
Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Remarks
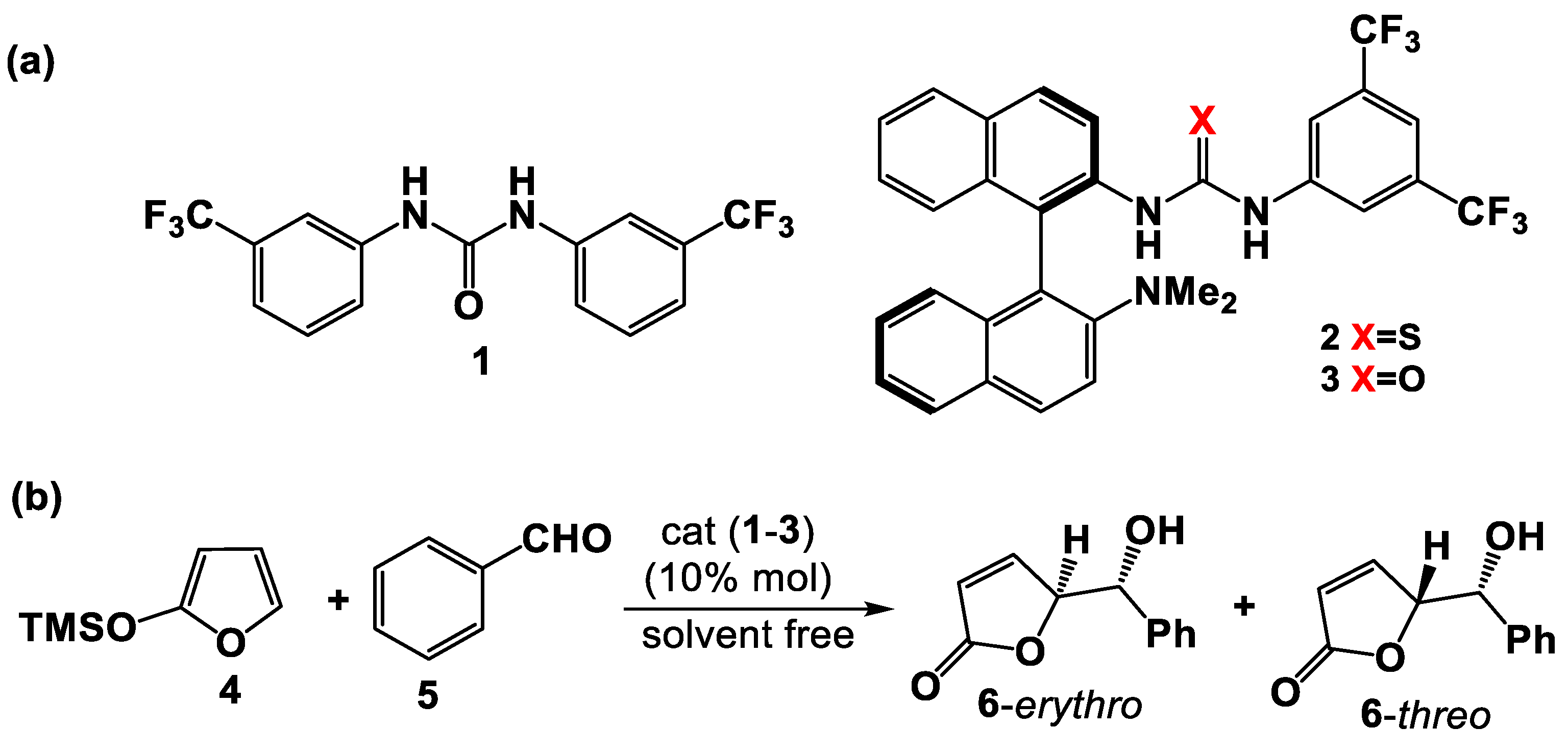
2.2. General Procedure for the Enantioselective Organocatalyzed Vinylogous Addition of TMSOF to Benzaldehyde
3. Results and Discussion
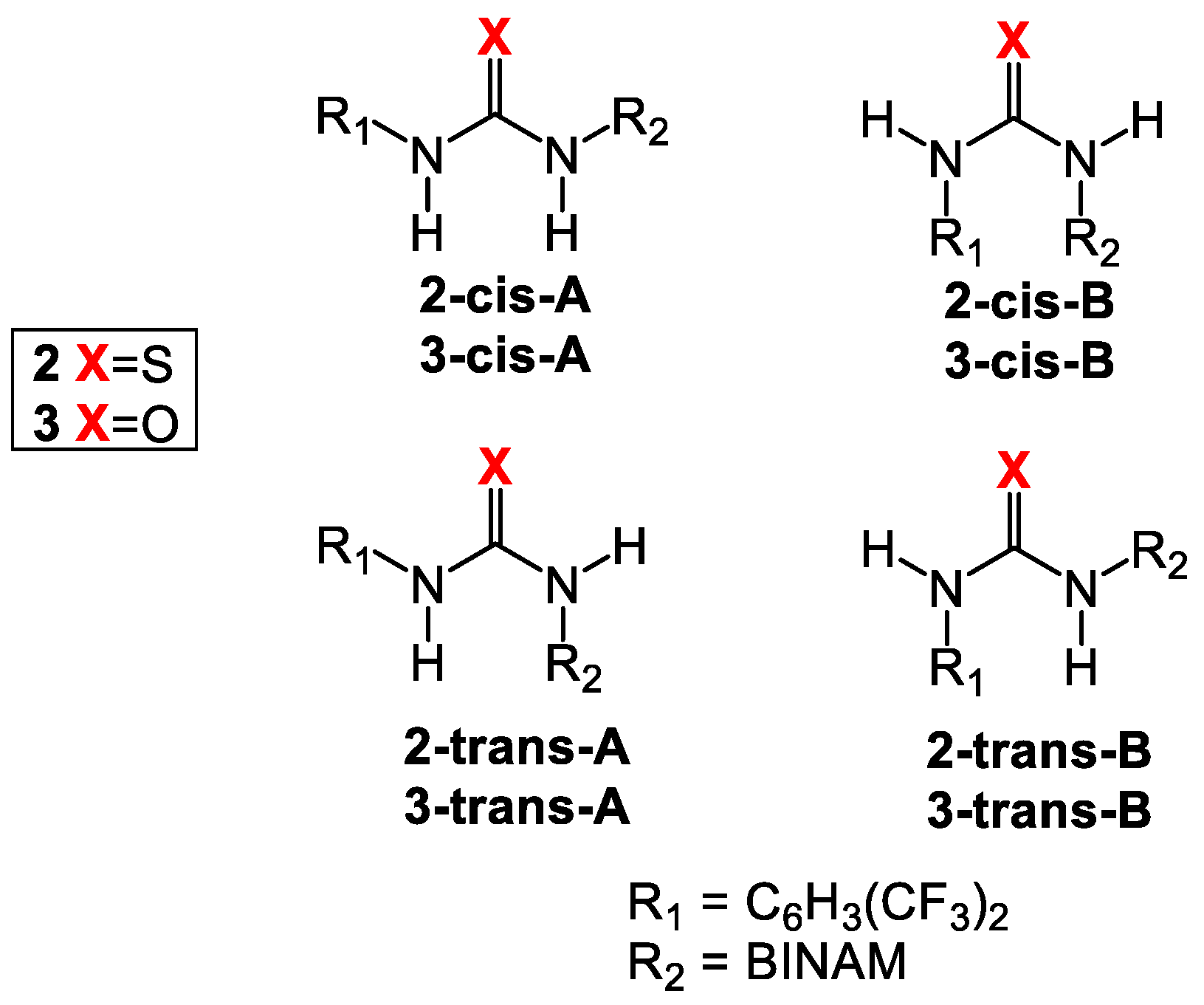
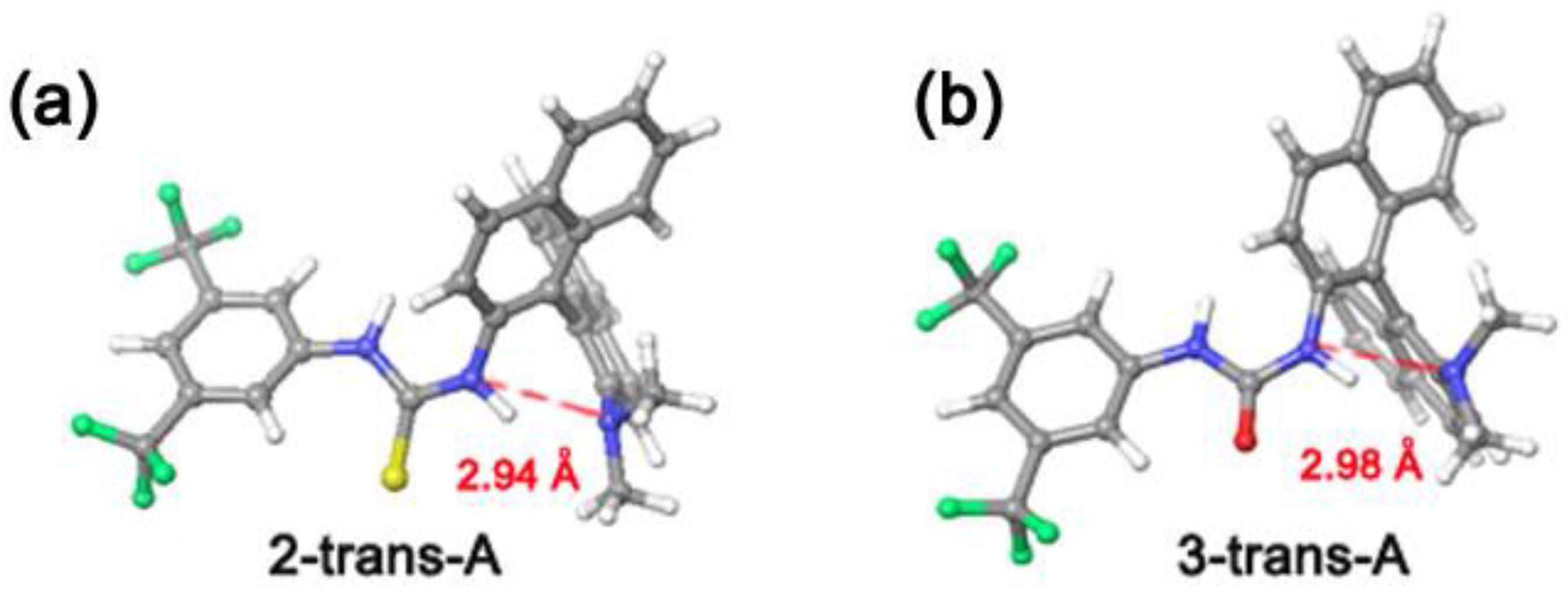
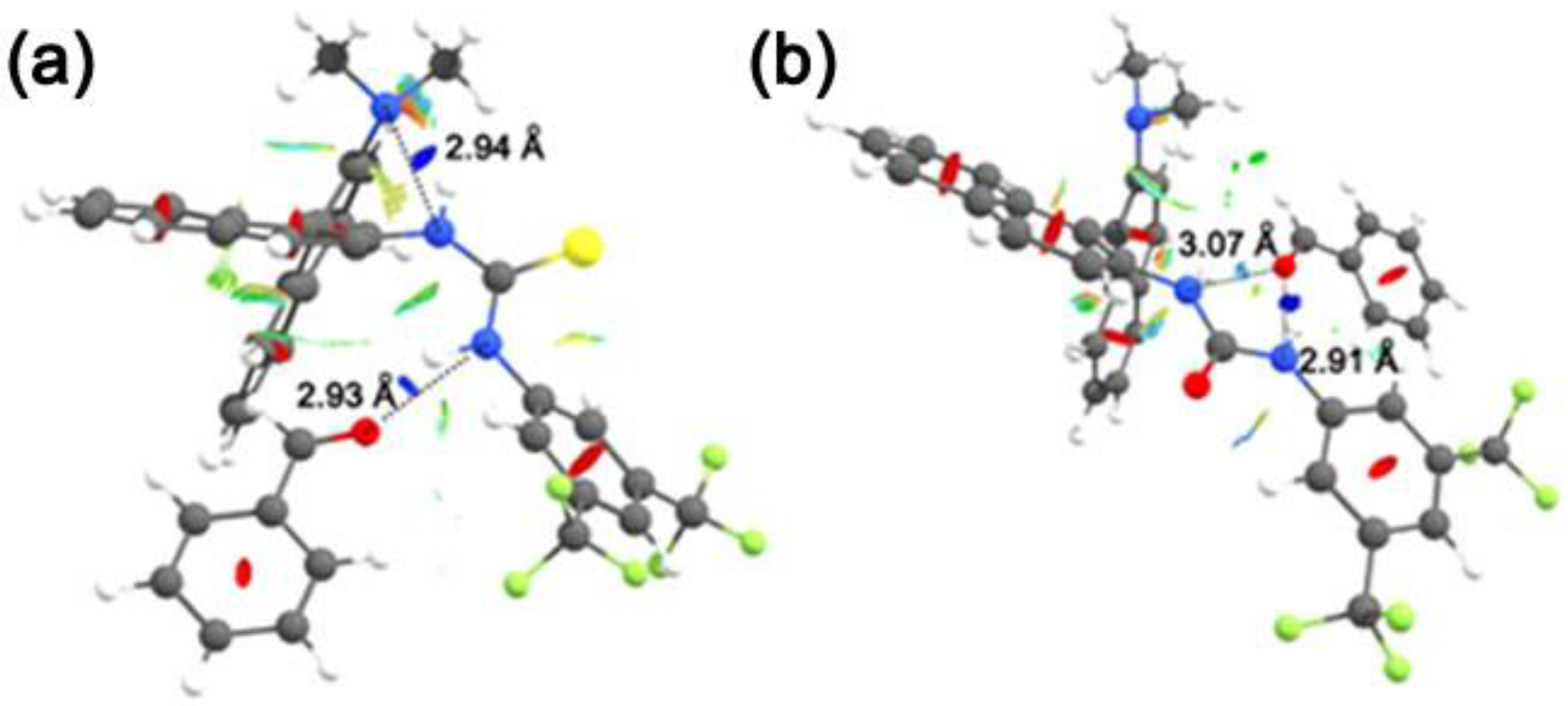
3.1. Mono(thio)urea Catalysts 2 and 3
3.2. Bis(thio)urea Catalysts 7 and 8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Breslow, R. Artificial Enzymes. Science 1982, 218, 532–537. [Google Scholar] [CrossRef]
- McMillan, D.W.C. The advent and development of organocatalysis. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef]
- Purse, B.W.; Rebek, J., Jr. Functional cavitands: Chemical reactivity in structured environments. Proc. Natl. Acad. Sci. USA 2005, 102, 10777–10782. [Google Scholar] [CrossRef]
