
An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2 †


Department of Biochemistry and Molecular Biology, Faculty of Life Sciences, Bangabandhu Sheikh Mujibur Rahman Science and Technology University, Gopalganj 8100, Bangladesh
†
Presented at the 25th International Electronic Conference on Synthetic Organic Chemistry, 15–30 November 2021, Available online: https://ecsoc-25.sciforum.net/ .
Chem. Proc. 2022, 8(1), 5; https://doi.org/10.3390/ecsoc-25-11721
Published: 14 November 2021
(This article belongs to the Proceedings of The 25th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:CDKs are pivotal mediators essential for the cellular cycle progression. CDKs have relatively constant levels, and their activity is regulated by cyclins, proteins whose concentrations fluctuate during each cell cycle. Consequently, more CDK family members were found that occupy crucial functions in a variety of processes. Moreover, CKS2 is a member of the CDK family, which has been implicated in several malignancies as an oncogene. Additionally, CKS2 is engaged in many biological processes, including the cell cycle transition. CKS2 may act synergistically to promote embryonic development and somatic cell division. Current CDK2 drugs, however, also suppress CDK1, posing a toxicity risk. Investigators demonstrated that the potential conformational maps of cyclin-free CDK1 and CDK2 exhibit slight but substantial differences. The CDK1 unique characteristics may be used to distinguish it from other CDKs in prospective cancer treatment design. Computational-based in silico docking investigations were performed to uncover promising CDK1/Cks2 (6GU7) inhibitors utilizing the Maestro tool. Curcumin, quercetin, withanolide, and genistein were selected against the protein CDK1/Cks2 for protein–ligand XP docking. The physicochemical, lipophilicity, water-solubility, pharmacokinetics, drug-likeness, medicinal chemistry, and toxicological properties were analyzed using SwissADME and pkCSM of the selected ligands. Curcumin exerted an excellent docking score complexed with 6GU7 compared to other ligands. The revealed hit may be a potent inhibitor of 6GU7. However, it will require to be assessed extensively in vivo and in vitro experimental models.
1. Introduction
CDKs are serine/threonine kinases that require a governing subunit component known as a cyclin to function. CDKs, MAPKs, Gsk3β, members of the DYRK family, and CDK-like kinases all contribute to the CMGC group of kinases (called after the initials of several members), together with MAPKs, Gsk3β, and CDK-like kinases [1]. In closely similar kinases, including MAPKs, substrate sophistication is imparted via docking sites distinct from the catalytic region, but CDKs are defined by their reliance on distinct protein subunits that include different sequences essential for enzymatic activity [2]. CDK family members undertake a plethora of activities in the cell, including cell cycle and transcription monitoring, and differentiation in particular cell types [3,4]. CDK function imbalance is closely attributed to atypical cell progression, and as a consequence, numerous members of the CDK family were targeted as anticancer therapeutic targets [5,6]. Moreover, CDKs are proteins that influence cell cycle progression and are consequently promising targets in cancer. The activity of CDKs is regulated by their interaction with cyclin-dependent kinases, phosphatases, and particular inhibitors. Multiple CDK complexes operate at distinct stages [7]. CDK1 is a crucial regulator of the cell cycle commencement and progression through mitosis. Prior investigation has established that loss of CDK1 function or abnormal CDK1 expression is associated with G2 phase arrest and various tumor forms, confirming CDK1 as a therapeutic candidate. As a result, there was a spike in attention to developing potent CDK1 inhibitors as promising chemotherapeutic agents [4]. CKS2 belongs to the CDK family that was pinpointed as an oncogene in a variety of cancers.
Furthermore, CKS2 is also involved in the cell cycle transition in a variety of biological functions. CKS2 may pointedly enhance embryonic progression and somatic cell division [8]. However, mounting data suggested that CKS2 may play a role in tumor development [9]. Curcumin, a polyphenol derived from Curcuma longa, has garnered global prominence for its biological properties, including antioxidant, antimicrobial, anti-inflammatory, and antiviral, the most well-documented of which are its anticancer activities, which are currently under investigation [10,11,12]. It is demonstrated that curcumin plays a crucial role in cell signaling cascades implicated in cancer growth and proliferation that curcumin targets. Curcumin was shown to affect enzymes, growth factors, kinase, transcription factors, inflammatory cytokines, antiapoptotic (by downregulation), and proapoptotic (by upregulation) proteins [13,14]. However, quercetin is the most abundant flavonoid flavonol. Quercetin is found in various fruits and vegetables and is among the most prevalent flavonols in the western diet [15,16]. Anticancer properties of quercetin comprise its potency to induce cell death, autophagy, and apoptosis via regulation of the Wnt/-catenin, PI3K/Akt/mTOR, and MAPK/ERK1/2 pathways [17,18,19]. Moreover, withanolides are a broad class of steroidal lactones abundant in Solanaceae plants that were shown to have anticancer properties [20,21]. Withanolide was shown to inhibit and/or constrain tumor development in humans [22,23]. Genistein is an isoflavone found in soy that has various molecular actions, including the suppression of inflammation, the stimulation of apoptosis, and the regulation of metabolic pathways and steroidal hormone receptors [24,25]. Because these molecular changes influence carcinogenesis, obesity, cancer progression, metabolic syndromes, and osteoporosis, genistein is crucial for protecting and controlling common diseases. Genistein is a chemotherapeutic agent that suppresses metastasis in several kinds of cancer by modifying apoptosis, angiogenesis, and the cell cycle [26,27,28].
2. Materials and Methods
2.1. Receptor Preparation
The crystal structure of the target protein CDK1/Cks2 (PDB id: 6GU7) with the inhibitor-bound structure was obtained from the protein data bank (https://www.rcsb.org/, accessed on 10 March 2021) [29]. The Protein Preparation Wizard (PPW) tool [30] of the Maestro v.11.2 (Maestro, Schrödinger, LLC, New York, NY, USA) was performed for protein preparation. The PPW tool includes three-step stratagems, including importing and refining, reviewing and modifying, and optimizing and minimizing the protein. In the first instance, the protein (PDB id: 6GU7) was pre-processed by appending hydrogen atoms, eliminating displeased water molecules afar 5 Å from the hetero group and generating het states applying Epik [31] at pH 7.0 (+/−2). Allocating the RMSD of 0.30 Å through the OPLS3e force fields [32], the protein was minimized.
