
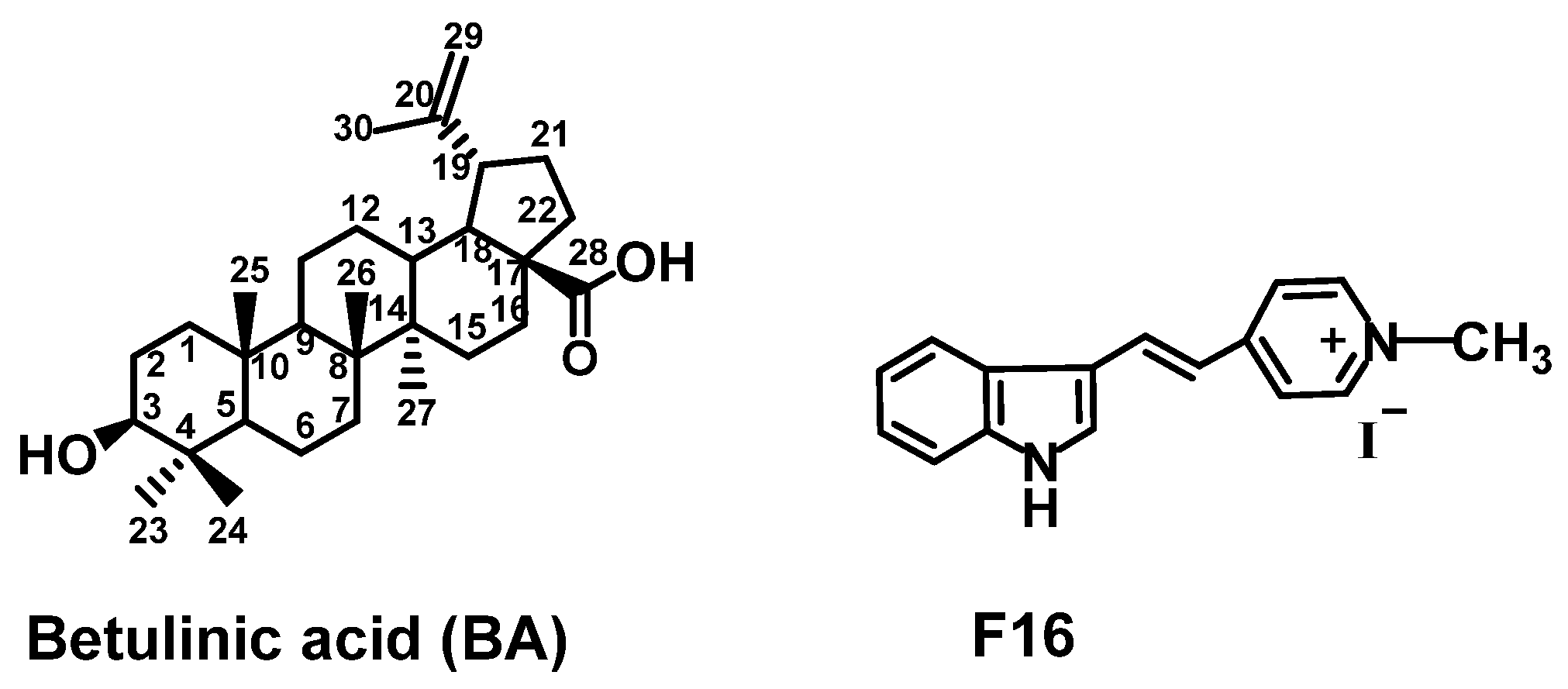
Effective Synthesis of a Novel Betulinic Acid Conjugate with Mitochondria-Targeting Cation F16 †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
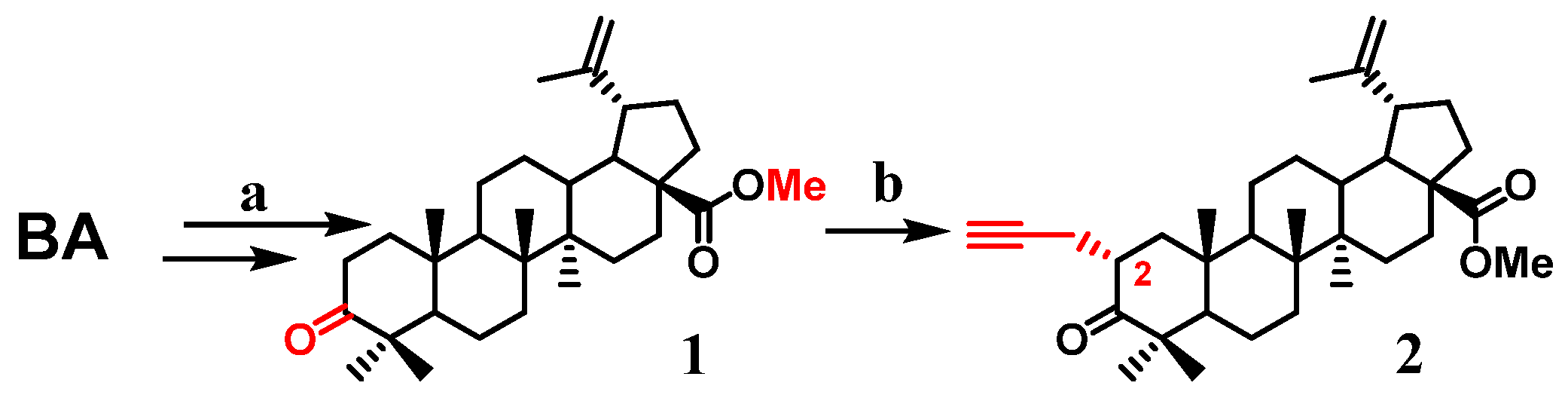
2.1. Chemistry
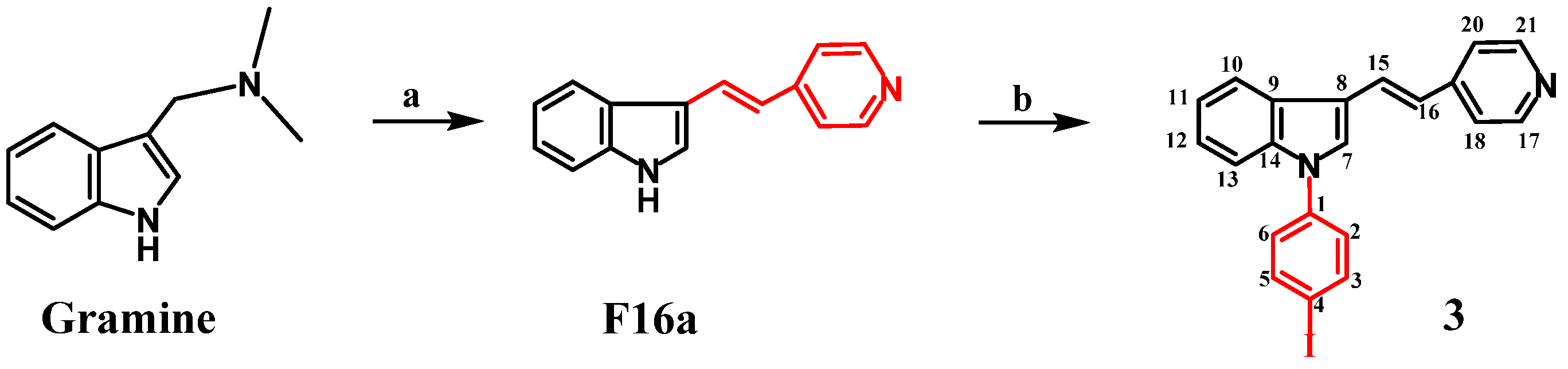
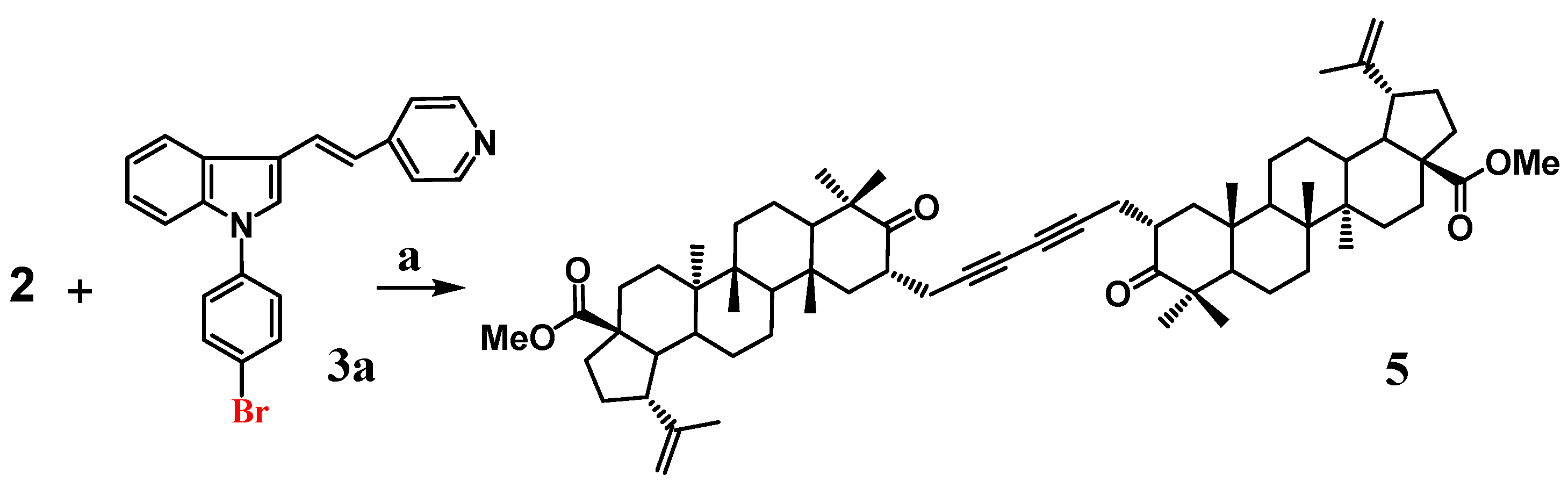
2.1.1. Synthesis of 1-Iodo-4-{(E)-4-[2-(1H-indol-3-yl)vinyl]-pyridine}phenyl (3)
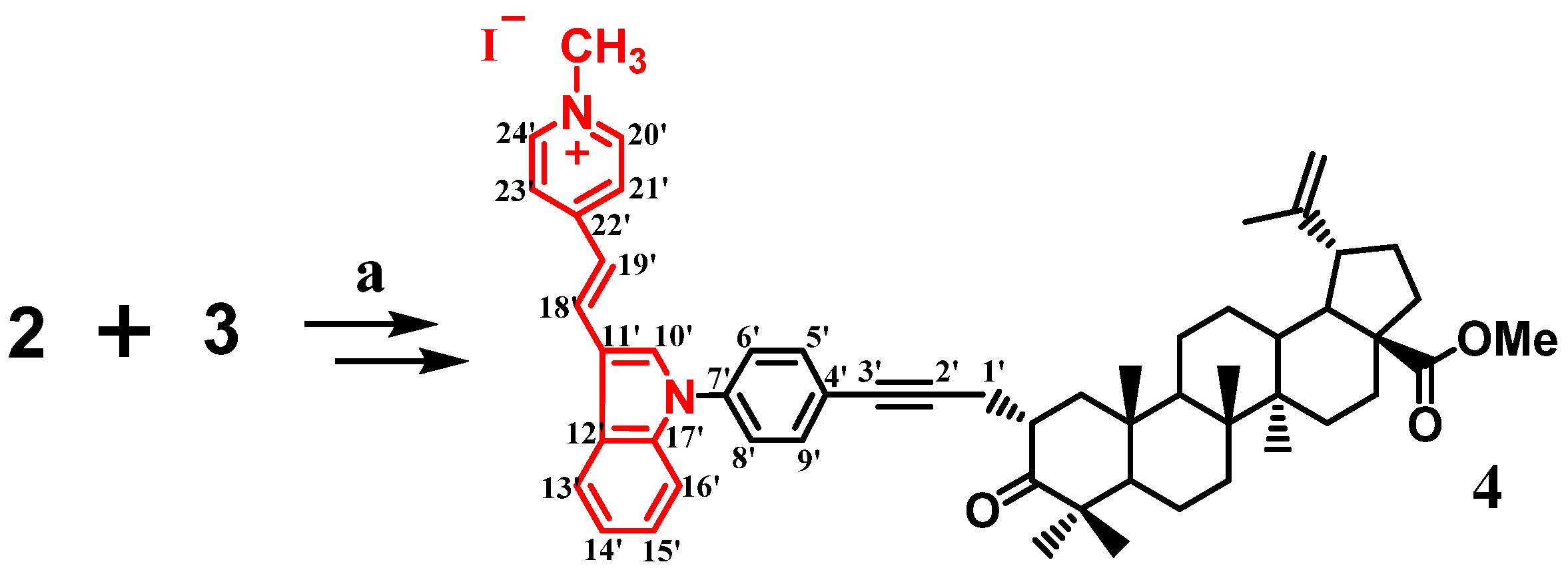
2.1.2. Synthesis of Methyl 2α-{[(E)-4-(1H-indol-3-yl-vinyl)-N-methyl-pyridinium iodide]phenylpropynyl}-3-oxolup-20(29)en-28-oate (4)
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kessler, J.H.; Mullauer, F.B.; Roo, G.M.; Medema, J.P. Broad in vitro efficacy of plant-derived betulinic acid against cell lines derived from the most prevalent human cancer types. Cancer Lett. 2007, 251, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, J.; Chen, Y. Betulinic acid and the pharmacological effects of tumor suppression (Review). Mol. Med. Rep. 2016, 14, 4489–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Discov. Today 2009, 14, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kroemer, G. Mitochondria as therapeutic targets for the treatment of malignant disease. Antioxid. Redox Signal. 