- Meeuwissen, J.; Reek, J.N.H. Supramolecular catalysis beyond enzyme mimics. Nat. Chem. 2010, 2, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Gambaro, S.; Talotta, C.; Della Sala, P.; Soriente, A.; De Rosa, M.; Gaeta, C.; Neri, P.J. Kinetic and Thermodynamic Modulation of Dynamic Imine Libraries Driven by the Hexameric Resorcinarene Capsule. Am. Chem. Soc. 2020, 142, 14914–14923. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, G.; Forgan, R.S. (Eds.) Reactivity in Confined Spaces. In Monographs in Supramolecular Chemistry; The Royal Society of Chemistry: London, UK, 2021. [Google Scholar]
- Iuliano, V.; Della Sala, P.; Talotta, C.; De Rosa, M.; Soriente, A.; Gaeta, C.; Neri, P. Supramolecular control on reactivity and selectivity inside the confined space of H-bonded hexameric capsules. Curr. Opin. Colloid Interface 2023, 65, 101692. [Google Scholar] [CrossRef]
- Borsato, G.; Rebek, J., Jr.; Scarso, A. From Selective Nanocatalysts and Nanoscience; Zecchina, A., Bordiga, S., Groppo, E.E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 105–168. [Google Scholar]
- Catti, L.; Zhang, Q.; Tiefenbacher, K. Advantages of Catalysis in Self-Assembled Molecular Capsules. Chem. Eur. J. 2016, 22, 9060–9066. [Google Scholar] [CrossRef]
- Hong, C.M.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Self-Assembled Tetrahedral Hosts as Supramolecular Catalysts. Acc. Chem. Res. 2018, 51, 2447–2455. [Google Scholar] [CrossRef]
- Ren, Y.; Tao, L.; Li, C.; Jayakumar, S.; Li, H.; Yang, Q. Development of Efficient Solid Chiral Catalysts with Designable Linkage for Asymmetric Transfer Hydrogenation of Quinoline Derivatives. Chin. J. Catal. 2021, 42, 1576–1585. [Google Scholar] [CrossRef]
