. Medicine 2019, 98, e15323. [Google Scholar] [CrossRef]
- Montes-Moreno, S.; King, R.L.; Oschlies, I.; Ponzoni, M.; Goodlad, J.R.; Dotlic, S.; Traverse-Glehen, A.; Ott, G.; Ferry, J.A.; Calaminici, M. Update on lymphoproliferative disorders of the gastrointestinal tract: Disease spectrum from indolent lymphoproliferations to aggressive lymphomas. Virchows Arch. 2020, 476, 667–681. [Google Scholar] [CrossRef]
- Kohri, M.; Tsukasaki, K.; Akuzawa, Y.; Tanae, K.; Takahashi, N.; Saeki, T.; Okamura, D.; Ishikawa, M.; Maeda, T.; Kawai, N.; et al. Peripheral T-cell lymphoma with gastrointestinal involvement and indolent T-lymphoproliferative disorders of the gastrointestinal tract. Leuk. Res. 2020, 91, 106336. [Google Scholar] [CrossRef] [PubMed]
- Soon, G.; Wang, S. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract in a renal transplant patient: Diagnostic pitfalls and clinical challenges. Pathology 2017, 49, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, K.; Kim, K.H.; Bang, H.I.; Yoon, S.Y.; Choi, I.H. Diagnostic challenges of indolent peripheral T cell lymphoma: A case report and literature review. Medicine 2020, 99, e22657. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, K.; Fuchigami, T.; Nakamura, S.; Daimaru, Y.; Ohshima, K.; Sakai, Y.; Ichimaru, T. Primary gastrointestinal T-cell lymphoma resembling multiple lymphomatous polyposis. Gastroenterology 1996, 111, 778–782. [Google Scholar] [CrossRef]
- Saggini, A.; Baciorri, F.; Di Prete, M.; Zizzari, A.G.; Anemona, L. Oral manifestation of indolent T-cell lymphoproliferative disorder of the gastrointestinal tract: A potential diagnostic pitfall. J. Cutan. Pathol. 2020, 47, 494–496. [Google Scholar] [CrossRef]
- Nagaishi, T.; Yamada, D.; Suzuki, K.; Fukuyo, R.; Saito, E.; Fukuda, M.; Watabe, T.; Tsugawa, N.; Takeuchi, K.; Yamamoto, K.; et al. Indolent T cell lymphoproliferative disorder with villous atrophy in small intestine diagnosed by single-balloon enteroscopy. Clin. J. Gastroenterol. 2019, 12, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Goto, R.; Kawamura, N.; Watanabe, M.; Koshizuka, Y.; Shiratori, S.; Ara, M.; Honda, S.; Mitsuhashi, T.; Matsuno, Y.; Shimamura, T.; et al. Post-transplant indolent T cell lymphoproliferative disorder in living donor liver transplantation: A case report. Surg. Case Rep. 2020, 6, 147. [Google Scholar] [CrossRef]
- Takahashi, N.; Tsukasaki, K.; Kohri, M.; Akuzawa, Y.; Saeki, T.; Okamura, D.; Ishikawa, M.; Maeda, T.; Kawai, N.; Matsuda, A.; et al. Indolent T-cell lymphoproliferative disorder of the stomach successfully treated by radiotherapy. J. Clin. Exp. Hematop. 2020, 60, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, L.-G.; Zhang, X.-Y.; Wang, L.-L.; Zhang, L.; Xiao, Y.-J.; Xing, X.-M.; Lin, D.-L. Indolent T cell lymphoproliferative disorder of the gastrointestinal tract: An uncommon case with lymph node involvement and the classic Hodgkin’s lymphoma. J. Gastrointest. Oncol. 2020, 11, 812–819. [Google Scholar] [CrossRef]
- Ranheim, E.A.; Jones, C.; Zehnder, J.L.; Warnke, R.; Yuen, A. Spontaneously relapsing clonal, mucosal cytotoxic T-cell lymphoproliferative disorder: Case report and review of the literature. Am. J. Surg. Pathol. 2000, 24, 296–301. [Google Scholar] [CrossRef]
