
Exploring Quercetin Hydrate’s Potential as an Antiviral Treatment for Oropouche Virus

 ,
,  ,
,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Chemical
2.2. Evaluation of Cell Cytotoxicity
2.3. Dose Response Assay
2.4. Virus Plaque Assay
2.5. Protein Modeling
2.6. Quercetin Hydrate Exhaustive Docking
2.7. Molecular Dynamics Simulation
2.8. MM/PBSA Analysis
2.9. QM Binding Analysis Calculations
3. Results
3.1. Cytotoxicity and Antiviral Activity
3.2. OROV Gc Protein Modeling
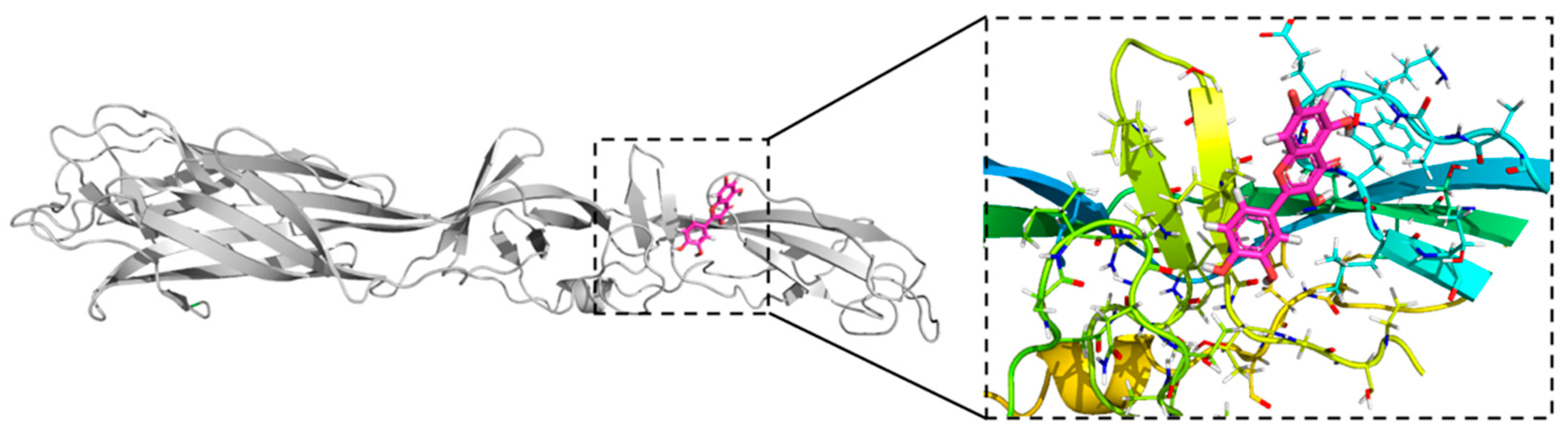
3.3. Quercetin Hydrate Docking in OROV Gc Protein
3.4. OROV Gc Protein MD Simulations and MM/PBSA Analysis
3.5. Quantum Interactions Analysis of OROV Gc and Quercetin Hydrate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Files, M.A.; Hansen, C.A.; Herrera, V.C.; Schindewolf, C.; Barrett, A.D.T.; Beasley, D.W.C.; Bourne, N.; Milligan, G.N. Baseline Mapping of Oropouche Virology, Epidemiology, Therapeutics, and Vaccine Research and Development. NPJ Vaccines 2022, 7, 38. [Google Scholar] [CrossRef]
- Sakkas, H.; Bozidis, P.; Franks, A.; Papadopoulou, C. Oropouche Fever: A Review. Viruses 2018, 10, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos, P.F.; Travassos Da Rosa, J.F.; Guerreiro, S.C.; Dégallier, N.; Travassos Da Rosa, E.S.; Travassos Da Rosa, A.P. Primeiro Registro de Epidemias Causadas Pelo Vírus Oropouche Nos Estados Do Maranhão e Goiás, Brasil. Rev. Inst. Med. Trop. São Paulo 1989, 31, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, R.M. Orthobunyaviruses: Recent Genetic and Structural Insights. Nat. Rev. Microbiol. 2014, 12, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Bowden, T.A.; Bitto, D.; McLees, A.; Yeromonahos, C.; Elliott, R.M.; Huiskonen, J.T. Orthobunyavirus Ultrastructure and the Curious Tripodal Glycoprotein Spike. PLoS Pathog. 2013, 9, e1003374. [Google Scholar] [CrossRef] [Green Version]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An Overview of Protein Structure Prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Costa, F.G.; da Silva Neto, B.R.; Gonçalves, R.L.; da Silva, R.A.; de Oliveira, C.M.A.; Kato, L.; dos Santos Freitas, C.; Giannini, M.J.S.M.; de Fátima da Silva, J.; de Almeida Soares, C.M.; et al. Alkaloids as Inhibitors of Malate Synthase from Paracoccidioides Spp.: Receptor-Ligand Interaction-Based Virtual Screening and Molecular Docking Studies, Antifungal Activity, and the Adhesion Process. Antimicrob. Agents Chemother. 2015, 59, 5581–5594. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, I.R.; Parise, M.R.; Pereira, M.; da Silva, R.A. Prospecting for New Catechol-O-Methyltransferase (COMT) Inhibitors as a Potential Treatment for Parkinson’s Disease: A Study by Molecular Dynamics and Structure-Based Virtual Screening. J. Biomol. Struct. Dyn. 2021, 39, 5872–5891. [Google Scholar] [CrossRef] [PubMed]
