
On the Molecular Driving Force of Protein–Protein Association


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Solvent-Excluded Volume Effect
3. Theory Section
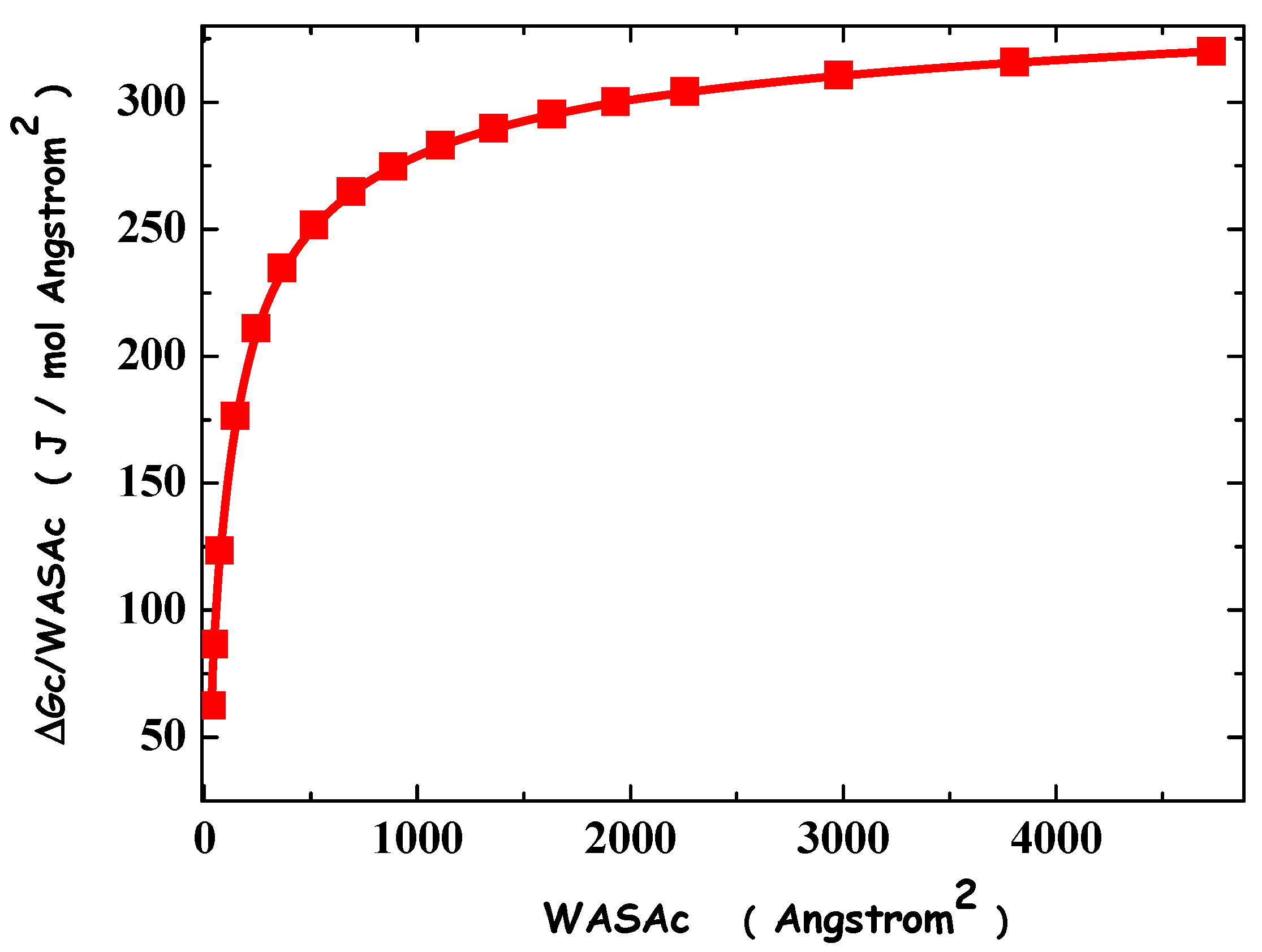
4. Results and Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, E.M.; Pellegrini, M.; Ng, H.L.; Rice, D.W.; Yeates, T.O.; Eisenberg, D. Detecting protein function and protein-protein interactions from genome sequences. Science 1999, 285, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Wodak, S.J.; Vlasblom, J.; Turinsky, A.L.; Pu, S. Protein-protein interaction networks: The puzzling riches. Curr. Opin. Struct. Biol. 2013, 23, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Knapp, K.; Le, K.N.; Schafer, N.P.; Safari, M.S.; Davtyan, A.; Wolynes, P.G.; Vekilov, P.G. Frustrated peptide chains at the fibril tip control the kinetics of growth of amyloid-beta fibrils. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Kumar, A.; Chakraborty, D.; Mugnai, M.L.; Straub, J.E.; Thirumalai, D. Sequence Determines the Switch in the Fibril Forming Regions in the Low-Complexity FUS Protein and Its Variants. J. Phys. Chem. Lett. 2021, 12, 9026–9032. [Google Scholar] [CrossRef] [PubMed]
- Ahlers, J.; Adams, E.M.; Bader, V.; Pezzotti, S.; Winklhofer, K.F.; Tatzelt, J.; Havenith, M. The key role of solvent in condensation: Mapping water in liquid-liquid phase-separated FUS. Biophys. J. 2021, 120, 1266–1275. [Google Scholar] [CrossRef]
- Chen, J.; Sawyer, N.; Regan, L. Protein-protein interactions: General trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 2013, 22, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379-IN374. [Google Scholar] [CrossRef]
- Miller, S.; Janin, J.; Lesk, A.M.; Chothia, C. Interior and surface of monomeric proteins. J. Mol. Biol. 1987, 196, 641–656. [Google Scholar] [CrossRef]
- Merlino, A.; Pontillo, N.; Graziano, G. A driving force for polypeptide and protein collapse. Phys. Chem. Chem. Phys. 2016, 19, 751–756. [Google Scholar] [CrossRef]
- Graziano, G. Contrasting the hydration thermodynamics of methane and methanol. Phys Chem. Chem. Phys. 2019, 21, 21418–21430. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Graziano, G. The Gibbs energy cost of cavity creation depends on geometry. J. Mol. Liq. 2015, 211, 1047–1051. [Google Scholar] [CrossRef]
- Luo, H.; Sharp, K. On the calculation of absolute macromolecular binding free energies. Proc. Natl. Acad. Sci. USA 2002, 99, 10399–10404. [Google Scholar] [CrossRef]
- Zhou, H.X.; Gilson, M.K. Theory of free energy and entropy in noncovalent binding. Chem. Rev. 2009, 109, 4092–4107. [Google Scholar] [CrossRef]
- Pan, A.C.; Jacobson, D.; Yatsenko, K.; Sritharan, D.; Weinreich, T.M.; Shaw, D.E. Atomic-level characterization of protein-protein association. Proc. Natl. Acad. Sci. USA 2019, 116, 4244–4249. [Google Scholar] [CrossRef]
