
Wilms Tumor: Updates about Pathogenesis and New Possible Clinical Treatments of the Most Frequent Pediatric Urogenital Cancer: A Narrative Review

Abstract
:1. Introduction
1.1. General and Epidemiological Profiles
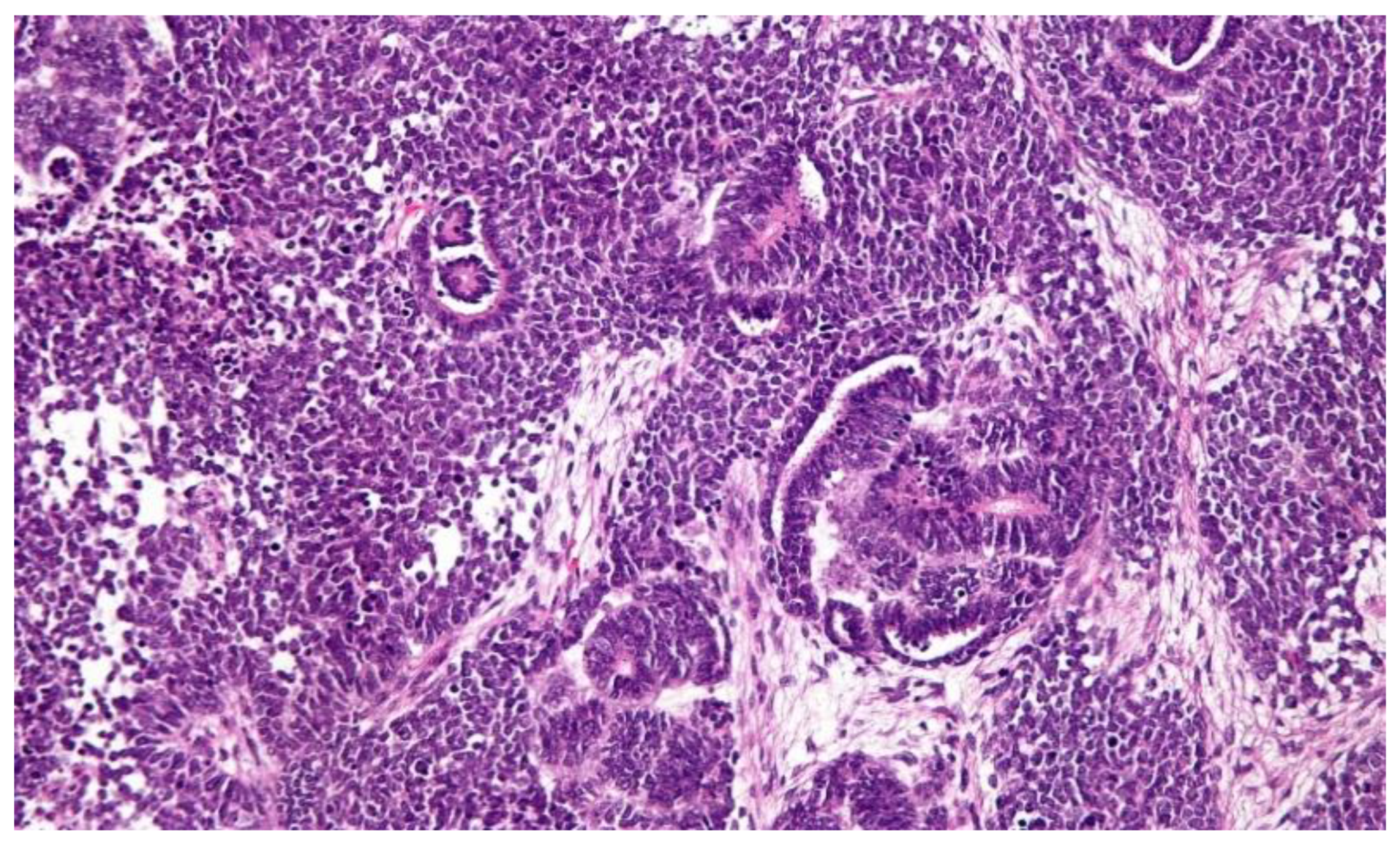
1.2. Pathology
1.3. Clinical Profiles
2. Objective and Aim
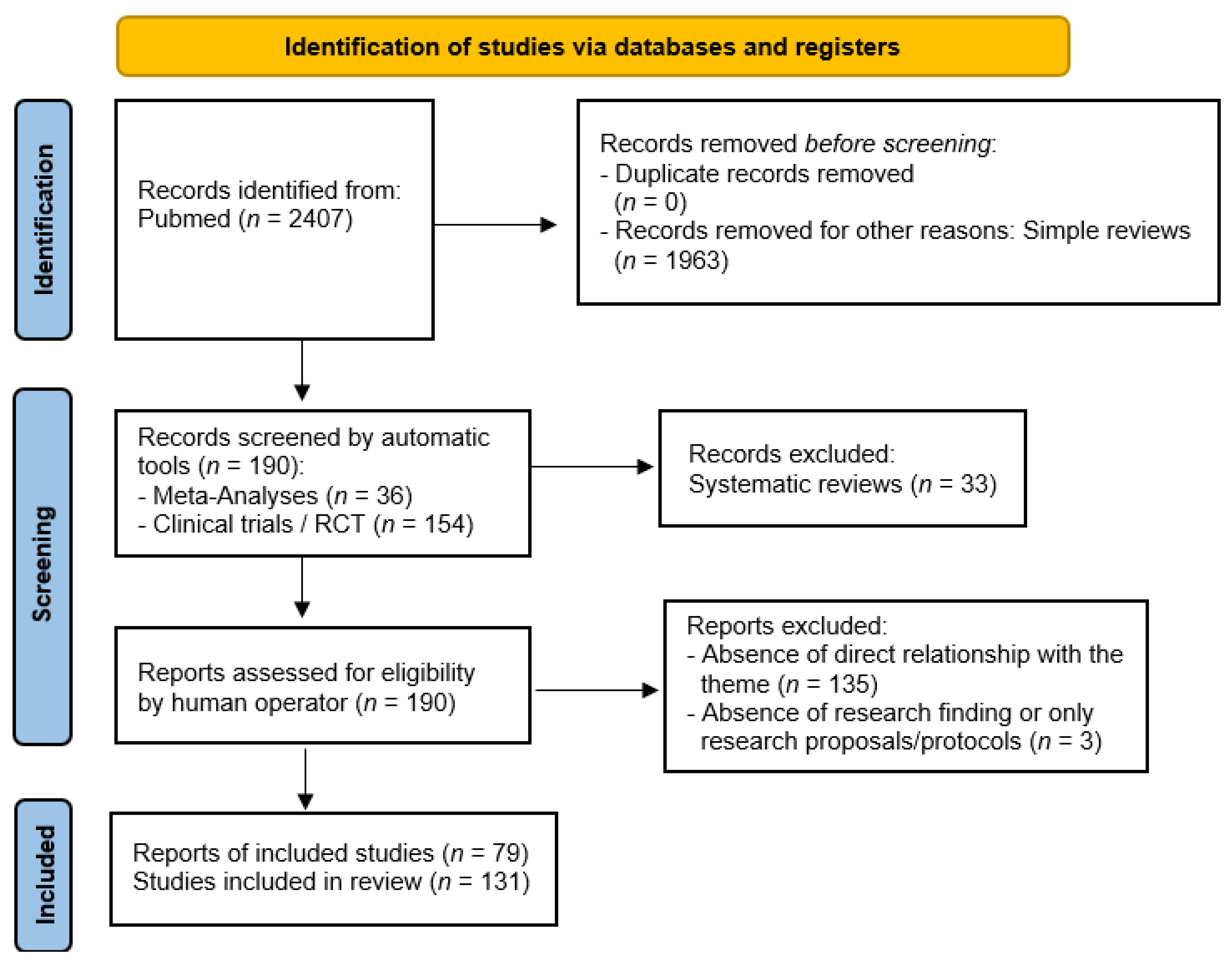
3. Materials and Method
4. Results
4.1. Etiology
4.2. Diagnosis
4.3. Therapy
4.4. Prognosis
4.5. Nutritional Implications
4.6. Psychological Implications
5. Discussion and Prospects
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spreafico, F.; Bellani, F.F. Wilms’ tumor: Past, present and (possibly) future. Expert Rev. Anticancer Ther. 2006, 6, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Welter, N.; Brzezinski, J.; Treece, A.; Chintagumpala, M.; Young, M.D.; Perotti, D.; Kieran, K.; Jongmans, M.C.J.; Murphy, A.J. The pathophysiology of bilateral and multifocal Wilms tumors: What we can learn from the study of predisposition syndromes. Pediatr. Blood Cancer 2022, 70, e29984. [Google Scholar] [CrossRef] [PubMed]
- Vujanic, G.M.; Parsons, L.N.; D’Hooghe, E.; Treece, A.L.; Collini, P.; Perlman, E.J. Pathology of Wilms’ tumour in International Society of Paediatric Oncology (SIOP) and Children’s oncology group (COG) renal tumour studies: Similarities and differences. Histopathology 2022, 80, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Gonzàlez-Arboleda, A.; Fernandez, N.; Garcìa-Perdomo, H.A. Genitourinary Tract Tumors in Children: An Update. Curr. Pediatr. Rev. 2022, 18, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.; Tang, D.; Gu, W.; Mao, J.; Shu, Q. Current treatment for Wilms tumor: COG and SIOP standards. World J. Pediatr. Surg. 2019, 2, e000038. [Google Scholar] [CrossRef]
- Ekenze, S.O.; Okafor, O.C.; Obasi, A.A.; Okafor, D.C.; Nnabugwu, I.I. Wilms tumor in Africa: A systematic review of management challenges and outcome in two decades (2000–2019). Pediatr. Blood Cancer 2020, 67, e28695. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.V.; Koenig, C.; Armstrong, A.E.; Brok, J.; de Camargo, B.; Mavinkurve-Groothuis, A.M.C. Advances in the clinical management of high-risk Wilms tumors. Pediatr. Blood Cancer 2023, 70, e30153. [Google Scholar] [CrossRef]
- Libes, J.; Hol, J.; Vallance, K.L.; van Tinteren, H.; Benedetti, D.J.; Ramirez Villar, G.L. Pediatric renal tumor epidemiology: Global perspectives, progress, and challenges. Pediatr. Blood Cancer 2023, 70, e30006. [Google Scholar] [CrossRef]
- Mohd, A.B.; Ghannam, R.A.; Mohd, O.B.; Elayan, R.; Albakri, K.; Huneiti, N.; Daraghmeh, F.; Al-Khatatbeh, E.; Al-Thnaibat, M. Etiologies, Gross Appearance, Histopathological Patterns, Prognosis, and Best Treatments for Subtypes of Renal Carcinoma: An Educational Review. Cureus 2022, 14, e32338. [Google Scholar] [CrossRef]
- Takahashi, R.; Asanuma, H.; Mizuno, R.; Oya, M. Current clinical perspective of urological oncology in the adolescent and young adult gen-eration. Int. J. Clin. Oncol. 2023, 28, 28–40. [Google Scholar] [CrossRef]
- Perrotta, G. Port-Site Metastasis (PSM): Definition, clinical contexts and possible preventive actions to reduce risk. J. Surg. Surg. Res. 2021, 7, 088–092. [Google Scholar]
- Quarello, P.; Carli, D.; Biasoni, D.; Gerocarni Nappo, S.; Morosi, C.; Cotti, R.; Garelli, E.; Zucchetti, G.; Spadea, M.; Tirtei, E.; et al. Implications of an Underlying Beckwith–Wiedemann Syndrome for Wilms Tumor Treatment Strategies. Cancers 2023, 15, 1292. [Google Scholar] [CrossRef] [PubMed]
- Craver, R.; Stark, M.; Moss, S.; Long, S.; Prasad, P.; Roth, C.C. WAGR, Sex Reversal, Bilateral Gonadoblastomas, and Intralobar Nephrogenic Rests: Uncertainties of Pre-Biopsy Chemotherapy in a High Risk Syndrome for Nephroblastoma. Fetal. Pediatr. Pathol. 2023, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Feng, S.J.; Jin, X.; Wang, K.J. A patient with Denys-Drash syndrome (DDS) underwent renal allotransplantation with preserved autologous kidney. Asian J. Surg. 2023, 46, 1313–1314. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.N.; Rhee, D.; Tracy, E.T.; Aldrink, J.H.; Baertschiger, R.M.; Lautz, T.B.; Glick, R.D.; Rodeberg, D.A.; Ehrlich, P.F.; Christison-Lagay, E. Pediatric solid tumors and associated cancer predisposition syndromes: Workup, management, and surveillance. A summary from the APSA Cancer Committee. J. Pediatr. Surg. 2022, 57, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xiong, Q.-W.; Wang, J.-H.; Peng, W.-X. Roles of lncRNAs in childhood cancer: Current landscape and future perspectives. Front. Oncol. 2023, 13, 1060107. [Google Scholar] [CrossRef]
- Hont, A.B.; Dumont, B.; Sutton, K.S.; Anderson, J.; Kentsis, A.; Drost, J. The tumor microenvironment and immune targeting therapy in pe-diatric renal tumors. Pediatr. Blood Cancer 2022, 70, e30110. [Google Scholar] [CrossRef]
- Bánki, T.; Drost, J.; van den Heuvel-Eibrink, M.M.; Mavinkurve-Groothuis, A.M.C.; de Krijger, R.R. Somatic, Genetic and Epigenetic Changes in Nephrogenic Rests and Their Role in the Transformation to Wilms Tumors, a Systematic Review. Cancers 2023, 15, 1363. [Google Scholar] [CrossRef]
- Bu, Q.; He, H.; Fan, D.; Lyu, J.; Pan, Z.; You, H. Association between loss of heterozygosity of chromosome 16q and survival in Wilms’ tumor: A meta-analysis. Pathol. Res. Pract. 2018, 214, 1772–1777. [Google Scholar] [CrossRef]
- Lizhi, L.; Rongdong, H.; Shaohua, H.; Yingquan, K.; Huihuang, X.; Shan, L.; Kunbin, T.; Di, X. Association Between TP53 Mutation and Prognosis in Wilms Tumor: A Meta-Analysis. Fetal Pediatr. Pathol. 2021, 40, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhuo, Z.; Yang, L.; Zhao, P.; Zhang, J.; Zhou, H.; He, J.; Li, P. HMGA2 gene polymorphisms and Wilms tumor susceptibility in Chinese children: A four-center case-control study. Biotechnol. Appl. Biochem. 2020, 67, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Feulefack, J.; Sergi, C. Exposure to pesticides and pediatric Wilms’ tumor. A meta-analysis on pre-conception and pregnancy parental exposure with an IARC/WHO commentary. Hum. Exp. Toxicol. 2022, 41, 36289056. [Google Scholar] [CrossRef] [PubMed]