- Margolskee, E.; Jobanputra, V.; Lewis, S.K.; Alobeid, B.; Green, P.H.R.; Bhagat, G. Indolent Small Intestinal CD4+ T-cell Lymphoma Is a Distinct Entity with Unique Biologic and Clinical Features. PLoS ONE 2013, 8, e68343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, A.M.; Bailey, N.G.; Bonnett, M.; Jaffe, E.S.; Chan, W.C. Disease Progression in a Patient With Indolent T-Cell Lymphoproliferative Disease of the Gastrointestinal Tract. Int. J. Surg. Pathol. 2019, 27, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Zivny, J.; Banner, B.F.; Agrawal, S.; Pihan, G.; Barnard, G.F. CD4+T-cell lymphoproliferative disorder of the gut clinically mimicking celiac sprue. Dig. Dis. Sci. 2004, 49, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Svrcek, M.; Garderet, L.; Sebbagh, V.; Rosenzwajg, M.; Parc, Y.; Lagrange, M.; Bennis, M.; Lavergne-Slove, A.; Fléjou, J.-F.; Fabiani, B. Small intestinal CD4+ T-cell lymphoma: A rare distinctive clinicopathological entity associated with prolonged survival. Virchows Arch. 2007, 451, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.M.; Warnke, R.A.; Hu, Q.; Gaulard, P.; Copie-Bergman, C.; Alkan, S.; Wang, H.-Y.; Cheng, J.X.; Bacon, C.M.; Delabie, J.; et al. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood 2013, 122, 3599–3606. [Google Scholar] [CrossRef] [Green Version]
- Malamut, G.; Meresse, B.; Kaltenbach, S.; Derrieux, C.; Verkarre, V.; Macintyre, E.; Ruskone–Fourmestraux, A.; Fabiani, B.; Radford–Weiss, I.; Brousse, N.; et al. Small Intestinal CD4+ T-Cell Lymphoma Is a Heterogenous Entity With Common Pathology Features. Clin. Gastroenterol. Hepatol. 2014, 12, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Soderquist, C.R.; Patel, N.; Murty, V.V.; Betman, S.; Aggarwal, N.; Young, K.H.; Xerri, L.; Leeman-Neill, R.; Lewis, S.K.; Green, P.H.; et al. Genetic and phenotypic characterization of indolent T-cell lymphoproliferative disorders of the gastrointestinal tract. Haematologica 2020, 105, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Sena Teixeira Mendes, L.; DAttygalle, A.; Cunningham, D.; Benson, M.; Andreyev, J.; Gonzales-de-Castro, D.; Wotherspoon, A. CD4-positive small T-cell lymphoma of the intestine presenting with severe bile-acid malabsorption: A supportive symptom control approach. Br. J. Haematol. 2014, 167, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, Y.; Inada, K.-I.; Morita, K.; Suzuki, T. T-cell lymphomas diffusely involving the intestine: Report of two rare cases. Jpn. J. Clin. Oncol. 1996, 26, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. (Eds.) World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Lyon, France, 2016. [Google Scholar]
- Vega, F.; Chang, C.-C.; Schwartz, M.R.; Preti, H.A.; Younes, M.; Ewton, A.; Verm, R.; Jaffe, E.S. Atypical NK-cell proliferation of the gastrointestinal tract in a patient with antigliadin antibodies but not celiac disease. Am. J. Surg. Pathol. 2006, 30, 539–544. [Google Scholar] [CrossRef]
- Mansoor, A.; Pittaluga, S.; Beck, P.L.; Wilson, W.H.; Ferry, J.A.; Jaffe, E.S. NK-cell enteropathy: A benign NK-cell lymphoproliferative disease mimicking intestinal lymphoma: Clinicopathologic features and follow-up in a unique case series. Blood 2011, 117, 1447–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Yokoyama, M.; Ishizawa, S.; Terui, Y.; Nomura, K.; Marutsuka, K.; Nunomura, M.; Fukushima, N.; Yagyuu, T.; Nakamine, H.; et al. Lymphomatoid gastropathy: A distinct clinicopathologic entity of self-limited pseudomalignant NK-cell proliferation. Blood 2010, 116, 5631–5637. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Megahed, N.; Takata, K.; Asano, N.; Niwa, Y.; Hirooka, Y.; Goto, H. A case of lymphomatoid gastropathy: An indolent CD56-positive atypical gastric lymphoid proliferation, mimicking aggressive NK/T cell lymphomas. Pathol. Res. Pract. 2011, 207, 786–789. [Google Scholar] [CrossRef] [PubMed]