2011, 15, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic acid, a natural compound with potent anti-cancer effects. Anticancer Drugs 2010, 21, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R. Betulinic acid and its derivatives: A patent review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Dubinin, M.V.; Belosludtsev, K.N. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. J. Pers. Med. 2021, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Xiao, Y.; Fu, B.; Qin, Z. TPP-based mitocans: A potent strategy for anticancer drug design. RSC Med. Chem. 2020, 11, 858–875. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; Ros, T.; Hurd, T.R.; Smith, R.A.J.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry 2005, 70, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Hoenke, S.; Serbian, I.; Deigner, H.-P.; Csuk, R. Mitocanic di- and triterpenoid rhodamine B conjugates. Molecules 2020, 25, 5443. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; Berardi, M.J.; Scorrano, L.; Korsmeyer, S.J.; Leder, P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell 2002, 2, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; Leder, P. F16, a mitochondriotoxic compound, triggers apoptosis or necrosis depending on the genetic background of the target carcinoma cell. Cancer Res. 2004, 64, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.B.; Zhao, Z.L.; Liu, T.; Xie, G.J.; Jin, C.; Deng, T.G.; Sun, Y.; Li, X.; Hu, X.X.; Zhang, X.B.; et al. A multimitochondrial anticancer agent that selectively kills cancer cells and overcomes drug resistance. Chem. Med. Chem. 2017, 12, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Fan, X.