- Doganis, D.; Katsimpris, A.; Panagopoulou, P.; Bouka, P.; Bouka, E.; Moschovi, M.; Polychronopoulou, S.; Papakonstantinou, E.; Tragiannidis, A.; Katzilakis, N.; et al. Maternal lifestyle characteristics and Wilms tumor risk in the offspring: A systematic review and meta-analysis. Cancer Epidemiol. 2020, 67, 101769. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Busch, M.; Beier, M.; Duhme, C.; de Torres, C.; Mora, J.; Brandt, A.; Royer, H.-D. Wilms tumor cells with WT1 mutations have characteristic features of mesenchymal stem cells and express molecular markers of paraxial mesoderm. Hum. Mol. Genet. 2010, 19, 1651–1668. [Google Scholar] [CrossRef]
- Brzezinski, J.; Choufani, S.; Romao, R.; Shuman, C.; Chen, H.; Cunanan, J.; Bagli, D.; Grant, R.; Lorenzo, A.; Weksberg, R. Clinically and biologically relevant subgroups of Wilms tumour defined by genomic and epigenomic analyses. Br. J. Cancer 2021, 124, 437–446. [Google Scholar] [CrossRef]
- Anvar, Z.; Acurzio, B.; Roma, J.; Cerrato, F.; Verde, G. Origins of DNA methylation defects in Wilms tumors. Cancer Lett. 2019, 457, 119–128. [Google Scholar] [CrossRef]
- Welter, N.; Furtwangler, R.; Schneider, G.; Graf, N.; Schenk, J.-P. Tumor predisposition syndromes and nephroblastoma: Early diagnosis with imaging. Radiologie 2022, 62, 1033–1042. [Google Scholar] [CrossRef]
- Jedrzejewski, G.; Wozniak, M.M.; Pawelec, A.; Matera, A.; Kunach, M.; Madej, T.; Wieczorek, A.P.; Nowakowska, K. Ultrasound screening for neoplasms in children up to 6 years old. Medicine 2016, 95, e5124. [Google Scholar] [CrossRef]
- van der Beek, J.N.; Artunduaga, M.; Schenk, J.; Eklund, M.J.; Smith, E.A.; Lederman, H.M.; Warwick, A.B.; Littooij, A.S.; Khanna, G. Similarities and controversies in imaging of pediatric renal tumors: A SIOP-RTSG and COG collaboration. Pediatr. Blood Cancer 2022, 70, e30080. [Google Scholar] [CrossRef]
- Uslu, L.; Donig, J.; Link, M.; Rosenberg, J.; Quon, A.; Daldrup-Link, H.E. Value of 18F-FDG PET and PET/CT for Evaluation of Pediatric Malignancies. J. Nucl. Med. 2015, 56, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Madanat-Harjuoja, L.M.; Renfro, L.A.; Klega, K.; Tornwall, B.; Thorner, A.R.; Nag, A. Circulating Tumor DNA as a Biomarker in Patients with Stage III and IV Wilms Tumor: Analysis From a Children’s Oncology Group Trial, AREN0533. J. Clin. Oncol. 2022, 40, 3047–3056. [Google Scholar] [CrossRef] [PubMed]
- Walz, A.L.; Maschietto, M.; Crompton, B.; Evageliou, N.; Dix, D.; Tytgat, G.; Gessler, M.; Gisselsson, D.; Daw, N.C.; Wegert, J. Tumor biology, biomarkers, and liquid biopsy in pediatric renal tumors. Pediatr. Blood Cancer 2023, 70, e30130. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Dai, R.; Li, X.; Liu, F. Genetic variation frequencies in Wilms’ tumor: A meta-analysis and systematic review. Cancer Sci. 2016, 107, 690–699. [Google Scholar] [CrossRef]
- Chagtai, T.; Zill, C.; Dainese, L.; Wegert, J.; Savola, S.; Popov, S.; Mifsud, W.; Vujanić, G.; Sebire, N.; Le Bouc, Y.; et al. Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated with Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J. Clin. Oncol. 2016, 34, 3195–3203. [Google Scholar] [CrossRef]
- Vujanic, G.M.; Gessler, M.; Ooms, A.H.A.G.; Collini, P.; Coulomb-L’Hermine, A.; D’Hooghe, E.; De Krijger, R.R.; Perotti, D.; Pritchard-Jones, K.; Vokuhl, C.; et al. The UMBRELLA SIOP–RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat. Rev. Urol. 2018, 15, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, J.B. Precursor lesions of Wilms tumor: Clinical and biological implications. Med. Pediatr. Oncol. 1993, 21, 158–168. [Google Scholar] [CrossRef]
- Emir, S. Wilms tumor with intravascular tumor thrombus. Transl. Pediatr. 2014, 3, 29–33. [Google Scholar] [CrossRef]
- National Cancer Institute (NIH). Wilms Tumor and Other Childhood Kidney Tumors Treatment (PDQ®)–Patient Version. Available online: https://www.cancer.gov/types/kidney/patient/wilms-treatment-pdq#section/_29 (accessed on 23 April 2023).
- Palmisani, F.; Kovar, H.; Kager, L.; Amann, G.; Metzelder, M.; Bergmann, M. Systematic review of the immunological landscape of Wilms tumors. Mol. Ther. Oncolytics 2021, 22, 454–467. [Google Scholar] [CrossRef]
- Abdelhafeez, A.H.; Reljic, T.; Kumar, A.; Banu, T.; Cox, S.; Davidoff, A.M.; Elgendy, A.; Ghandour, K.; Gerstle, J.T.; Karpelowsky, J.; et al. Evidence-based surgical guidelines for treating children with Wilms tumor in low-resource settings. Pediatr. Blood Cancer 2022, 69, e29906. [Google Scholar] [CrossRef]
- Sforza, S.; Palmieri, V.E.; Raspollini, M.R.; Roviello, G.; Mantovani, A.; Basso, U.; Affinita, M.C.; D’Angelo, A.; Antonuzzo, L.; Carini, M.; et al. Robotic approach with neoadjuvant chemotherapy in adult Wilms’ tumor: A feasibility study report and a systematic review of the literature. Asian J. Urol. 2023, 10, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Malek, M.M.; Behr, C.A.; Aldrink, J.H.; Dasgupta, R.; Heaton, T.E.; Gehred, A.; Lautz, T.B.; Baertschiger, R.M.; Christison-Lagay, E.R.; Tracy, E.T.; et al. Minimally invasive surgery for pediatric renal tumors: A systematic review by the APSA Cancer Committee. J. Pediatr. Surg. 2020, 55, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Li, K.; Dong, K.; Xiao, X.; Yao, W.; Liu, G. Clinical features, treatment, and outcomes of bilateral Wilms’ tumor: A systematic review and meta-analysis. J. Pediatr. Surg. 2018, 53, 2465–2469. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.; Irtan, S.; Bergeron, C.; Pritchard-Jones, K. Bilateral Wilms tumour: A review of clinical and molecular features. Expert Rev. Mol. Med. 2017, 19, e8. [Google Scholar] [CrossRef]
- Ehrlich, P.; Chi, Y.Y.; Chintagumpala, M.M.; Hoffer, F.A.; Perlman, E.J.; Kalapurakal, J.A. Results of the First Prospective Multi-institutional Treatment Study in Children with Bilateral Wilms Tumor (AREN0534): A Report from the Children’s Oncology Group. Ann. Surg. 2017, 266, 470–478. [Google Scholar] [CrossRef]