2.2. Ligand Preparation
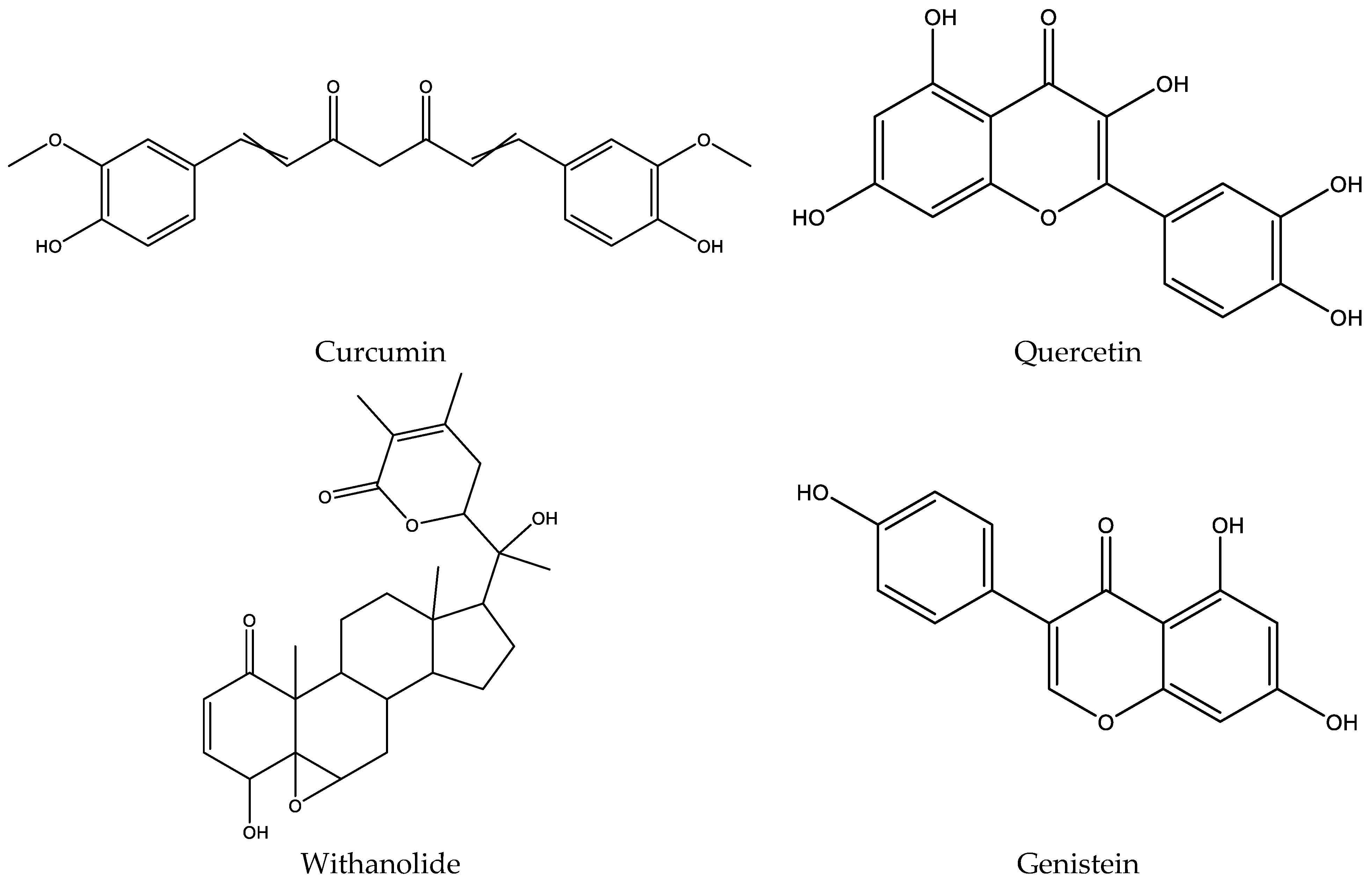
The ligands (Figure 1), curcumin (PubChem CID: 969516), quercetin (PubChem CID: 5280343), withanolide (PubChem CID: 53477765), and genistein (PubChem CID: 5280961) were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/, accessed on 10 March 2021)). The LigPrep tool (LigPrep, Schrödinger, LLC, New York, NY, USA) was used to convert to three-dimensional form and the production of potential tautomers and conformers. The LigPrep tool was performed at neutral ionization and the OPLS3e force field for minimizing the ligands.
2.3. Molecular Docking Study
All docking studies were performed using the GLIDE program [33] of Maestro (v.11.2), which identifies good associations between the ligand and the protein. A grid was constructed at the site of a co-crystallized ligand employing the Receptor Grid Generation tool. This grid reflects the features of the focused protein and the curvature utilized to produce a more comprehensive ligand, poses assessment. The Extra Precision (XP) docking mode was performed during the protein–ligand docking procedure.