- Hariono, M.; Choi, S.B.; Roslim, R.F.; Nawi, M.S.; Tan, M.L.; Kamarulzaman, E.E.; Mohamed, N.; Yusof, R.; Othman, S.; Abd Rahman, N.; et al. Thioguanine-Based DENV-2 NS2B/NS3 Protease Inhibitors: Virtual Screening, Synthesis, Biological Evaluation and Molecular Modelling. PLoS ONE 2019, 14, e0210869. [Google Scholar] [CrossRef]
- Saivish, M.V.; Menezes, G.D.L.; Da Costa, V.G.; Nebo, L.; Da Silva, G.C.D.; Pacca, C.C.; Marques, R.E.; Nogueira, M.L.; Da Silva, R.A. Structural Insights into Plasticity and Discovery of Flavonoid Allosteric Inhibitors of Flavivirus NS2B–NS3 Protease. Biophysica 2023, 3, 71–92. [Google Scholar] [CrossRef]
- Saivish, M.V.; Menezes, G.D.L.; Da Silva, R.A.; Fontoura, M.A.; Shimizu, J.F.; Da Silva, G.C.D.; Teixeira, I.D.S.; Mistrão, N.F.B.; Hernandes, V.M.; Rahal, P.; et al. Antiviral Activity of Quercetin Hydrate against Zika Virus. Int. J. Mol. Sci. 2023, 24, 7504. [Google Scholar] [CrossRef]
- Polyakov, I.; Nemukhin, A. Computational Modeling of the Neurofibromin-Stimulated Guanosine Triphosphate Hydrolysis by the KRas Protein. Biophysica 2023, 3, 373–384. [Google Scholar] [CrossRef]
- Ye, N.; Caruso, F.; Rossi, M. Mechanistic Insights into the Inhibition of SARS-CoV-2 Main Protease by Clovamide and Its Derivatives: In Silico Studies. Biophysica 2021, 1, 377–404. [Google Scholar] [CrossRef]
- Bezerra, K.S.; Fulco, U.L.; Esmaile, S.C.; Lima Neto, J.X.; Machado, L.D.; Freire, V.N.; Albuquerque, E.L.; Oliveira, J.I.N. Ribosomal RNA–Aminoglycoside Hygromycin B Interaction Energy Calculation within a Density Functional Theory Framework. J. Phys. Chem. B 2019, 123, 6421–6429. [Google Scholar] [CrossRef]
- Campos, D.M.O.; Bezerra, K.S.; Esmaile, S.C.; Fulco, U.L.; Albuquerque, E.L.; Oliveira, J.I.N. Intermolecular Interactions of Cn-716 and Acyl-KR-Aldehyde Dipeptide Inhibitors against Zika Virus. Phys. Chem. Chem. Phys. 2020, 22, 15683–15695. [Google Scholar] [CrossRef] [PubMed]
- Ferreira Schopf, P.; Zanella, I.; Cordeiro, M.N.D.S.; Ruso, J.M.; González-Durruthy, M.; Ortiz Martins, M. Nanomarker for Early Detection of Alzheimer’s Disease Combining Ab Initio DFT Simulations and Molecular Docking Approach. Biophysica 2021, 1, 76–86. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An Overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Khan, A.; Ahmad, I.; Alghamdi, S.; Rajab, B.S.; Babalghith, A.O.; Alshahrani, M.Y.; Islam, S.; Islam, M.R. Flavonoids a Bioactive Compound from Medicinal Plants and Its Therapeutic Applications. BioMed Res. Int. 2022, 2022, 1–9. [Google Scholar] [CrossRef]
- Borghetti, G.S.; Carini, J.P.; Honorato, S.B.; Ayala, A.P.; Moreira, J.C.F.; Bassani, V.L. Physicochemical Properties and Thermal Stability of Quercetin Hydrates in the Solid State. Thermochim. Acta 2012, 539, 109–114. [Google Scholar] [CrossRef]