- Chintagumpala, M.M.; Perlman, E.J.; Tornwall, B.; Chi, Y.; Kim, Y.; Hoffer, F.A.; Kalapurakal, J.A.; Warwick, A.B.; Shamberger, R.C.; Khanna, G.; et al. Outcomes based on histopathologic response to preoperative chemotherapy in children with bilateral Wilms tumor: A prospective study (COG AREN0534). Cancer 2022, 128, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Gurria, J.P.; Malek, M.M.; Heaton, T.E.; Gehred, A.; Lautz, T.B.; Rhee, D.S.; Tracy, E.T.; Grant, C.N.; Baertshiger, R.M.; Bruny, J.; et al. Minimally invasive surgery for abdominal and thoracic neuroblastic tumors: A systematic review by the APSA Cancer committee. J. Pediatr. Surg. 2020, 55, 2260–2272. [Google Scholar] [CrossRef]
- Ehrlich, P.F. Commentary Re: Lymph node sampling in Wilms tumor—Getting risk based therapy right. J. Pediatr. Surg. 2020, 55, 2676. [Google Scholar] [CrossRef]
- Sandberg, J.K.; Chi, Y.-Y.; Smith, E.A.; Servaes, S.; Hoffer, F.A.; Mullen, E.A.; Perlman, E.J.; Tornwall, B.; Ehrlich, P.F.; Geller, J.I.; et al. Imaging Characteristics of Nephrogenic Rests Versus Small Wilms Tumors: A Report From the Children’s Oncology Group Study AREN03B2. AJR Am. J. Roentgenol. 2020, 214, 987–994. [Google Scholar] [CrossRef]
- Ehrlich, P.F.; Chi, Y.-Y.; Chintagumpala, M.M. Results of Treatment for Patients with Multicentric or Bilaterally Predisposed Unilateral Wilms Tumor (AREN0534): A report from the Children’s Oncology Group. Cancer 2020, 126, 3516–3525. [Google Scholar] [CrossRef]
- Higgins, M.; Curigliano, G.; Dieras, V.; Kuemmel, S.; Kunz, G.; Fasching, P.A.; Campone, M.; Bachelot, T.; Krivorotko, P.; Chan, S.; et al. Safety and immunogenicity of neoadjuvant treatment using WT1-immunotherapeutic in combination with standard therapy in patients with WT1-positive Stage II/III breast cancer: A randomized Phase I study. Breast Cancer Res. Treat. 2017, 162, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Gómez, F.M.; Patel, P.A.; Stuart, S.; Roebuck, D.J. Systematic review of ablation techniques for the treatment of malignant or aggressive benign lesions in children. Pediatr. Radiol. 2014, 44, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel-Eibrink, M.M.; Hol, J.A.; Pritchard-Jones, K.; van Tinteren, H.; Furtwängler, R.; Verschuur, A.C.; Vujanic, G.M.; Leuschner, I.; Brok, J.; Rübe, C.; et al. Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP–RTSG 2016 protocol. Nat. Rev. Urol. 2017, 14, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pineda, I.; Garcìa-Cantòn, J.A.; Pérez-Bertolez, S.; Tuduri, I.; Ramìrez, G.; Marquez, C.; de Augustìn, J.C. Fine-needle aspiration cytopathology in the diagnosis of Wilms tumor. Clin. Transl. Oncol. 2011, 13, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Grundy, P.E.; Breslow, N.E.; Li, S.; Perlman, E.; Beckwith, J.B.; Ritchey, M.L.; Shamberger, R.C.; Haase, G.M.; D’Angio, G.J.; Donaldson, M.; et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favora-ble-histology Wilms tumor: A report from the National Wilms Tumor Study Group. J. Clin. Oncol. 2005, 23, 7312–7321. [Google Scholar] [CrossRef] [PubMed]
- Dome, J.S.; Cotton, C.A.; Perlman, E.J.; Breslow, N.E.; Kalapurakal, J.A.; Ritchey, M.L.; Grundy, P.E.; Malogolowkin, M.; Beckwith, J.B.; Shamberger, R.C.; et al. Treatment of anaplastic histology Wilms’ tumor: Results from the fifth national wilms’ tumor study. J. Clin. Oncol. 2006, 24, 2352–2358. [Google Scholar] [CrossRef] [PubMed]
- Phelps, H.M.; Kaviany, S.; Borinstein, S.C.; Lovvorn III, H.N. Biological Drivers of Wilms Tumor Prognosis and Treatment. Children 2018, 5, 145. [Google Scholar] [CrossRef]
- Daw, N.C.; Chi, Y.-Y.; Kalapurakal, J.A.; Kim, Y.; Hoffer, F.A.; Geller, J.I.; Perlman, E.J.; Ehrlich, P.F.; Mullen, E.A.; Warwick, A.B.; et al. Activity of Vincristine and Irinotecan in Diffuse Anaplastic Wilms Tumor and Therapy Outcomes of Stage II to IV Disease: Results of the Children’s Oncology Group AREN0321 Study. J. Clin. Oncol. 2020, 38, 1558–1568. [Google Scholar] [CrossRef]
- Gellerm, J.I.; Hong, A.L.; Vallance, K.L.; Evageliou, N.; Aldrink, J.H.; Cost, N.G.; Treece, A.L.; Renfro, L.A.; Mullen, E.A.; COG Renal Tumor Committee. Children’s Oncology Group’s 2023 blueprint for research: Renal tumors. Pediatr. Blood Cancer 2023, 70 (Suppl. S6), e30586. [Google Scholar] [CrossRef]
- Morris, L.; Squire, R.; Sznajder, B.; van Tinteren, H.; Godzinski, J.; Powis, M. Optimal neoadjuvant chemotherapy duration in Wilms tumour with intravascular thrombus: A literature review and evidence from SIOP WT 2001 trial. Pediatr. Blood Cancer 2019, 66, e27930. [Google Scholar] [CrossRef]
- Popov, S.D.; Sebire, N.J.; Vujanic, G.M. Chapter 1—Wilms’ Tumour—Histology and Differential Diagnosis. In Wilms Tumor; van den Heuvel-Eibrink, M.M., Ed.; Codon Publications: Brisbane, Australia, 2016. [Google Scholar]
- Pater, L.; Melchior, P.; Rube, C.; Cooper, B.T.; McAleer, M.F.; Kalapurakal, J.A.; Paulino, A.C. Wilms tumor. Pediatr. Blood Cancer 2021, 68 (Suppl. S2), e28257. [Google Scholar] [CrossRef]
- Malogolowkin, M.; Spreafico, F.; Dome, J.S.; van Tinteren, H.; Pritchard-Jones, K.; van den Heuvel-Eibrink, M.M. Incidence and outcomes of patients with late recurrence of Wilms’ tumor. Pediatr. Blood Cancer 2013, 60, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Tahbaz, R.; Schmid, M.; Merseburger, A.S. Prevention of kidney cancer incidence and recurrence: Lifestyle, medication and nutrition. Curr. Opin. Urol. 2018, 28, 62–79. [Google Scholar] [CrossRef]
- Geets, E.; Meuwissen, M.; Van Hul, W. Clinical, molecular genetics and therapeutic aspects of syndromic obesity. Clin. Genet. 2019, 95, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y. Wilms’ tumor management. Curr. Opin. Urol. 2005, 15, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.K.; Suson, K.D. Syndromic Wilms tumor: A review of predisposing conditions, surveillance and treatment. Transl. Androl. Urol. 2020, 9, 2370–2381. [Google Scholar] [CrossRef]
- Perrotta, G. Anxiety disorders: Definitions, contexts, neural correlates and strategic therapy. J. Neur. Neurosci. 2019, 6, 046. [Google Scholar]