2.4. ADMET Analysis
3. Results and Discussion
3.1. Analyzing Molecular Docking Results and Binding Interactions
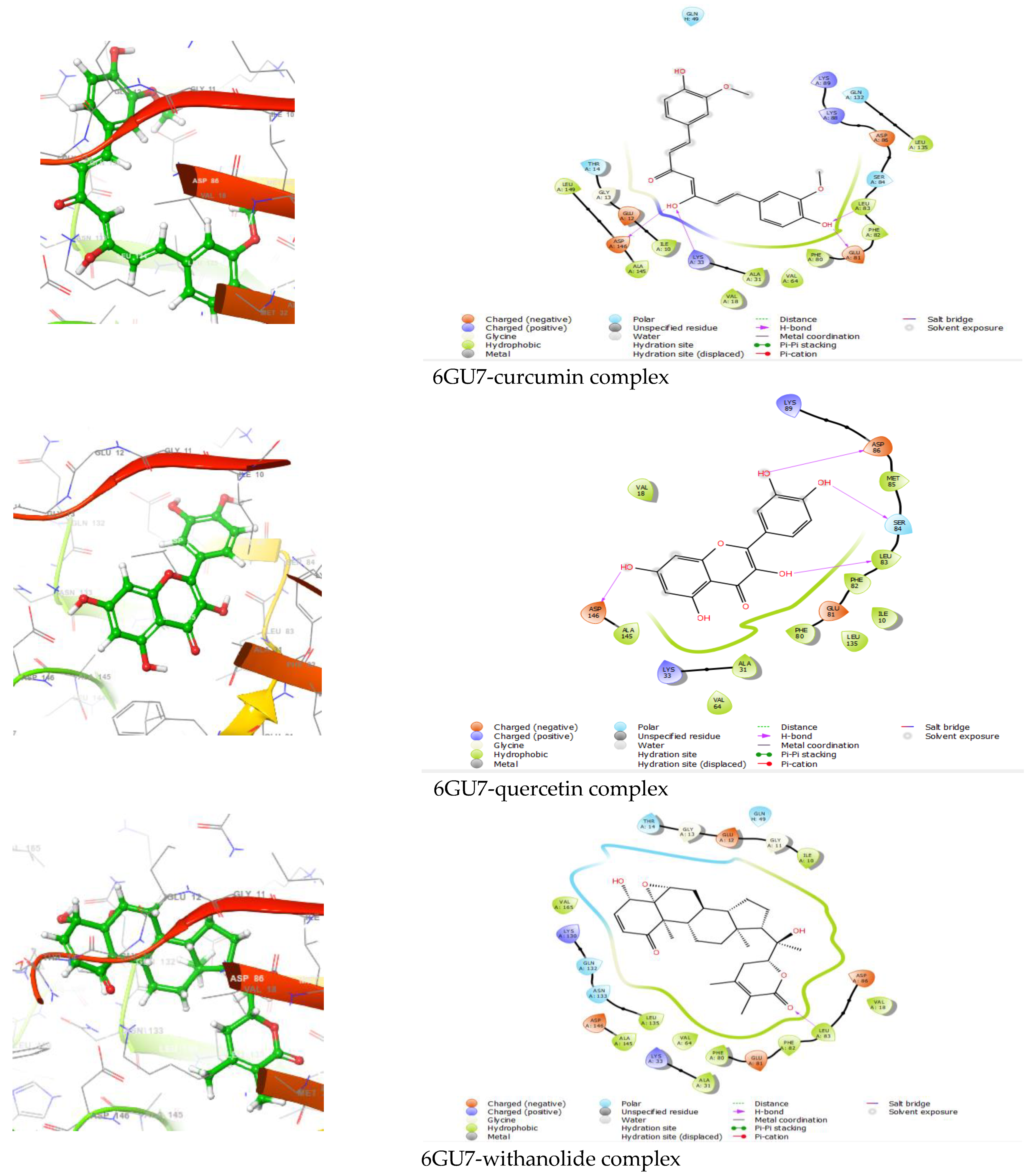
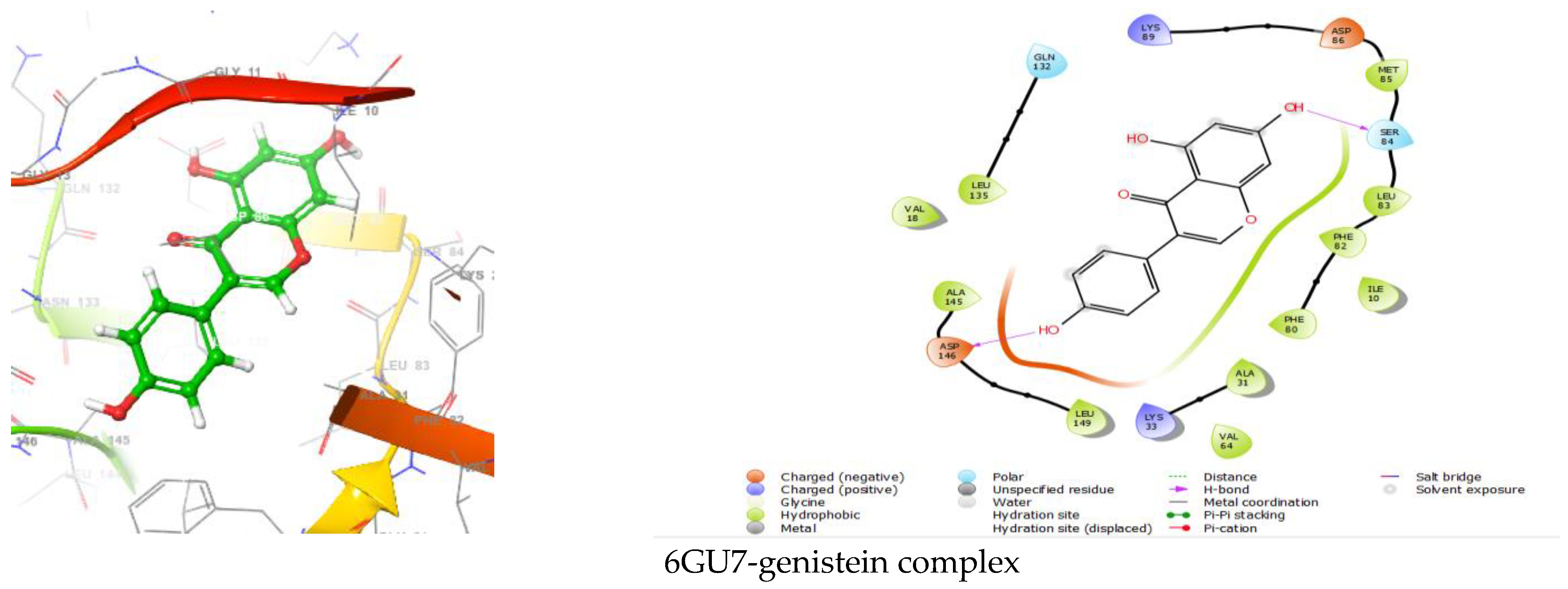
Pharmaceutical research has effectively integrated various molecular modeling techniques into several drug development programs to explore complicated biological and chemical processes. Combining computational and experimental techniques has proven highly beneficial in identifying and developing innovative, promising chemicals [37,38]. Frequently employed in contemporary drug design, molecular docking techniques investigate the conformations of ligands inside macromolecular target binding sites. Additionally, this technique calculates the free energy of ligand–receptor interaction by examining critical events engaged in the intermolecular interaction mechanism [39,40]. The XP molecular docking (grid box size as of 10 Å × 10 Å × 10 Å) was performed using the GLIDE program. The XP docking measured the docking scores of the 6GU7-curcumin complex, 6GU7-quercetin complex, 6GU7-withanolide complex, and 6GU7-genistein complex as of −9.419 kcal/mol, −8.709 kcal/mol, −7.174 kcal/mol, and −6.301 kcal/mol, respectively (Table 1, Figure 2).