- McElroy, M.K.; Read, W.L.; Harmon, G.S.; Weidner, N. A unique case of an indolent CD56-positive T-cell lymphoproliferative disorder of the gastrointestinal tract: A lesion potentially misdiagnosed as natural killer/T-cell lymphoma. Ann. Diagn. Pathol. 2011, 15, 370–375. [Google Scholar] [CrossRef]
- Isom, J.A.; Arroyo, M.R.; Reddy, D.; Joshi-Guske, P.; Al-Quran, S.Z.; Li, Y.; Allan, R.W. NK cell enteropathy: A case report with 10 years of indolent clinical behaviour. Histopathology 2018, 73, 345–350. [Google Scholar] [CrossRef]
- Wang, R.; Kariappa, S.; Toon, C.W.; Varikatt, W. NK-cell enteropathy, a potential diagnostic pitfall of intestinal lymphoproliferative disease. Pathology 2019, 51, 338–340. [Google Scholar] [CrossRef]
- Dargent, J.-L.; Tinton, N.; Trimech, M.; de Leval, L. Lymph node involvement by enteropathy-like indolent NK-cell proliferation. Virchows Arch. 2021, 478, 1197–1202. [Google Scholar] [CrossRef]
- Terai, T.; Sugimoto, M.; Uozaki, H.; Kitagawa, T.; Kinoshita, M.; Baba, S.; Yamada, T.; Osawa, S.; Sugimoto., K. Lymphomatoidgastropathy mimicking extranodal NK/T cell lymphoma, nasal type: A case report. World J. Gastroenterol. 2012, 18, 2140–2144. [Google Scholar] [CrossRef]
- Koh, J.; Go, H.; Lee, W.A.; Jeon, Y.K. Benign Indolent CD56-Positive NK-Cell Lymphoproliferative Lesion Involving Gastrointestinal Tract in an Adolescent. Korean J. Pathol. 2014, 48, 73. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, Y.; Matsuzono, E.; Yokoyama, F.; Ohara, Y.; Sugai, N.; Seki, H.; Miura, A.; Fujita, J.; Suzuki, J.; Fujisawa, T.; et al. A case of lymphomatoid gastropathy: A self-limited pseudomalignant natural killer (NK)-cell proliferative disease mimicking NK/T-cell lymphomas. Clin. J. Gastroenterol. 2013, 6, 287–290. [Google Scholar] [CrossRef]
- Takata, K.; Noujima-Harada, M.; Miyata-Takata, T.; Ichimura, K.; Sato, Y.; Miyata, T.; Naruse, K.; Iwamoto, T.; Tari, A.; Masunari, T.; et al. Clinicopathologic Analysis of 6 Lymphomatoid Gastropathy Cases. Am. J. Surg. Pathol. 2015, 39, 1259–1266. [Google Scholar] [CrossRef]
- Xia, D.; A Morgan, E.; Berger, D.; Pinkus, G.S.; Ferry, J.A.; Zukerberg, L.R. NK-Cell Enteropathy and Similar Indolent Lymphoproliferative Disorders. Am. J. Clin. Pathol. 2018, 151, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Fujishima, F.; Ichinohasama, R.; Imatani, A.; Asano, N.; Harigae, H. A case of benign natural killer cell proliferative disorder of the stomach (gastric manifestation of natural killer cell lymphomatoid gastroenteropathy) mimicking extranodal natural killer/T-cell lymphoma. Leuk. Lymphoma 2011, 52, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Park, J.S.; Jeong, S.H.; Yim, H.; Han, J.H. Indolent NK cell proliferative lesion mimicking NK/T cell lymphoma in the gallbladder. Hum. Pathol. Case Rep. 2016, 5, 39–42. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Gupta, G.K.; Yao, J.; Jang, Y.J.; Xi, L.; Baik, J.; Sigler, A.; Kumar, A.; Moskowitz, A.J.; Arcila, M.E.; et al. Recurrent somatic JAK3 mutations in NK-cell enteropathy. Blood 2019, 134, 986–991. [Google Scholar] [CrossRef]
- Jahnsen, F.L.; Farstad, I.N.; Aanesen, J.P.; Brandtzaeg, P. Phenotypic distribution of T cells in human nasal mucosa differs from that in the gut. Am. J. Respir. Cell Mol. Biol. 1998, 18, 392–401. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Spencer, J. Mouse and human intestinal immunity: Same ballpark, different players; Different rules, same score. Mucosal Immunol. 2011, 4, 148–157. [Google Scholar] [CrossRef]