- Nash, R.P.; Loiselle, M.M.; Stahl, J.L.; Conklin, J.L.; Rose, T.L.; Hutto, A.; Evon, D.M.; Flythe, J.E.; Burker, E.J. Post-Traumatic Stress Disorder and Post-Traumatic Growth following Kidney Transplantation. Kidney360 2022, 3, 1590–1598. [Google Scholar] [CrossRef]
- Perrotta, G. Depressive disorders: Defi nitions, contexts, differential diagnosis, neural correlates and clinical strategies. Arch. Depress. Anxiety 2019, 5, 9–33. [Google Scholar] [CrossRef]
- Schuyler, M.; Geller, D.A. Childhood Obsessive-Compulsive Disorder. Psychiatr. Clin. N. Am. 2023, 46, 89–106. [Google Scholar] [CrossRef]
- Leibenluft, E.; Rich, B.A. Pediatric bipolar disorder. Annu. Rev. Clin. Psychol. 2008, 4, 163–187. [Google Scholar] [CrossRef]
- Perrotta, G. Psychological trauma: Definition, clinical contexts, neural correlations and therapeutic approaches. Curr. Res. Psychiatry Brain Disord. 2019, 2019, CRPBD-100006. [Google Scholar]
- Perrotta, G. The reality plan and the subjective construction of one’s perception: The strategic theoretical model among sensations, perceptions, defence mechanisms, needs, personal constructs, beliefs system, social influences and systematic errors. J. Clin. Res. Rep. 2019, 1. [Google Scholar] [CrossRef]
- Perrotta, G. The “Acceptance” in the Elaboration of Mourning in Oncological Diseases: Definition, Theoretical Models and Practical Applications: Needs Analysis and Subjective Oncological Reality. Biomed. J. Sci. Tech. Res. 2019, 21, 1. [Google Scholar] [CrossRef]
- Knoerl, R.; Mazzola, E.; Woods, H.; Buchbinder, E.; Frazier, L.; LaCasce, A.; Li, B.T.; Luskin, M.R.; Phillips, C.S.; Thornton, K.; et al. Exploring the Feasibility of a Mindfulness-Music Therapy Intervention to Improve Anxiety and Stress in Adolescents and Young Adults with Cancer. J. Pain Symptom Manag. 2022, 63, e357–e363. [Google Scholar] [CrossRef]
- Hasanah, I.; Nursalam, N.; Krisnana, I.; Ramdani, W.F.; Haikal, Z.; Rohita, T. Psychoneuroimmunological Markers of Psychological Intervention in Pediatric Cancer: A Systematic Review and New Integrative Model. Asian Nurs. Res. 2023, 17, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Melesse, T.G.; Chau, J.P.C.; Nan, M.A. Effectiveness of psychosocial interventions on health outcomes of children with cancer: A systematic review of randomised controlled trials. Eur. J. Cancer Care 2022, 31, e13695. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Cheng, C.; Williams, J.; Shaw, T.I.; Pinto, E.M.; Dieseldorff-Jones, K.; Brzezinski, J.; Renfro, L.A.; Tornwall, B.; Huff, V.; et al. The Genetic and Epigenetic Features of Bilateral Wilms Tumor Predisposition: A Report from the Children’s Oncology Group AREN18B5-Q Study. Res. Sq. 2023, rs.3.rs-2675436. [Google Scholar]
- Sanatkar, S.A.; Heidari, A.; Arya, S.; Ghasemi, M.; Rezaei, N. The Potential Role of Immunotherapy in Wilms’ Tumor: Opportunities and Challenges. Curr. Pharm. Des. 2023, 29, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Shi, B.; Zhu, K.; Zhong, X.; Lai, D.; Wang, J.; Tou, J. Bioinformatical analysis of the key differentially expressed genes for screening potential biomarkers in Wilms tumor. Sci. Rep. 2023, 13, 15404. [Google Scholar] [CrossRef]
- Bouty, A.; Blanc, T.; Leclair, M.D.; Lavrand, F.; Faure, A.; Binet, A.; Rod, J.; O’Brien, M.; Sarnacki, S.; Nightingale, M.; et al. Minimally invasive surgery for unilateral Wilms tumors: Multicenter retrospective analysis of 50 transperitoneal laparoscopic total nephrectomies. Pediatr. Blood Cancer 2020, 67, e28212. [Google Scholar] [CrossRef]
- Gavens, E.; Arul, G.S.; Pachl, M. A single centre matched pair series comparing minimally invasive and open surgery for the resection of pediatric renal tumours. Surg. Oncol. 2020, 35, 498–503. [Google Scholar] [CrossRef]
- Dix, D.B.; Seibel, N.L.; Chi, Y.-Y.; Khanna, G.; Gratias, E.; Anderson, J.R.; Mullen, E.A.; Geller, J.I.; Kalapurakal, J.A.; Paulino, A.C.; et al. Treatment of Stage IV Favorable Histology Wilms Tumor with Lung Metastases: A Report From the Children’s Oncology Group AREN0533 Study. J. Clin. Oncol. 2018, 36, 1564–1570. [Google Scholar] [CrossRef]
- Irtan, S.; Messahel, B.; Moroz, V.; Taylor, R.E.; Grundy, R.; Kelsey, A.; Vujanic, G.; Pritchard-Jones, K. Outcomes of non-anaplastic stage III and ‘inoperable’ Wilms tumour treated in the UKW3 trial. Radiother. Oncol. 2019, 131, 1–7. [Google Scholar] [CrossRef]
- Vujanic, G.M.; D’Hooghe, E.; Popov, S.D.; Sebire, N.J.; Kelsey, A. The effect of preoperative chemotherapy on histological subtyping and staging of Wilms tumors: The United Kingdom Children’s Cancer Study Group (UKCCSG) Wilms tumor trial 3 (UKW3) experience. Pediatr. Blood Cancer 2019, 66, e27549. [Google Scholar] [CrossRef] [PubMed]
- Joannon, P.; Becker, A.; Kabalan, P.; Concha, E.; Beresi, V.; Salgado, C.; Martínez, P.; Olate, P.; Arriagada, M.; Espinoza, F.; et al. Results of Therapy for Wilms Tumor and Other Malignant Kidney Tumors: A Report from the Chilean Pediatric National Cancer Program (PINDA). J. Pediatr. Hematol. Oncol. 2016, 38, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Hol, J.A.; van den Heuvel-Eibrink, M.M.; Graf, N.; Pritchard-Jones, K.; Brok, J.; van Tinteren, H.; Howell, L.; Verschuur, A.; Bergeron, C.; Kager, L.; et al. Irinotecan for relapsed Wilms tumor in pediatric patients: SIOP experience and review of the literature—A report from the SIOP Renal Tumor Study Group. Pediatr. Blood Cancer 2018, 65, 29077255. [Google Scholar] [CrossRef]
- Kim, A.; Widemann, B.C.; Krailo, M.; Jayaprakash, N.; Fox, E.; Weigel, B. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Tsuboi, A.; Kagawa, N.; Chiba, Y.; Izumoto, S.; Kinoshita, M.; Kijima, N.; Oka, Y.; Morimoto, S.; Nakajima, H.; et al. Wilms tumor 1 peptide vaccination combined with temozolomide against newly diagnosed glioblastoma: Safety and impact on immunological response. Cancer Immunol. Immunother. 2015, 64, 707–716. [Google Scholar] [CrossRef]