3.2. ADMET Analysis
In the field of efficient medication, a potent molecule must approach its target in the body in a bioactive state and remain there long enough for the predicted physiological activities to transpire. Drug development progressively incorporates ADMET screening early in the discovery phase, when the number of candidate compounds is large, but the availability of physical samples is restricted. In this situation, computer models are viable substitutes for experimentation [41,42]. Investigators implement the existing SwissADME [35] web platform that provides free access to a reservoir of rapid yet reliable prognostic models for pharmacokinetics, physicochemical characteristics, drug-likeness, and medicinal chemistry pleasantness, including proprietary methods the iLOGP [43], BOILED-Egg [44], and Bioavailability Radar to assist in their drug development accomplishments. Moreover, the pkCSM employs the utilization of graph-based identifications to anticipate pharmacokinetic characteristics. These reflect the tiny molecule and are exploited to train prediction algorithms [36,45].
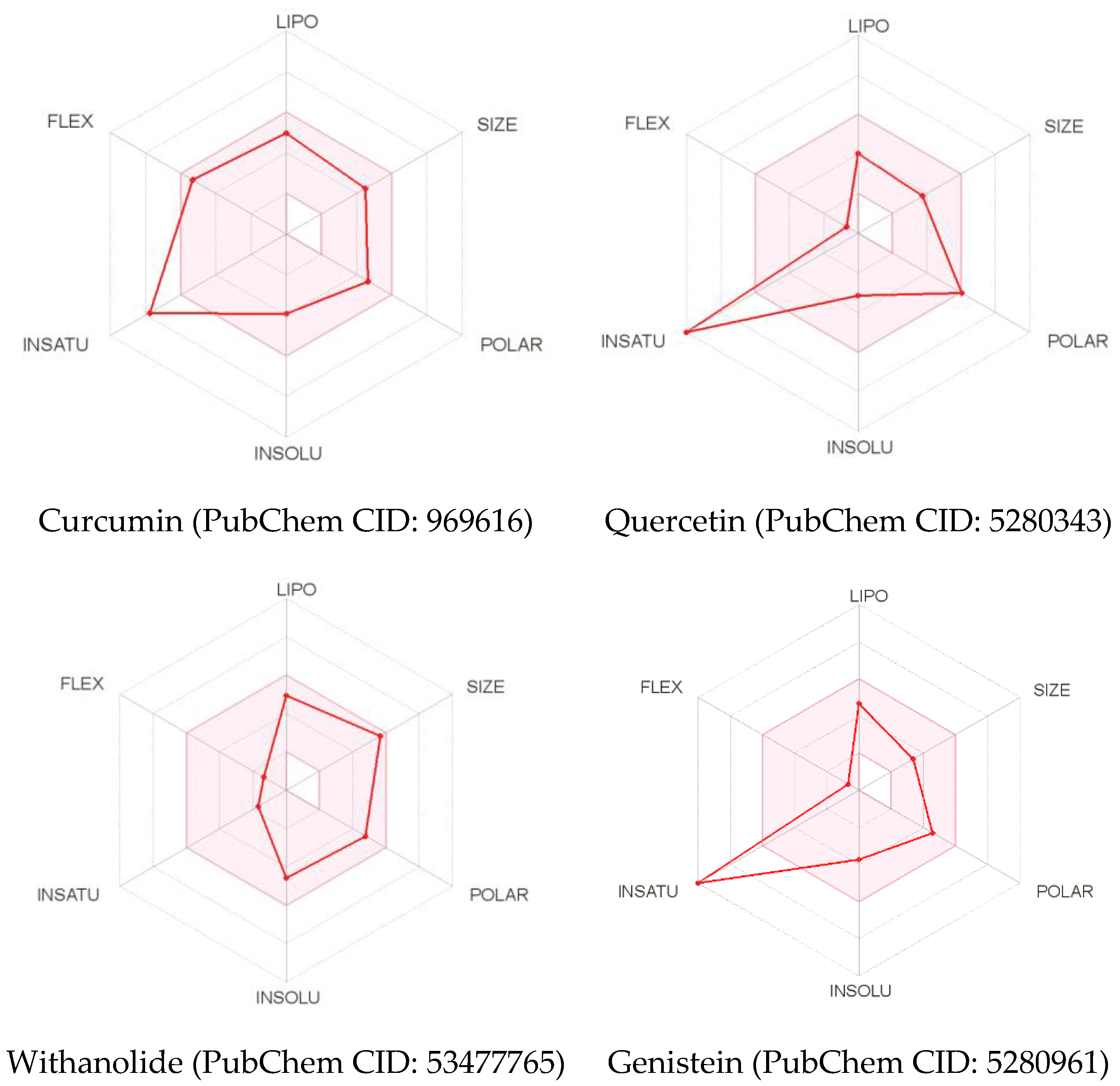
Physicochemical properties refer to the inherent physical and chemical features of a substance. Curcumin, quercetin, withanolide, and genistein have a molecular weight of 368.38 g/mol, 302.24 g/mol, 470.60 g/mol 270.24 g/mol with the number of heavy atoms of 27, 22, 34, and 20, respectively (Table 2). Curcumin and withanolide contain the fraction Csp3 values of 0.14 and 0.79, whereas quercetin and genistein remain nil. It is observed that selected ligands, except withanolide, proclaim a breach in unsaturation (0.25 < Fraction Csp3 < 1) (Figure 3). Moreover, all ligands represent outstanding flexibility (0 < rotatable bonds < 9). Curcumin, quercetin, withanolide, and genistein contain H-bond acceptors as of 6, 7, 6, and 5, respectively. Quercetin displays a violation in H-bond donors (<5) and in polarity (20 Å2 < TPSA < 130 Å2) compared to other ligands.