-Y.; Yang, L.-Y.; He, H.; Huang, R.; Jiang, F.-L. Conjugated 5-fluorouracil with mitochondria-targeting lipophilic cation: Design, synthesis and biological evaluation. Med. Chem Commun. 2016, 7, 2016–2019. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Davletshin, E.V.; Tukhbatullin, A.A.; D’yakonov, V.A.; Yunusbaeva, M.M.; Dzhemileva, L.U.; Dzhemilev, U.M. Pentacyclic triterpene acid conjugated with mitochondria-targeting cation F16: Synthesis and evaluation of cytotoxic activities. Med. Chem. Res. 2021, 30, 940–951. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Gubaidullin, R.R.; Galimshina, Z.R.; Nedopekina, D.A.; Odinokov, V.N. Effective synthesis of novel C(2)-propargyl derivatives of betulinic and ursolic acids and their conjugation with β-d-glucopyranoside azides via click chemistry. Tetrahedron 2016, 72, 1249–1256. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Wang, L.-Y.; Zhai, G.-H.; Wen, Z.-Y.; Zhang, Z.-X. Microwave-assisted solvent-free synthesis of some dimethine cyanine dyes, spectral properties and TD-DFT/PCM calculations. Bull. Korean Chem. Soc. 2007, 28, 2382–2388. [Google Scholar] [CrossRef]