- Materska, M. Quercetin and Its Derivatives: Chemical Structure and Bioactivity—A Review. Pol. J. Food Nutr. Sci. 2008, 58, 407–413. [Google Scholar]
- Kumar, R.; Vijayalakshmi, S.; Nadanasabapathi, S. Health Benefits of Quercetin. Def. Life Sci. J. 2017, 2, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.-T.; Ding, C.; Zhou, N.; Xu, C. Quercetin Protects Gastric Epithelial Cell from Oxidative Damage In Vitro and In Vivo. Eur. J. Pharmacol. 2015, 754, 115–124. [Google Scholar] [CrossRef]
- Angeloni, C.; Spencer, J.P.E.; Leoncini, E.; Biagi, P.L.; Hrelia, S. Role of Quercetin and Its in Vivo Metabolites in Protecting H9c2 Cells against Oxidative Stress. Biochimie 2007, 89, 73–82. [Google Scholar] [CrossRef]
- Lesjak, M.; Beara, I.; Simin, N.; Pintać, D.; Majkić, T.; Bekvalac, K.; Orčić, D.; Mimica-Dukić, N. Antioxidant and Anti-Inflammatory Activities of Quercetin and Its Derivatives. J. Funct. Foods 2018, 40, 68–75. [Google Scholar] [CrossRef]
- Gormaz, J.; Quintremil, S.; Rodrigo, R. Cardiovascular Disease: A Target for the Pharmacological Effects of Quercetin. Curr. Top. Med. Chem. 2015, 15, 1735–1742. [Google Scholar] [CrossRef]
- Papakyriakopoulou, P.; Velidakis, N.; Khattab, E.; Valsami, G.; Korakianitis, I.; Kadoglou, N.P. Potential Pharmaceutical Applications of Quercetin in Cardiovascular Diseases. Pharmaceuticals 2022, 15, 1019. [Google Scholar] [CrossRef]
- Jeong, J.-H.; An, J.Y.; Kwon, Y.T.; Rhee, J.G.; Lee, Y.J. Effects of Low Dose Quercetin: Cancer Cell-Specific Inhibition of Cell Cycle Progression. J. Cell. Biochem. 2009, 106, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, L.; Pinti, M.; Nasi, M.; Montagna, J.P.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cooper, E.L.; Cossarizza, A. Quercetin and Cancer Chemoprevention. Evid. Based Complement. Altern. Med. 2011, 2011, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bachmetov, L.; Gal-Tanamy, M.; Shapira, A.; Vorobeychik, M.; Giterman-Galam, T.; Sathiyamoorthy, P.; Golan-Goldhirsh, A.; Benhar, I.; Tur-Kaspa, R.; Zemel, R. Suppression of Hepatitis C Virus by the Flavonoid Quercetin Is Mediated by Inhibition of NS3 Protease Activity: Quercetin Suppresses HCV through Inhibition of NS3. J. Viral Hepat. 2012, 19, e81–e88. [Google Scholar] [CrossRef]
- Cheng, Z.; Sun, G.; Guo, W.; Huang, Y.; Sun, W.; Zhao, F.; Hu, K. Inhibition of Hepatitis B Virus Replication by Quercetin in Human Hepatoma Cell Lines. Virol. Sin. 2015, 30, 261–268. [Google Scholar] [CrossRef]