- Froloff, N.; Windemuth, A.; Honig, B. On the calculation of binding free energies using continuum methods: Application to MHC class I protein-peptide interactions. Protein Sci. 1997, 6, 1293–1301. [Google Scholar] [CrossRef]
- Jackson, R.M.; Sternberg, M.J. A continuum model for protein-protein interactions: Application to the docking problem. J. Mol. Biol. 1995, 250, 258–275. [Google Scholar] [CrossRef]
- Ben-Naim, A. Temperature and Pressure Dependence of the Hydrophobic Interactions. In Hydrophobic Interactions; Springer: Boston, MA, USA, 1980; pp. 185–258. [Google Scholar]
- Graziano, G. Dimerization thermodynamics of large hydrophobic plates: A scaled particle theory study. J. Phys. Chem. B 2009, 113, 11232–11239. [Google Scholar] [CrossRef] [PubMed]
- Setny, P.; Baron, R.; McCammon, J.A. How Can Hydrophobic Association Be Enthalpy Driven? J. Chem. Theory Comput. 2010, 6, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. Molecular driving forces of the pocket–ligand hydrophobic association. Chem. Phys. Lett. 2012, 533, 95–99. [Google Scholar] [CrossRef]
- Pierotti, R.A. A scaled particle theory of aqueous and nonaqueous solutions. Chem. Rev. 1976, 76, 717–726. [Google Scholar] [CrossRef]
- Wallqvist, A.; Berne, B.J. Molecular Dynamics Study of the Dependence of Water Solvation Free Energy on Solute Curvature and Surface Area. J. Phys. Chem. 1995, 99, 2885–2892. [Google Scholar] [CrossRef]
- Underwood, R.; Tomlinson-Phillips, J.; Ben-Amotz, D. Are long-chain alkanes hydrophilic? J. Phys. Chem. B 2010, 114, 8646–8651. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Sternberg, M.J. Side-chain conformational entropy in protein folding. Protein Sci. 1995, 4, 2247–2251. [Google Scholar] [CrossRef] [PubMed]
- Samanta, U.; Bahadur, R.P.; Chakrabarti, P. Quantifying the accessible surface area of protein residues in their local environment. Protein Eng. 2002, 15, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.V.; Janin, J. The price of lost freedom: Entropy of bimolecular complex formation. Protein Eng. Des. Sel. 1989, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics. Chem. Rev. 1999, 99, 2161–2200. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapuano, R.; Graziano, G. On the Molecular Driving Force of Protein–Protein Association. Biophysica 2022, 2, 240-247. https://doi.org/10.3390/biophysica2030023
Rapuano R, Graziano G. On the Molecular Driving Force of Protein–Protein Association. Biophysica. 2022; 2(3):240-247. https://doi.org/10.3390/biophysica2030023
Chicago/Turabian StyleRapuano, Roberta, and Giuseppe Graziano. 2022. "On the Molecular Driving Force of Protein–Protein Association" Biophysica 2, no. 3: 240-247. https://doi.org/10.3390/biophysica2030023