- He, H.; Li, D.-W.; Yang, L.-Y.; Fu, L.; Zhu, X.-J.; Wong, W.-K.; Jiang, F.-L.; Liu, Y. A novel bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Sci. Rep. 2015, 5, 13543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubaidullin, R.R.; Khalitova, R.R.; Nedopekina, D.A.; Spivak, A.Y. Homo- and cross coupling of C-2 propargyl substituted triterpenoic acids: Synthesis of novel symmetrical and unsymmetrical triterpene 1,3-diynes. ChemistrySelect 2018, 3, 13526–13529. [Google Scholar] [CrossRef]
- Xiang, C.; Li, D.W.; Qi, Z.D.; Jiang, F.L.; Ge, Y.S.; Liu, Y. Synthesis of F16 conjugated with 5-fluorouracil and biophysical investigation of its interaction with bovine serum albumin by a spectroscopic and molecular modeling approach. Luminescence 2012, 28, 865–872. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nedopekina, D.; Davletshin, E.; Spivak, A. Effective Synthesis of a Novel Betulinic Acid Conjugate with Mitochondria-Targeting Cation F16. Chem. Proc. 2022, 8, 97. https://doi.org/10.3390/ecsoc-25-11638
Nedopekina D, Davletshin E, Spivak A. Effective Synthesis of a Novel Betulinic Acid Conjugate with Mitochondria-Targeting Cation F16. Chemistry Proceedings. 2022; 8(1):97. https://doi.org/10.3390/ecsoc-25-11638
Chicago/Turabian StyleNedopekina, Darya, Eldar Davletshin, and Anna Spivak. 2022. "Effective Synthesis of a Novel Betulinic Acid Conjugate with Mitochondria-Targeting Cation F16" Chemistry Proceedings 8, no. 1: 97. https://doi.org/10.3390/ecsoc-25-11638