Lipophilicity is traditionally defined by the partition coefficient between n-octanol and water (log Po/w) [46]. Numerous computer techniques for estimating log Po/w have been devised with varying degrees of effectiveness on various chemical combinations. Several predictors are frequently used to choose the best reliable techniques for a particular chemical combination or create a consensus approximation [47]. Curcumin, quercetin, withanolide, and genistein have the iLOGP [43] values of 3.27, 1.63, 3.62, 1.91, respectively. All ligands, including curcumin, quercetin, withanolide, and genistein represent sublime lipophilicity (−0.7 < XLOGP3 < +5) [48]. The WLOGP values for the completely atomistic approach centered on Wildman and Crippen’s fragmental methodology [49] determined for curcumin, quercetin, withanolide, and genistein are 3.15, 1.99, 3.50, 2.58, respectively. The MLOGP is a prototypical topological strategy based on a linear connection [50,51]. The MLOGP values for curcumin, quercetin, withanolide, and genistein are 1.47, −0.56, 2.75, 0.52, respectively. The hybrid technique SILICOS-IT describes the topological descriptors [51] for curcumin, quercetin, withanolide, and genistein as 4.04, 1.54, 3.78, and 2.52, respectively.
Possessing a soluble molecule simplifies several drug development processes, most notably handling and formulation [52]. Moreover, for discovery initiatives aimed at oral delivery, solubility is a critical factor affecting absorption [53]. Additionally, a medication intended for parenteral administration must be highly soluble in water to provide an adequate amount of active components in such a tiny volume of pharmaceutical dose [54]. The Log S (ESOL) values [55] are −3.94, −3.16, −4.59, −3.72; and the solubility as of 4.22 × 10−2 mg/mL (1.15 × 10−4 mol/L), 2.11 × 10−1 mg/mol (6.98 × 10−4 mol/L), 1.21 × 10−2 mg/mL (2.56 ×105 mol/L), and 5.11 × 10−2 mg/mL (1.89 × 10−4 mol/L) for curcumin, quercetin, withanolide, and genistein, respectively. Curcumin, quercetin, and genistein are anticipated as soluble, whereas withanolide is predicted as moderately soluble.
Pharmacokinetics is the study of how medicines enter into, circulate through, and exit the body. The way an individual reacts to a specific medicine is determined by the substance’s inherent pharmacological characteristics at the site of action [56,57,58]. Both passive human gastrointestinal (GI) absorption and blood–brain barrier (BBB) penetration estimates are made using the BOILED-Egg model as a conclusion [59]. All those selected have high GI absorption levels with negative BBB permeant features. Understanding which compounds are substrates or non-substrates for the permeability glycoprotein (P-gp, recommended to be the most crucial representative of the ABC-transporters) is critical for evaluating active efflux across biological membranes, for example, from the gastrointestinal wall to the lumen or from the brain [60]. P-gp plays a critical function in protecting the CNS against xenobiotics [61]. Additionally, P-gp is abundantly expressed in some tumor cells, resulting in multidrug-resistant cancers [62]. The estimated P-gp substrate values for curcumin, quercetin, withanolide, and genistein were reported as unfavorable. The skin permeability coefficient (Kp) values for curcumin, quercetin, withanolide, and genistein are −6.28 cm/s, −7.05 cm/s, −6.96 cm/s, and −6.05 cm/s, respectively, therefore, all the selected ligands that were assessed have less skin permeation propensity [63].
Drug-likeness evaluates qualitatively the potential of a chemical becoming an oral drug in terms of bioavailability. The drug-likeness was determined via structural or physicochemical evaluations of compounds in development that had advanced sufficiently to be presumed, oral drug candidates. This concept is frequently used to filter chemical libraries to eliminate compounds with characteristics that are most likely contradictory to a satisfactory pharmacokinetic profile [64,65]. All ligands contain positive Lipinski values [66] without any violation, whereas withanolide has a Ghose value [67] in negative with one violation (Num. of atoms > 70) compared to the rest. Moreover, Curcumin, quercetin, withanolide, and genistein contain Veber [68], Egan [69], and Muegge [70] values in the positive. Surprisingly all selected ligands contain the same bioavailability score [71] of 0.55 each.
The Medicinal Chemistry section is intended to assist medicinal chemists in their regular drug development initiatives. Even though curcumin, withanolide, and genistein contain no alert for PAINS assessment [72], quercetin has an alert for catechol-A. Curcumin has two alerts for Brenk evaluation [73], whereas quercetin and withanolide have a single alert for each, and genistein has no alert compared to others. Both quercetin and genistein positively impact Lead-likeness [74], whereas curcumin and withanolide have two and a single violation, respectively. A significant attribute of CADD operations is identifying the most favorable simulated molecules for synthesization and submission to biological assessments or other investigations. In this evaluation procedure, synthetic accessibility (SA) is a vital component to consider [75,76]. Curcumin, quercetin, withanolide, and genistein contain the SA values of 2.97, 3.23, 6.85, and 2.87.
The AMES toxicity test is an extensively used strategy for determining the mutagenesis potential of a substance using bacteria. A positive test demonstrates that the chemical is mutagenic and hence has the potential to cause cancer [77,78,79]. Indeed, curcumin, quercetin, withanolide, and genistein have no mutagenesis potential. The MRTD represents an approximation of a chemical’s hazardous dosage threshold in humans [80,81]. Curcumin has the lowest MRTD (0.081), whereas quercetin, withanolide, and genistein have elevated MRTD compared to each other. Surprisingly, all ligands, including curcumin, quercetin, withanolide, and genistein, do not show hepatotoxicity, skin sensitization, or hERG I and II inhibitor effects. It is imperative to evaluate the hazardous potential of a compound. The lethal dose estimates (LD50) are a common method for determining the acute toxicity of various compounds. LD50 is the dose of a compound that causes 50% death of a set of test animals when administered all at once [82]. Curcumin has the lowest LD50 value (1.833 mol/kg) compared to quercetin (2.471 mol/kg), withanolide (2.831 mol/kg), and genistein (2.268 mol/kg). Moreover, withanolide has a diminished LOAEL value compared to curcumin, quercetin, and genistein.