- Koido, S.; Homma, S.; Okamoto, M.; Takakura, K.; Mori, M.; Yoshizaki, S. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin. Cancer Res. 2014, 20, 4228–4239. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, M.; Kostic, T.; Virijevic, M.; Karan-Djurasevic, T.; Vukovic, N.S.; Pavlovic, S.; Tosic, N. The influence of Wilms’ tumor 1 gene expression level on prognosis and risk stratification of acute promyelocytic leukemia patients. Int. J. Lab. Hematol. 2020, 42, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Jacobsohn, D.A.; Loken, M.R.; Fei, M.; Adams, A.; Brodersen, L.E.; Logan, B.R.; Ahn, K.W.; Shaw, B.E.; Kletzel, M.; Olszewski, M.; et al. Outcomes of Measurable Residual Disease in Pediatric Acute Myeloid Leukemia before and after Hematopoietic Stem Cell Transplant: Validation of Difference from Normal Flow Cytometry with Chimerism Studies and Wilms Tumor 1 Gene Expression. Biol. Blood Marrow Transplant. 2018, 24, 2040–2046. [Google Scholar] [CrossRef]
- Shibasaki, Y.; Seki, Y.; Tanaka, T.; Miyakoshi, S.; Fuse, K.; Kozakai, T.; Kobayashi, H.; Ushiki, T.; Abe, T.; Yano, T.; et al. The association of level of reduction of Wilms’ tumor gene 1 mRNA transcript in bone marrow and outcome in acute myeloid leukemia patients. Leuk. Res. 2015, 39, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, M.; Miyashita, M.; Yamagishi, Y.; Ota, S. Phase I/II Pilot Study of Wilms’ Tumor 1 Peptide-Pulsed Dendritic Cell Vaccination Combined with Conventional Chemotherapy in Patients with Head and Neck Cancer. Ther. Apher. Dial. 2019, 23, 279–288. [Google Scholar] [CrossRef]
- Kreutmair, S.; Pfeifer, D.; Waterhouse, M.; Takács, F.; Graessel, L.; Döhner, K.; Duyster, J.; Illert, A.L.; Frey, A.-V.; Schmitt, M.; et al. First-in-human study of WT1 recombinant protein vaccination in elderly patients with AML in remission: A single-center experience. Cancer Immunol. Immunother. 2022, 71, 2913–2928. [Google Scholar] [CrossRef]
- Sakai, K.; Shimodaira, S.; Maejima, S.; Udagawa, N.; Sano, K.; Higuchi, Y.; Koya, T.; Ochiai, T.; Koide, M.; Uehara, S.; et al. Dendritic cell–based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J. Neurosurg. 2015, 123, 989–997. [Google Scholar] [CrossRef]
- Long, J.; Fang, S.; Dai, Q.; Liu, X.; Zhu, W.; Wang, S. The Wilms Tumor-1 (WT1) rs16754 polymorphism is a prognostic factor in acute myeloid leukemia (AML): A meta-analysis. Oncotarget 2016, 7, 32079–32087. [Google Scholar] [CrossRef]
- Nishida, S.; Koido, S.; Takeda, Y.; Homma, S.; Komita, H.; Takahara, A.; Morita, S.; Ito, T.; Morimoto, S.; Hara, K.; et al. Wilms tumor gene (WT1) peptide–based cancer vaccine combined with gemcitabine for patients with advanced pancreatic cancer. J. Immunother. 2014, 37, 105–114. [Google Scholar] [CrossRef]
- Saito, S.; Yanagisawa, R.; Yoshikawa, K.; Higuchi, Y.; Koya, T.; Yoshizawa, K.; Tanaka, M.; Sakashita, K.; Kobayashi, T.; Kurata, T.; et al. Safety and tolerability of allogeneic dendritic cell vaccination with induction of Wilms tumor 1–Specific T cells in a pediatric donor and pediatric patient with relapsed leukemia: A case report and review of the literature. Cytotherapy 2015, 17, 330–335. [Google Scholar] [CrossRef]
- Tsuboi, A.; Hashimoto, N.; Fujiki, F.; Morimoto, S.; Kagawa, N.; Nakajima, H.; Hosen, N.; Nishida, S.; Nakata, J.; Morita, S.; et al. A phase I clinical study of a cocktail vaccine of Wilms’ tumor 1 (WT1) HLA class I and II peptides for recurrent malignant glioma. Cancer Immunol. Immunother. 2019, 68, 331–340. [Google Scholar] [CrossRef]
- Takagi, M.; Ogawa, C.; Iehara, T.; Aoki-Nogami, Y.; Ishibashi, E.; Imai, M.; Kimura, T.; Nagata, M.; Yasuhara, M.; Masutani, M.; et al. First phase 1 clinical study of olaparib in pediatric patients with refractory solid tumors. Cancer 2022, 128, 2949–2957. [Google Scholar] [CrossRef]
- Hirabayashi, K.; Yanagisawa, R.; Saito, S.; Higuchi, Y.; Koya, T.; Sano, K.; Koido, S.; Okamoto, M.; Sugiyama, H.; Nakazawa, Y.; et al. Feasibility and Immune Response of WT1 Peptide Vaccination in Combination with OK-432 for Paediatric Solid Tumors. Anticancer Res. 2018, 38, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, R.; Koizumi, T.; Koya, T.; Sano, K.; Koido, S.; Nagai, K.; Kobayashi, M.; Okamoto, M.; Sugiyama, H.; Shimodaira, S. WT1-pulsed Dendritic Cell Vaccine Combined with Chemotherapy for Resected Pancreatic Cancer in a Phase I Study. Anticancer Res. 2018, 38, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, X.; Cui, P.; Piao, C.; Xiao, M.; Liu, X.; Wang, Y.; Wu, X.; Liu, J.; Yang, L. Phase I/II clinical trial of a Wilms’ tumor 1-targeted dendritic cell vaccination-based immunotherapy in patients with advanced cancer. Cancer Immunol. Immunother. 2019, 68, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, S.; Kitago, M.; Sakurai, T.; Matsuda, T.; Fujita, T.; Higuchi, H.; Taguchi, J.; Takeuchi, H.; Itano, O.; Aiura, K.; et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015, 106, 397–406. [Google Scholar] [CrossRef]
- Brayer, J.; Lancet, J.E.; Powers, J.; List, A.; Balducci, L.; Komrokji, R.; Pinilla-Ibarz, J. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am. J. Hematol. 2015, 90, 602–607. [Google Scholar] [CrossRef]
- Ota, S.; Miyashita, M.; Yamagishi, Y.; Ogasawara, M. Baseline immunity predicts prognosis of pancreatic cancer patients treated with WT1 and/or MUC1 peptide-loaded dendritic cell vaccination and a standard chemotherapy. Hum. Vaccines Immunother. 2021, 17, 5563–5572. [Google Scholar] [CrossRef]
- Ito, Z.; Kan, S.; Bito, T.; Horiuchi, S.; Akasu, T.; Yoshida, S.; Kajihara, M.; Hokari, A.; Saruta, M.; Yoshida, N.; et al. Predicted Markers of Overall Survival in Pancreatic Cancer Patients Receiving Dendritic Cell Vaccinations Targeting WT1. Oncology 2019, 97, 135–148. [Google Scholar] [CrossRef]
- Kyi, C.; Doubrovina, E.; Zhou, Q.; Kravetz, S.; Iasonos, A.; Aghajanian, C.; Sabbatini, P.; Spriggs, D.; O’Reilly, R.J.; O’cearbhaill, R.E. Phase I dose escalation safety and feasibility study of autologous WT1-sensitized T cells for the treatment of patients with recurrent ovarian cancer. J. Immunother. Cancer 2021, 9, e002752. [Google Scholar] [CrossRef]