- Chiow, K.H.; Phoon, M.C.; Putti, T.; Tan, B.K.H.; Chow, V.T. Evaluation of Antiviral Activities of Houttuynia Cordata Thunb. Extract, Quercetin, Quercetrin and Cinanserin on Murine Coronavirus and Dengue Virus Infection. Asian Pac. J. Trop. Med. 2016, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, A.E.; Kuster, R.M.; Yamamoto, K.A.; Salles, T.S.; Campos, R.; De Meneses, M.D.; Soares, M.R.; Ferreira, D. Quercetin and Quercetin 3-O-Glycosides from Bauhinia longifolia (Bong.) Steud. Show Anti-Mayaro Virus Activity. Parasites Vectors 2014, 7, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanunza, E.; Iampietro, M.; Distinto, S.; Corona, A.; Quartu, M.; Maccioni, E.; Horvat, B.; Tramontano, E. Quercetin Blocks Ebola Virus Infection by Counteracting the VP24 Interferon-Inhibitory Function. Antimicrob. Agents Chemother. 2020, 64, e00530-20. [Google Scholar] [CrossRef]
- Gravina, H.D.; Tafuri, N.F.; Silva Júnior, A.; Fietto, J.L.R.; Oliveira, T.T.; Diaz, M.A.N.; Almeida, M.R. In Vitro Assessment of the Antiviral Potential of Trans-Cinnamic Acid, Quercetin and Morin against Equid Herpesvirus 1. Res. Vet. Sci. 2011, 91, e158–e162. [Google Scholar] [CrossRef]
- Kim, C.H.; Kim, J.-E.; Song, Y.-J. Antiviral Activities of Quercetin and Isoquercitrin Against Human Herpesviruses. Molecules 2020, 25, 2379. [Google Scholar] [CrossRef]
- Choi, H.J.; Song, J.H.; Park, K.S.; Kwon, D.H. Inhibitory Effects of Quercetin 3-Rhamnoside on Influenza A Virus Replication. Eur. J. Pharm. Sci. 2009, 37, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Kim, J.-H.; Lee, C.-H.; Ahn, Y.-J.; Song, J.-H.; Baek, S.-H.; Kwon, D.-H. Antiviral Activity of Quercetin 7-Rhamnoside against Porcine Epidemic Diarrhea Virus. Antivir. Res. 2009, 81, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatima, I.; Ahmad, S.; Alamri, M.A.; Mirza, M.U.; Ul Qamar, M.T.; Rehman, A.; Shahid, F.; Alatawi, E.A.; Alkhayl, F.F.A.; Al-Megrin, W.A.; et al. Discovery of Rift Valley Fever Virus Natural Pan-Inhibitors by Targeting Its Multiple Key Proteins through Computational Approaches. Sci. Rep. 2022, 12, 9260. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hutter, J. Car-Parrinello Molecular Dynamics: Car-Parrinello Molecular Dynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 604–612. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Zhang, D.W.; Zhang, J.Z.H. Molecular Fractionation with Conjugate Caps for Full Quantum Mechanical Calculation of Protein–Molecule Interaction Energy. J. Chem. Phys. 2003, 119, 3599–3605. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Car, R. Introduction to Density-Functional Theory and Ab-Initio Molecular Dynamics. Quant. Struct.-Act. Relatsh. 2002, 21, 97–104. [Google Scholar] [CrossRef]
- Albuquerque, E.L.; Fulco, U.; Freire, V.; Caetano, E.; Lyra, M.; de Moura, F. DNA-Based Nanobiostructured Devices: The Role of Quasiperiodicity and Correlation Effects. Phys. Rep. 2014, 535, 139–209. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Albuquerque, A.C.C.; Bezerra, K.S.; De Fátima Vianna, J.; Batista, S.O.; De Lima Neto, J.X.; De Oliveira Campos, D.M.; Oliveira, J.I.N.; Galvão, D.S.; Fulco, U.L. In Silico Evaluation of the Binding Energies of Androgen Receptor Agonists in Wild-Type and Mutational Models. J. Phys. Chem. B 2023, 127, 5005–5017. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Harvey, A. Natural Products in Drug Discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [Green Version]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An Analysis of FDA-Approved Drugs: Natural Products and Their Derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef]