4. Conclusions
CDK1 is an essential mediator of the initiation and advancement of the cell cycle during mitosis. CKS2 is a member of the CDK family, which was implicated in several malignancies as an oncogene. Because CDK1 alone or in tandem with other treatment options was connected to powerful anticancer effects, it was postulated that CDK1 may be the preferred CDK benchmark for cancer treatment. The present investigation used an in silico strategy targeting potential inhibitors against the CDK1/Cks2 protein (6GU7) for advancements in cancer treatment. Curcumin, quercetin, withanolide, and genistein were selected as promising candidates for XP molecular docking against 6GU7 with the Maestro program. The SwissADME and the pkCSM anticipated the ADMET properties of the selected ligands. Analyzing the different binding interactions, curcumin showed a high binding affinity with 6GU7 compared to quercetin, withanolide, and genistein. However, in vivo and in vitro investigations are required to evaluate the current study.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ecsoc-25-11721/s1.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
CDK: Cyclin-dependent kinase; MAPKs: Mitogen-activated protein kinases; Gsk3β: Glycogen synthase kinase-3 beta; DYRK: Dual-specificity tyrosine-regulated kinase; CDK1: Cyclin-dependent kinase-1; CKS2: Cyclin-dependent kinases regulatory subunit 2: OPLS3e: Optimized potentials for liquid simulation_3e; RMSD: Root-mean-square deviation; GLIDE: Grid-based Ligand Docking with Energetics; ADMET: Absorption, Distribution, Metabolism, Excretion, and Toxicity; CNS: Central nervous system; BBB: Blood–brain barrier; PAINS: Pan assay interference compounds; MRTD: Maximum recommended tolerated dose.
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Wang, Q.; Su, L.; Liu, N.; Zhang, L.; Xu, W.; Fang, H. Cyclin Dependent Kinase 1 Inhibitors: A Review of Recent Progress. Curr. Med. Chem. 2011, 18, 2025–2043. [Google Scholar] [CrossRef]
- Martinsson-Ahlzén, H.-S.; Liberal, V.; Grünenfelder, B.; Chaves, S.R.; Spruck, C.H.; Reed, S.I. Cyclin-Dependent Kinase-Associated Proteins Cks1 and Cks2 Are Essential during Early Embryogenesis and for Cell Cycle Progression in Somatic Cells. Mol. Cell. Biol. 2008, 28, 5698–5709. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Lin, H.; Zhang, Z. CKS2 in human cancers: Clinical roles and current perspectives (Review). Mol. Clin. Oncol. 2015, 3, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Giordano, A.; Tommonaro, G. Curcumin and cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yu, J.; Cui, R.; Lin, J.; Ding, X. Curcumin in Treating Breast Cancer: A Review. J. Lab. Autom. 2016, 21, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V. Curcumin as a clinically-promising anticancer agent: Pharmacokinetics and drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 953–972. [Google Scholar] [CrossRef] [PubMed]
- Devassy, J.G.; Nwachukwu, I.D.; Jones, P.J.H. Curcumin and cancer: Barriers to obtaining a health claim. Nutr. Rev. 2015, 73, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Wei, Y.; Lee, R.J.; Zhao, L. Liposomal curcumin and its application in cancer Physical property. Int. J. Nanomed. 2017, 12, 6027–6044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Farias, M.; Carrasco-Pozo, C. The anti-cancer effect of quercetin: Molecular implications in cancer metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef]
- Tang, S.M.; Deng, X.T.; Zhou, J.; Li, Q.P.; Ge, X.X.; Miao, L. Pharmacological basis and new insights of quercetin action in respect to its anticancer effects. Biomed. Pharmacother. 2020, 121, 109604. [Google Scholar] [CrossRef]
- Maurya, A.K.; Vinayak, M. Quercetin regresses Dalton’s lymphoma growth via suppression of PI3K/AKT signaling leading to upregulation of p53 and decrease in energy metabolism. Nutr. Cancer 2015, 67, 354–363. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Alsahli, M.A.; Almatroudi, A.; Verma, A.K.; Aloliqi, A.; Allemailem, K.S.; Khan, A.A.; Rahmani, A.H. Potential therapeutic targets of quercetin, a plant flavonol, and its role in the therapy of various types of cancer through the modulation of various cell signaling pathways. Molecules 2021, 26, 1315. [Google Scholar] [CrossRef]
- Jayaprakasam, B.; Zhang, Y.; Seeram, N.P.; Nair, M.G. Growth inhibition of human tumor cell lines by withanolides from Withania somnifera leaves. Life Sci. 2003, 74, 125–132. [Google Scholar] [CrossRef]
- Wang, H.C.; Tsai, Y.L.; Wu, Y.C.; Chang, F.R.; Liu, M.H.; Chen, W.Y.; Wu, C.C. Withanolides-induced breast cancer cell death is correlated with their ability to inhibit heat protein 90. PLoS ONE 2012, 7, e37764. [Google Scholar] [CrossRef] [PubMed]
- Palliyaguru, D.L.; Singh, S.V.; Kensler, T.W. Withania somnifera: From prevention to treatment of cancer. Mol. Nutr. Food Res. 2016, 60, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, R.; Khalil, R.; Green, R.; Mohapatra, S.S.; Mohapatra, S. Withania somnifera (Ashwagandha) and withaferin a: Potential in integrative oncology. Int. J. Mol. Sci. 2019, 20, 5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukund, V.; Mukund, D.; Sharma, V.; Mannarapu, M.; Alam, A. Genistein: Its role in metabolic diseases and cancer. Crit. Rev. Oncol. Hematol. 2017, 119, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; Nabavi, S.M. Genistein and Cancer: Current Status, Challenges, and future directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef]
- Ji, Z.; Huo, C.; Yang, P. Genistein inhibited the proliferation of kidney cancer cells via CDKN2a hypomethylation: Role of abnormal apoptosis. Int. Urol. Nephrol. 2020, 52, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Pavese, J.M.; Farmer, R.L.; Bergan, R.C. Inhibition of cancer cell invasion and metastasis by genistein. Cancer Metastasis Rev. 2010, 29, 465–482. [Google Scholar] [CrossRef] [Green Version]