- Cosorich, I.; McGuire, H.M.; Warren, J.; Danta, M.; King, C. CCR9 expressing T helper and T follicular helper cells exhibit site-specific identities during inflammatory disease. Front. Immunol. 2019, 10, 2899. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2009, 20, 4–12. [Google Scholar] [CrossRef]
- Peine, M.; Rausch, S.; Helmstetter, C.; Fröhlich, A.; Hegazy, A.N.; Kühl, A.A.; Grevelding, C.G.; Höfer, T.; Hartmann, S.; Löhning, M. Stable T-bet+GATA-3+ Th1/Th2 Hybrid Cells Arise In Vivo, Can Develop Directly from Naive Precursors, and Limit Immunopathologic Inflammation. PLoS Biol. 2013, 11, e1001633. [Google Scholar] [CrossRef] [Green Version]
- Hegazy, A.N.; Peine, C.; Helmstetter, C.; Panse, I.; Fröhlich, A.; Bergthaler, A.; Flatz, L.; Pinschewer, D.D.; Radbruch, A.; Löhning, M. Interferons Direct Th2 Cell Reprogramming to Generate a Stable GATA-3+T-bet+ Cell Subset with Combined Th2 and Th1 Cell Functions. Immunity 2010, 32, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Harland, K.; Kedzierska, K.; Kelso, A. Exposure of human CD8+ T cells to type-2 cytokines impairs division and differentiation and induces limited polarization. Front. Immunol. 2018, 9, 1141. [Google Scholar] [CrossRef] [PubMed]
- Priatel, J.J.; Utting, O.; Teh, H.-S. TCR/self-antigen interactions drive double-negative T cell peripheral expansion and differentiation into suppressor cells. J. Immunol. 2001, 167, 6188–6194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, D.; Hedrich, C.M. TCRαβ+CD3+CD4−CD8− (double negative) T cells in autoimmunity. Autoimmun. Rev. 2018, 17, 422–430. [Google Scholar] [CrossRef]
- Pahar, B.; Lackner, A.A.; Veazey, R.S. Intestinal double-positive CD4+CD8+ T cells are highly activated memory cells with an increased capacity to produce cytokines. Eur. J. Immunol. 2006, 36, 583–592. [Google Scholar] [CrossRef]
- Nascimbeni, M.; Shin, E.-C.; Chiriboga, L.; Kleiner, D.; Rehermann, B. Peripheral CD4(+)CD8(+) T cells are differentiated effector memory cells with antiviral functions. Blood 2004, 104, 478–486. [Google Scholar] [CrossRef]
- Keir, M.E.; Francisco, L.M.; Sharpe, A.H. PD-1 and its ligands in T-cell immunity. Curr. Opin. Immunol. 2007, 19, 309–314. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Miao, N.; Sun, T. Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 2019, 156, 29. [Google Scholar] [CrossRef] [Green Version]
- Laâbi, Y.; Gras, M.P.; Carbonnel, F.; Brouet, J.C.; Berger, R.; Larsen, C.J.; Tsapis, A. A new gene, BCM, on chromosome 16 is fused to the interleukin 2 gene by a t(4;16)(q26;p13) translocation in a malignant T cell lymphoma. EMBO J. 1992, 11, 3897–3904. [Google Scholar] [CrossRef]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; Van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettersperger, J.; Montcuquet, N.; Malamut, G.; Guegan, N.; Lastra, S.L.; Gayraud, S.; Reimann, C.; Vidal, E.; Cagnard, N.; Villarese, P.; et al. Interleukin-15-Dependent T-Cell-like Innate Intraepithelial Lymphocytes Develop in the Intestine and Transform into Lymphomas in Celiac Disease. Immunity 2016, 45, 610–625. [Google Scholar] [CrossRef] [PubMed]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 411–433. [Google Scholar] [CrossRef] [PubMed]
- Myer, V.E.; Fan, X.C.; Steitz, J.A. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 1997, 16, 2130–2139. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Del Gatto–Konczak, F.; Wu, Z.; Karin, M. Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science 1998, 280, 1945–1949. [Google Scholar] [CrossRef]