- Fukuda, K.; Funakoshi, T.; Sakurai, T.; Nakamura, Y.; Mori, M.; Tanese, K.; Tanikawa, A.; Taguchi, J.; Fujita, T.; Okamoto, M.; et al. Peptide-pulsed dendritic cell vaccine in combination with carboplatin and paclitaxel chemotherapy for stage IV melanoma. Melanoma Res. 2017, 27, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Green, D.M.; Lange, J.M.; Qu, A.; Peterson, S.M.; Kalapurakal, J.A.; Stokes, D.C.; Grigoriev, Y.A.; Takashima, J.R.; Norkool, P.; Friedman, D.L.; et al. Pulmonary disease after treatment for wilms tumor: A report from the national wilms tumor long-term follow-up study. Pediatr. Blood Cancer 2013, 60, 1721–1726. [Google Scholar] [CrossRef]
- Maslak, P.G.; Dao, T.; Bernal, Y.; Chanel, S.M.; Zhang, R.; Frattini, M.; Rosenblat, T.; Jurcic, J.G.; Brentjens, R.J.; Arcila, M.E.; et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018, 2, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Coosemans, A.; Vanderstraeten, A.; Tuyaerts, S.; Verschuere, T.; Moerman, P.; Berneman, Z.; Vergote, I.; Amant, F.; Van Gool, S.W. Wilms’ Tumor Gene 1 (WT1)—Loaded dendritic cell immunotherapy in patients with uterine tumors: A phase I/II clinical trial. Anticancer Res. 2013, 33, 5495–5500. [Google Scholar] [PubMed]
- Nishida, S.; Ishikawa, T.; Egawa, S.; Koido, S.; Yanagimoto, H.; Ishii, J.; Kanno, Y.; Kokura, S.; Yasuda, H.; Oba, M.S.; et al. Combination Gemcitabine and WT1 Peptide Vaccination Improves Progression-Free Survival in Advanced Pancreatic Ductal Adenocarcinoma: A Phase II Randomized Study. Cancer Immunol. Res. 2018, 6, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Oji, Y.; Hashimoto, N.; Tsuboi, A.; Murakami, Y.; Iwai, M.; Kagawa, N.; Chiba, Y.; Izumoto, S.; Elisseeva, O.; Ichinohasama, R.; et al. Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int. J. Cancer 2016, 139, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Nakata, J.; Nakae, Y.; Kawakami, M.; Morimoto, S.; Motooka, D.; Hosen, N.; Fujiki, F.; Nakajima, H.; Hasegawa, K.; Nishida, S.; et al. Wilms tumour 1 peptide vaccine as a cure-oriented post-chemotherapy strategy for patients with acute myeloid leukaemia at high risk of relapse. Br. J. Haematol. 2018, 182, 287–290. [Google Scholar] [CrossRef]
- Uttenthal, B.; Martinez-Davila, I.; Ivey, A.; Craddock, C.; Chen, F.; Virchis, A.; Kottaridis, P.; Grimwade, D.; Khwaja, A.; Stauss, H.; et al. Wilms’ Tumour 1 (WT1) peptide vaccination in patients with acute myeloid leukaemia induces short-livedWT1-specific immune responses. Br. J. Haematol. 2014, 164, 366–375. [Google Scholar] [CrossRef]
- Lu, J.; Gu, Y.; Li, Q.; Zhong, H.; Wang, X.; Zheng, Z. Wilms’ tumor 1 (WT1) as a prognosis factor in gynecological cancers: A meta-analysis. Medicine 2018, 97, e11485. [Google Scholar] [CrossRef]
- Ueda, Y.; Ogura, M.; Miyakoshi, S.; Suzuki, T.; Heike, Y.; Tagashira, S. Phase 1/2 study of the WT1 peptide cancer vaccine WT4869 in patients with myelodysplastic syndrome. Cancer Sci. 2017, 108, 2445–2453. [Google Scholar] [CrossRef]
- Israyelyan, A.; Goldstein, L.; Tsai, W.; Aquino, L.; Forman, S.J.; Nakamura, R.; Diamond, D.J. Real-time assessment of relapse risk based on the WT1 marker in acute leukemia and myelodysplastic syndrome patients after hematopoietic cell transplantation. Bone Marrow Transplant. 2015, 50, 26–33. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Fu, K.; Hu, J.; Zhao, Z.; Fu, W.; Liu, G.-C. Meta-analysis of the effect of preoperative chemotherapy on Wilms’ tumor. J. BUON 2018, 23, 211–217. [Google Scholar]
- Koshinaga, T.; Takimoto, T.; Okita, H.; Tanaka, Y.; Inoue, E.; Oue, T. Blastemal predominant type Wilms tumor in Japan: Japan Children’s Cancer Group. Pediatr. Int. 2019, 61, 351–357. [Google Scholar] [CrossRef]
- Trink, A.; Kanter, I.; Pode-Shakked, N.; Urbach, A.; Dekel, B.; Kalisky, T. Geometry of Gene Expression Space of Wilms’ Tumors From Human Patients. Neoplasia 2018, 20, 871–881. [Google Scholar] [CrossRef]
- Margolin, E.J.; Martina, L.A.P.; Miles, C.H.; Wenske, S.; McKiernan, J.M.; DeCastro, G.J.; Hyams, E.S.; Drake, C.G.; Lim, E.A.; Stein, M.N.; et al. Telemedicine in management of genitourinary malignancies: Patient and physician perspectives. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 480–486. [Google Scholar] [CrossRef]
- Hartman, A.; Pluijm, S.M.; Wijnen, M.; Neggers, S.J.; Clemens, E.; Pieters, R.; Heuvel-Eibrink, M.M.v.D. Health-related fitness in very long-term survivors of childhood cancer: A cross-sectional study. Pediatr. Blood Cancer 2018, 65, e26907. [Google Scholar] [CrossRef]
- Yin, Y.; Cao, H.; Zou, H. Influence of psychological nursing intervention in the recovery of children with Wilms’ tumor. Minerva Pediatr. 2019, 71, 545–548. [Google Scholar] [CrossRef]
- Perrotta, G. The intestinal microbiota: Towards a multifactorial integrative model. Eubiosis and dysbiosis in morbid physical and psychological conditions. Arch. Clin. Gastroenterol. 2021, 7, 024–035. [Google Scholar]
- Bhutani, N.; Kajal, P.; Sharma, U. Many faces of Wilms Tumor: Recent advances and future directions. Ann. Med. Surg. 2021, 64, 102202. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Chemotherapy-Induced Change | Histological Features (% of Viable Tumor) | ||
|---|---|---|---|---|
| Blastema | Epithelium | Stroma | ||
| Completely necrotic | 100 | 0 | 0 | 0 |
| Regressive | >66 | 0–100 | 0–100 | 0–100 |
| Mixed | <66 | 0–65 | 0–65 | 0–65 |
| Mixed | <66 | 11–65 | 0–89 | 0–89 |
| Epithelial | <66 | 0–10 | 66–100 | 0–33 |
| Stromal | <66 | 0–10 | 0–33 | 66–100 |
| Blastemal | <66 | 66–100 | 0–33 | 0–33 |
| Type | COG | SIOP |
|---|---|---|
| Surgery | Primary surgery before chemotherapy is recommended. For resectable tumors, preoperative or intraoperative biopsy is not performed, whereas in radical nephrectomy and lymph nodes, harvesting is performed through a transabdominal incision. To prevent tumor leakage, en bloc resection can be performed. The resection of a primary renal tumor should be considered even if at a stage IV disease (with metastases); renal-sparing surgery is not recommended, except in children with a solitary kidney, a predisposition to bilateral tumors or a horseshoe kidney, or in infants with Denys–Drash or Frasier syndrome (in order to delay the need for dialysis). | Radical nephrectomy of the tumor, performed after preoperative chemotherapy, is recommended. Lymph node sampling is important for staging, and the sampling of seven locoregional lymph nodes is necessary for accurate staging. Nephron-sparing surgery is used for nonsyndromic unilateral Wilms tumors, provided the following clinical conditions are met: (a) small tumor volume (<300 mL); (b) the expectation of substantial residual renal function in patients who have never had lymph node involvement. |