- Mukund, V. Genistein: Its Role in Breast Cancer Growth and Metastasis. Curr. Drug Metab. 2020, 21, 6–10. [Google Scholar] [CrossRef]
- Wood, D.J.; Korolchuk, S.; Tatum, N.J.; Wang, L.Z.; Endicott, J.A.; Noble, M.E.M.; Martin, M.P. Differences in the Conformational Energy Landscape of CDK1 and CDK2 Suggest a Mechanism for Achieving Selective CDK Inhibition. Cell Chem. Biol. 2019, 26, 121–130.e5. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Walker, M. In Silico Methods for Predicting Drug Toxicity. Methods Mol. Biol. 2016, 1425, 63–83. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.G.; Santos, R.N.D.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Kaur, T.; Madgulkar, A.; Bhalekar, M.; Asgaonkar, K. Molecular Docking in Formulation and Development. Curr. Drug Discov. Technol. 2019, 16, 30–39. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Adv. Docking 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Norinder, U.; Bergström, C.A.S. Prediction of ADMET properties. ChemMedChem 2006, 1, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/ Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, S.; Leszczynski, J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov. 2020, 15, 1473–1487. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Molecular Nanomedicine Towards Cancer. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Moriguchi, I.; Shuichi, H.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1994, 17, 1460–1462. [Google Scholar] [CrossRef] [Green Version]
- Penzotti, J.E.; Landrum, G.A.; Putta, S. Building predictive ADMET models for early decisions in drug discovery. Curr. Opin. Drug Discov. Dev. 2004, 7, 7–61. [Google Scholar]
- Ritchie, T.J.; MacDonald, S.J.F.; Peace, S.; Pickett, S.D.; Luscombe, C.N. Increasing small molecule drug developability in sub-optimal chemical space. MedChemComm 2013, 4, 673–680. [Google Scholar] [CrossRef]
- Ottaviani, G.; Gosling, D.J.; Patissier, C.; Rodde, S.; Zhou, L.; Faller, B. What is modulating solubility in simulated intestinal fluids? Eur. J. Pharm. Sci. 2010, 41, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Waller, D.G.; Sampson, A.P. Pharmacokinetics. In Medical Pharmacology and Therapeutics, 5th ed.; Waller, D.G., Sampson, A.P., Eds.; Elsevier B.V.: Southampton, UK, 2018; pp. 33–62. [Google Scholar] [CrossRef]
- Saghir, S.A.; Ansari, R.A. Pharmacokinetics. Ref. Modul. Biomed. Sci. 2018, 1–9. [Google Scholar] [CrossRef]
- Turfus, S.C.; Delgoda, R.; Picking, D.; Gurley, B.J. Pharmacokinetics. In Pharmacognosy. Fundamentals, Applications and Strategies; Badal, S., Delgoda, R., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 495–512. [Google Scholar] [CrossRef]
- Paixão, P.; Gouveia, L.F.; Morais, J.A. Prediction of the human oral bioavailability by using in vitro and in silico drug related parameters in a physiologically based absorption model. Int. J. Pharm. 2012, 429, 84–98. [Google Scholar] [CrossRef]
- Montanari, F.; Ecker, G.F. Prediction of drug-ABC-transporter interaction—Recent advances and future challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef]
- Sharom, F.J. ABC Multidrug Transporters: Structure, Function and Role in Chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Edwards, D.A.; Langer, R. A linear theory of transdermal transport phenomena. J. Pharm. Sci. 1994, 83, 1315–1334. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Y.; Li, J.Y.; Hao, G.F.; Yang, G.F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Pedrys, A.; Duszkiewicz, R.; Kucia, U. Ligand Potency, Efficiency and Drug-likeness: A Story of Intuition, Misinterpretation and Serendipity. Curr. Protein Pept. Sci. 2019, 20, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Kurosawa, T.; Mikami, Y.; Nakamura, H. Prediction of synthetic accessibility based on commercially available compound databases. J. Chem. Inf. Model. 2014, 54, 3259–3267. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Zeiger, E. The test that changed the world: The Ames test and the regulation of chemicals. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 841, 43–48. [Google Scholar] [CrossRef]
- Föllmann, W.; Degen, G.; Oesch, F.; Hengstler, J.G. Ames Test, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Jain, A.K.; Singh, D.; Dubey, K.; Maurya, R.; Mittal, S.; Pandey, A.K. Models and Methods for In Vitro Toxicity, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Gad, S.C. Maximum Tolerated Dose, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Stampfer, H.G.; Gabb, G.M.; Dimmitt, S.B. Why maximum tolerated dose? Br. J. Clin. Pharmacol. 2019, 85, 2213–2217. [Google Scholar] [CrossRef]
- Gad, S.C. LD50/LC50 (Lethal Dosage 50/Lethal Concentration 50), 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
Figure 1.
The chemical structures of curcumin, quercetin, withanolide, and genistein.

Figure 2.
Interactions of 6GU7 with curcumin, quercetin, withanolide, and genistein.


Figure 3.
The physicochemical space for oral bioavailability. The bioavailability radar provides an initial assessment of drug-likeness of a molecule. The colored zone is the suitable physicochemical space for oral bioavailability. The comprehending outcomes for physicochemical space of curcumin (PubChem CID: 969616), quercetin (PubChem CID: 5280343), withanolide (PubChem CID: 53477765), and genistein (PubChem CID: 5280961) were illustrated following the parameters as of LIPO (Lipophility): −0.7 < XLOGP3 < +5; SIZE: 150 g/mol < MW < 500 g/mol; POLAR (Polarity): 20 Å2 < TPSA < 130 Å2; INSOLU (Insolubility): 0 < Log S (ESOL) < 6; INSATU (Unsaturation): 0.25 < Fraction Csp3 < 1; and FLEX (Flexibity): 0 < Num. rotatable bonds < 9.
Figure 3.