- Schwartz, F.H.; Cai, Q.; Fellmann, E.; Hartmann, S.; Mäyränpää, M.I.; Karjalainen-Lindsberg, M.-L.; Sundström, C.; Scholtysik, R.; Hansmann, M.-L.; Küppers, R. TET2 mutations in B cells of patients affected by angioimmunoblastic T-cell lymphoma. J. Pathol. 2017, 242, 129–133. [Google Scholar] [CrossRef]
- Soderquist, C.R.; Lewis, S.K.; Gru, A.A.; Vlad, G.; Williams, E.S.; Hsiao, S.; Mansukhani, M.M.; Park, D.C.; Bacchi, C.E.; Alobeid, B.; et al. Immunophenotypic Spectrum and Genomic Landscape of Refractory Celiac Disease Type II. Am. J. Surg. Pathol. 2021, 45, 905–916. [Google Scholar] [CrossRef]
- Cording, S.; Lhermitte, L.; Malamut, G.; Berrabah, S.; Trinquand, A.; Guegan, N.; Villarese, P.; Kaltenbach, S.; Meresse, B.; Khater, S.; et al. Oncogenetic landscape of lymphomagenesis in coeliac disease. Gut 2021, 71, 497–508. [Google Scholar] [CrossRef]
- Yu, X.; Vargas, J.; Green, P.H.; Bhagat, G. Innate Lymphoid Cells and Celiac Disease: Current Perspective. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 803–814. [Google Scholar] [CrossRef]
- Shi, F.-D.; Ljunggren, H.-G.; La Cava, A.; Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol. 2011, 11, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, C.; Chasson, L.; Luci, C.; Tomasello, E.; Geissmann, F.; Vivier, E.; Walzer, T. The trafficking of natural killer cells. Immunol. Rev. 2007, 220, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S.K.; Colonna, M. Innate lymphoid cells in mucosal immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, R.; Ring, K.; Williams, E.; Portell, C.; Jaffe, E.S.; Gru, A.A. An Enteropathy-like Indolent NK-Cell Proliferation Presenting in the Female Genital Tract. Am. J. Surg. Pathol. 2020, 44, 561. [Google Scholar] [CrossRef]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [Green Version]
- Van den Brand, M.; Rijntjes, J.; Möbs, M.; Steinhilber, J.; van der Klift, M.Y.; Heezen, K.C.; Kroeze, L.I.; Reigl, T.; Porc, J.; Darzentas, N.; et al. Next-Generation Sequencing–Based Clonality Assessment of Ig Gene Rearrangements: A Multicenter Validation Study by EuroClonality-NGS. J. Mol. Diagn. 2021, 23, 1105–1115. [Google Scholar] [CrossRef]
- Rossi, D.; Spina, V.; Bruscaggin, A.; Gaidano, G. Liquid biopsy in lymphoma. Haematologica 2019, 104, 648. [Google Scholar] [CrossRef] [Green Version]
- Freiche, V.; Cordonnier, N.; Paulin, M.V.; Huet, H.; Turba, M.E.; Macintyre, E.; Malamut, G.; Cerf-Bensussan, N.; Molina, T.J.; Hermine, O.; et al. Feline low-grade intestinal T cell lymphoma: A unique natural model of human indolent T cell lymphoproliferative disorder of the gastrointestinal tract. Lab. Investig. 2021, 101, 794–804. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soderquist, C.R.; Bhagat, G. Indolent T- and NK-Cell Lymphoproliferative Disorders of the Gastrointestinal Tract: Current Understanding and Outstanding Questions. Hemato 2022, 3, 219-231. https://doi.org/10.3390/hemato3010018
Soderquist CR, Bhagat G. Indolent T- and NK-Cell Lymphoproliferative Disorders of the Gastrointestinal Tract: Current Understanding and Outstanding Questions. Hemato. 2022; 3(1):219-231. https://doi.org/10.3390/hemato3010018
Chicago/Turabian StyleSoderquist, Craig R., and Govind Bhagat. 2022. "Indolent T- and NK-Cell Lymphoproliferative Disorders of the Gastrointestinal Tract: Current Understanding and Outstanding Questions" Hemato 3, no. 1: 219-231. https://doi.org/10.3390/hemato3010018