| Chemotherapy | Surgery is recommended as initial therapy before chemotherapy. Preoperative chemotherapy is indicated only in the following conditions: (a) with an inoperable Wilms tumor type; (b) with a solitary kidney; (c) with bilateral synchronous Wilms tumor; (d) a tumor thrombus in the inferior vena cava, extending above the level of the hepatic veins; (e) a tumor involving contiguous structures, whereby the removal of the kidney requires the removal of other organs (such as the spleen, pancreas or colon; (f) stage IV (with extensive pulmonary metastases). | Preoperative chemotherapy is recommended for all patients after diagnosis. For patients with unilateral localized tumors, a 4-week pretreatment is administered using vincristine (weekly) and dactinomycin (biweekly), while for patients with bilateral tumors, vincristine–dactinomycin is recommended for no more than 9 to 12 weeks (in some patients, doxorubicin is added as a reinforcer). Again, for patients with metastases, a regimen including 6 weeks of vincristine–#dactinomycin (as described above) and doxorubicin at weeks 1 and 5 is given. |
| Postoperative chemotherapy | It is recommended that postoperative chemo-therapy be used routinely in all patients with Wilms tumor except those at very low risk (those less than 2 years of age at diagnosis with a tumor, with favorable stage I histology, weighing less than 550 g, with sampling and with confirmed negative lymph nodes). | Postoperative chemotherapy is recommended in all patients with Wilms tumor except those with low-risk stage I tumors. |
| Postoperative radiation | Postoperative irradiation in the tumor bed is recommended for all patients with stage III cancer. | Radiation therapy of the whole abdomen is recommended for patients with intermediate-histology or high-risk tumors and with major tumor rupture preoperatively or intraoperatively, or with macroscopic peritoneal deposits. Lung radiotherapy is indicated for lung metastases without complete response until the 10th postoperative week. Patients with a complete response after induction chemotherapy with or without surgery do not need pulmonary radiotherapy. Patients with viable metastases at surgery or with high-risk histology require pulmonary radiation therapy. Whole-lung irradiation is recommended for patients who did not receive lung irradiation during first-line treatment, regardless of histology. |
| Recurrent WT | Wilms tumors with characteristic high recurrence are divided into three risk groups: (1) standard risk; (2) high risk; (3) very high risk. In the first case (1), surgery (when possible), radiotherapy and chemotherapy (alternating cycles of vincristine/do-xorubicin/cyclophosphamide and etoposide/cyclophosphamide) are used. In the second and third cases (2,3), chemotherapy (alternating cycles of cyclophosphamide/ethoposide and carboplatin/ethoposide), surgery and/or radiotherapy and hematopoietic stem cell transplantation are recommended. | Patients with Wilms tumor are classified into AA, BB and CC, but essentially nothing changes from the previous classification. For the former (AA), only vincristine and/or dactinomycin is used as first-line treatment (without radiotherapy), with a four-drug regimen (combinations of doxorubicin and/or cyclophosphamide and carboplatin and/or etoposide); for the second group (BB), an intensive reinduction regimen (including the combination of etoposide and carboplatin with phosphamide or cyclophosphamide) is administered, followed by high-dose melphalan and autologous stem cell rescue or two more rounds of reinduction; for the third group (CC), camptothecins (irinotecan or topotecan) or new biologic compounds are recommended. |
| Stage V—WT | Both the COG and SIOP recommend preoperative chemotherapy and resection for bilateral WT. Bilateral renal-sparing surgery can be performed in patients with synchronous bilateral WT. Renal parenchyma sparing may help preserve renal function in these children. Renal transplantation is recommended and is usually delayed for 1–2 years without evidence of relapse. The SIOP also suggests that preoperative chemotherapy should be limited to no longer than 12 weeks, with time intervals for evaluation fixed to 6 weeks. | |
| Accepted Chemotherapy Regimens for Wilms Tumor |
| |
| RISK | Pretreated Tumors | Primary Nephrectomy Tumors |
|---|---|---|
| Low risk | Mesoblastic nephroma | Mesoblastic nephroma |
| Cystic partially differentiated nephroblastoma | Cystic partially differentiated nephroblastoma | |
| Completely necrotic nephroblastoma | ||
| Intermediate risk | Nephroblastoma—epithelial type | Nonanaplastic nephron-blastoma and its variants |
| Nephroblastoma—stromal type | Nephroblastoma—focal anaplasia type | |
| Nephroblastoma—mixed type | ||
| Nephroblastoma—regressive type | ||
| Nephroblastoma—focal anaplasia type | ||
| High risk | Nephroblastoma—blastemal type | Nephroblastoma—diffuse anaplasia type |
| Nephroblastoma—diffuse anaplasia type | Clear cell sarcoma of the kidney | |
| Clear cell sarcoma of the kidney | Rhabdoid tumor of the kidney | |
| Rhabdoid tumor of the kidney |
| Keyword | Clinical Message |
|---|---|
| Etiology | Most of the scientific literature agrees that Wilms tumor develops as a result of a genetic mutation (WT1, located on the short arm of chromosome 11 at position 11p13), and therefore a preventive genetic analysis could exclude the risk of being a carrier. Testing positive for the genetic test does not indicate certainty that the disease may occur during childhood, but correlations related to other genetic portions are also being investigated (e.g., 11p15. 5, WT1, TRIM28 and REST, but also LOH1p, 16q, 1q and LOH11p15); however, in the case of a positive result, attention should also be paid to other circumstances considered favorable and related to the disease: exposure to pesticides (such as organophosphates), folic acid deficiency during pregnancy and maternal consumption of cigarettes and alcohol or living in unhealthy environments. |