- Butler, M.S.; Robertson, A.A.B.; Cooper, M.A. Natural Product and Natural Product Derived Drugs in Clinical Trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabavi, S.F.; Russo, G.L.; Daglia, M.; Nabavi, S.M. Role of Quercetin as an Alternative for Obesity Treatment: You Are What You Eat! Food Chem. 2015, 179, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-H.; Lee, S.; Son, H.-Y.; Park, S.-B.; Kim, M.-S.; Choi, E.-J.; Singh, T.S.K.; Ha, J.-H.; Lee, M.-G.; Kim, J.-E.; et al. Flavonoids Inhibit Histamine Release and Expression of Proinflammatory Cytokines in Mast Cells. Arch. Pharm. Res. 2008, 31, 1303–1311. [Google Scholar] [CrossRef]
- Ezzati, M.; Yousefi, B.; Velaei, K.; Safa, A. A Review on Anti-Cancer Properties of Quercetin in Breast Cancer. Life Sci. 2020, 248, 117463. [Google Scholar] [CrossRef]
- Barbosa, N.S.; Concha, J.O.; daSilva, L.L.P.; Crump, C.M.; Graham, S.C. Oropouche Virus Glycoprotein Topology and Cellular Requirements for Glycoprotein Secretion. J. Virol. 2023, 97, e01331-22. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AutoDock Vina Scores (kcal/mol) | ||||||
|---|---|---|---|---|---|---|
| Min. | 1st Quartile | Median | Mean | 3rd Quartile | Max. | SD & |
| −6.268 | −5.943 | −5.919 | −5.916 | −5.885 | −5.799 | 0.059 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menezes, G.d.L.; Saivish, M.V.; Sacchetto, L.; da Silva, G.C.D.; Teixeira, I.d.S.; Mistrão, N.F.B.; Nogueira, M.L.; Oliveira, J.I.N.; Bezerra, K.S.; da Silva, R.A.; et al. Exploring Quercetin Hydrate’s Potential as an Antiviral Treatment for Oropouche Virus. Biophysica 2023, 3, 485-500. https://doi.org/10.3390/biophysica3030032
Menezes GdL, Saivish MV, Sacchetto L, da Silva GCD, Teixeira IdS, Mistrão NFB, Nogueira ML, Oliveira JIN, Bezerra KS, da Silva RA, et al. Exploring Quercetin Hydrate’s Potential as an Antiviral Treatment for Oropouche Virus. Biophysica. 2023; 3(3):485-500. https://doi.org/10.3390/biophysica3030032
Chicago/Turabian StyleMenezes, Gabriela de Lima, Marielena Vogel Saivish, Lívia Sacchetto, Gislaine Celestino Dutra da Silva, Igor da Silva Teixeira, Natalia Franco Bueno Mistrão, Maurício Lacerda Nogueira, Jonas Ivan Nobre Oliveira, Katyanna Sales Bezerra, Roosevelt Alves da Silva, and et al. 2023. "Exploring Quercetin Hydrate’s Potential as an Antiviral Treatment for Oropouche Virus" Biophysica 3, no. 3: 485-500. https://doi.org/10.3390/biophysica3030032