The physicochemical space for oral bioavailability. The bioavailability radar provides an initial assessment of drug-likeness of a molecule. The colored zone is the suitable physicochemical space for oral bioavailability. The comprehending outcomes for physicochemical space of curcumin (PubChem CID: 969616), quercetin (PubChem CID: 5280343), withanolide (PubChem CID: 53477765), and genistein (PubChem CID: 5280961) were illustrated following the parameters as of LIPO (Lipophility): −0.7 < XLOGP3 < +5; SIZE: 150 g/mol < MW < 500 g/mol; POLAR (Polarity): 20 Å2 < TPSA < 130 Å2; INSOLU (Insolubility): 0 < Log S (ESOL) < 6; INSATU (Unsaturation): 0.25 < Fraction Csp3 < 1; and FLEX (Flexibity): 0 < Num. rotatable bonds < 9.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The XP docking scores with different binding interactions.
| Protein–Ligand Complex | Docking Score (kcal/mol) | H-Bond | Non-Bonding Interactions |
|---|---|---|---|
| 6GU7-curcumin | −9.419 | ASP146, LYS33, GLU81, LEU83 | Polar THR14, SER84, GLN132, GLN49 Hydrophobic LEU149, ILE10, ALA145, VAL18, ALA31, VAL64, PHE80, PHE82, LEU83, LEU135 Charged (Negative) GLU12, ASP146, GLU81, ASP86 Charged (Positive) LYS33, LYS88, LYS89 |
| 6GU7-quercetin | −8.709 | ASP146, LEU83, SER84, ASP86 | Polar SER84 Hydrophobic Val18, ALA145, VAL64, ALA31, PHE 80, LEU135, ILE10, PHE82, LEU83, MET85 Charged (Negative) ASP146, GLU81, ASP86 Charged (Positive) LYS33, LYS89 |
| 6GU7-withanolide | −7.174 | LEU83 | Polar GLN132, ASN133, THR14, GLN49 Hydrophobic VAL165, LEU135, ALA145, VAL64, PHE80, ALA31, PHE82, LEU83, VAL18, ILE10 Charged (Negative) ASP146, GLU81, ASP86, GLU12 Charged (Positive) LYS130, LYS33 |
| 6GU7-genistein | −6.301 | ASP146, SER 84 | Polar GLN132, SER84 Hydrophobic LEU135, VAL18, ALA145, LEU149, VAL64, VAL31, PHE80, ILE10, PHE82, LEU83, MET85 Charged (Negative) ASP146, ASP86 Charged (Positive) LYS33, LYS89 |
Table 2.
Interactions of 6GU7 with curcumin, quercetin, withanolide, and genistein.
| Curcumin | Quercetin | Withanolide | Genistein | |
|---|---|---|---|---|
| Physicochemical Properties | ||||
| Molecular weight (g/mol) | 368.38 | 302.24 | 470.60 | 270.24 |
| Heavy atoms | 27 | 22 | 34 | 20 |
| Fraction Csp3 | 0.14 | 0.00 | 0.79 | 0.00 |
| Rotatable bonds | 8 | 1 | 2 | 1 |
| H-bond acceptors | 6 | 7 | 6 | 5 |
| H-bond donors | 2 | 5 | 2 | 3 |
| TPSA (Å2) | 93.06 | 131.36 | 96.36 | 90.90 |
| Lipophilicity | ||||
| Log Po/w (iLOGP) | 3.27 | 1.63 | 3.62 | 1.91 |
| Log Po/w (XLOGP3) | 3.20 | 1.54 | 3.12 | 2.67 |
| Log Po/w (WLOGP) | 3.15 | 1.99 | 3.50 | 2.58 |
| Log Po/w (MLOGP) | 1.47 | −0.56 | 2.75 | 0.52 |
| Log Po/w (SILICOS-IT) | 4.04 | 1.54 | 3.78 | 2.52 |
| Water Solubility | ||||
| Log S (ESOL) | −3.94 | −3.16 | −4.59 | −3.72 |
| Solubility (mg/mL; mol/L) | 4.22 × 10−2; 1.15 × 10−4 | 2.11 × 10−1; 6.98 × 10−4 | 1.21 × 10−2; 2.56 × 10−5 | 5.11 × 10−2; 1.89 × 10−4 |
| Class | Soluble | Soluble | Moderately soluble | Soluble |
| Pharmacokinetics | ||||
| GI absorption | High | High | High | High |
| BBB permeant | No | No | No | No |
| P-gp substrate | No | No | No | No |
| Log Kp (skin permeation) (cm/s) | −6.28 | −7.05 | −6.96 | −6.05 |
| Druglikeness | ||||
| Lipinski | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Ghose | Yes | Yes | No; 1 violation: #atoms > 70 | Yes |
| Veber | Yes | Yes | Yes | Yes |
| Egan | Yes | Yes | Yes | Yes |
| Muegge | Yes | Yes | Yes | Yes |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 |
| Medicinal Chemistry | ||||
| PAINS | 0 alert | 1 alert: catechol_A | 0 alert | 0 alert |
| Brenk | 2 alerts: beta_keto_anhydride, michael_acceptor_1 | 1 alert: catechol | 1 alert: Three-membered_heterocycle | 0 alert |
| Leadlikeness | No; 2 violations: MW > 350, Rotors > 7 | Yes | No; 1 violation: MW > 350 | Yes |
| Synthetic accessibility | 2.97 | 3.23 | 6.85 | 2.87 |
| Toxicological Properties | ||||
| AMES toxicity | No | No | No | No |
| Max. tolerated dose (human) (log mg/kg/day) | 0.081 | 0.499 | 0.867 | 0.478 |
| hERG I inhibitor | No | No | No | No |
| hERG II inhibitor | No | No | No | No |
| Oral Rat Acute Toxicity (LD50) (mol/kg) | 1.833 | 2.471 | 2.831 | 2.268 |
| Oral Rat Chronic Toxicity (LOAEL) (log mg/kg_bw/day) | 2.228 | 2.612 | 1.776 | 2.189 |
| Hepatotoxicity | No | No | No | No |
| Skin Sensitization | No | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Saikat, A.S.M. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chem. Proc. 2022, 8, 5. https://doi.org/10.3390/ecsoc-25-11721
AMA Style
Saikat ASM. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chemistry Proceedings. 2022; 8(1):5. https://doi.org/10.3390/ecsoc-25-11721
Chicago/Turabian StyleSaikat, Abu Saim Mohammad. 2022. "An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2" Chemistry Proceedings 8, no. 1: 5. https://doi.org/10.3390/ecsoc-25-11721