| Diagnosis | The first level of investigation is always the objective examination (with palpation) and abdominal ultrasound. In the second level of proceeding, computed tomography and contrast will be used, both for in situ evaluation and for the vascular component, but this is better investigated with MRI, as it better defines the invasion of the great vessels, although the latter is rarely used in U.S. medical practice. The use of biomarkers, including the prognostic biomarker circulating tumor DNA, appears to be promising but still needs more investigation about its use and validity. |
| Therapy | Treatments are modulated according to clinical and anatomopathologic variables, as well as according to national protocols specific to each national health system. Generally, the initial treatment of unilateral Wilms tumor is primary surgical resection (with an approach that can be anticipated or delayed precisely according to each patient’s histologic and clinical outcomes) followed by adjuvant chemotherapy. The type of chemotherapy drug and duration of therapy depends on the histology and stage of the tumor. Chemotherapy regimens depend on the risk group but usually consist of actinomycin D (dactinomycin) and vincristine, with or without doxorubicin, or adriamycin. For more aggressive tumors, intensive multiagent chemotherapy regimens are used. Radiation therapy is given to children who have more advanced-stage disease (stage III and in the presence of distant metastases, usually lung, that do not easily regress with chemotherapy). In most cases, radical nephrectomy, i.e., the surgical removal of the affected kidney, combined with the resection of regional and para-aortic lymph nodes ipsilateral to the neoplasm, is performed; in bilateral tumors or patients with specific syndromes predisposing them to nephroblastoma, partial nephrectomy is preferred when possible; when possible, especially in cases of bilaterality, even partial preservation of the renal structure should be preferred, unless the clinical conditions do not allow it and the balance with the renal function to be preserved is compatible with possible tumor recurrence. One study finally showed that radiofrequency with cryoablation is also effective for this type of tumor. However, immunotherapy and cryotherapy are not yet generally approved therapies in scientific communities, such as the Children’s Oncology Group and the Renal Tumor Study Group of the International Society of Pediatric Oncology (SIOP-RTSG), because of the few studies still in the literature. |
| Prognosis | Wilms tumor is the most common renal tumor in childhood, and the prognosis is mainly related to the histologic appearance of the neoplasm, where the presence of anaplastic (undifferentiated) cells suggests a poorer prognosis. The prognosis of Wilms tumor also depends on the stage at diagnosis and the patient’s age (advanced age is associated with a worse prognosis). Cure rates for low-stage disease (localized to the kidneys) range from 85 to 95 percent; children with the more advanced disease also have a good prognosis: cure rates range from 60 percent (unfavorable histology) to 90 percent (favorable histology). The tumor can sometimes recur, usually within two years of diagnosis, although a cure is possible even in children with recurrent tumors; the problem of tumor recurrence, especially in the case of bilaterality, is a negative index that warrants closer and more prolonged monitoring over time. |
| Nutritional implications | The diagnosis of Wilms tumor implies a modification of the patient’s nutritional plan, depending on the symptoms and severity of the disease, such that generalization is impossible because each patient is a unique universe that requires careful analysis of all factors involved, starting with age and subjective history, following him or her through all stages of the disease process. |
| Psychological implications | The “cancer” event necessarily impacts the psychological profile of the patient affected by the disease, as well as the personal and relational life of his or her family members, also considering the patient’s average age (of childhood range). In the case of Wilms tumor, the final prognosis is good in most cases, especially if diagnosed in the early stages of the disease, and thus there is a greater chance of helping the patient and his family to overcome this complicated phase; however, the therapist must be prepared to handle the possible distress that is grafted onto one or more personality frameworks of the people involved, perhaps already dysfunctional or decompensated by other pathologies, including mental pathologies, in psychophysical comorbidity. Therefore, the need to support the patient and family, from the earliest stages of the illness, is central to the healing process (including through cycles of psychotherapy and parent training techniques), even and especially in the future perspective. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrotta, G.; Castellani, D. Wilms Tumor: Updates about Pathogenesis and New Possible Clinical Treatments of the Most Frequent Pediatric Urogenital Cancer: A Narrative Review. Surgeries 2023, 4, 678-697. https://doi.org/10.3390/surgeries4040064
Perrotta G, Castellani D. Wilms Tumor: Updates about Pathogenesis and New Possible Clinical Treatments of the Most Frequent Pediatric Urogenital Cancer: A Narrative Review. Surgeries. 2023; 4(4):678-697. https://doi.org/10.3390/surgeries4040064
Chicago/Turabian StylePerrotta, Giulio, and Daniele Castellani. 2023. "Wilms Tumor: Updates about Pathogenesis and New Possible Clinical Treatments of the Most Frequent Pediatric Urogenital Cancer: A Narrative Review" Surgeries 4, no. 4: 678-697. https://doi.org/10.3390/surgeries4040064