- Wang, B.; Jin, C.; Shao, S.; Yue, Y.; Zhang, Y.; Wang, S.; Chang, R.; Zhang, H.; Zhao, J.; Li, X. Electron-Deficient Cu Site Catalyzed Acetylene Hydrochlorination. Green Energy Environ. 2023, 8, 1128–1140. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Wang, B.; Yue, Y.; Zhao, J. Migration: A Neglected Potential Contribution of HCl-Oxidized Au(0). Molecules 2023, 28, 1600. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 12, 5713–5743. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Li, S.; Du, J.; Zhang, B.; Liu, Y.; Mei, Q.; Meng, Q.; Dong, M.; Du, J.; Zhao, Z.; Zheng, L.; et al. Selective Hydrogenation of 5-(Hydroxymethyl)Furfural to 5-Methylfurfural by Exploiting the Synergy between Steric Hindrance and Hydrogen Spillover. Acta Phys. Chim. Sin. 2022, 38, 2206019. [Google Scholar] [CrossRef]
- Wang, K.; Wang, B.; Liu, X.; Fan, H.; Liu, Y.; Li, C. Palladium-Catalyzed Enantioselective Linear Allylic Alkylation of Vinyl Benzoxazinanones: An Inner-Sphere Mechanism. Chin. J. Catal. 2021, 42, 1227–1237. [Google Scholar] [CrossRef]
- Cafeo, G.; De Rosa, M.; Kohnke, F.H.; Neri, P.; Soriente, A.; Valenti, L. Efficient organocatalysis with a calix[4]pyrrole derivative. Tetrahedron Lett. 2008, 49, 153–155. [Google Scholar] [CrossRef]
- Cafeo, G.; De Rosa, M.; Kohnke, F.H.; Soriente, A.; Talotta, C.; Valenti, L. Calixpyrrole Derivatives: “Multi Hydrogen Bond” Catalysts for γ-Butenolide Synthesis. Molecules 2009, 14, 2594–2601. [Google Scholar] [CrossRef]
- De Rosa, M.; Citro, L.; Soriente, A. The first organocatalytic addition of 2-trimethylsilyloxyfuran to carbonyl compounds: Hydrogen-bond catalysis in γ-butenolides synthesis. Tetrahedron Lett. 2006, 47, 8507–8510. [Google Scholar] [CrossRef]
- Smith, C.J.; Abbanat, D.; Bernan, V.S.; Maiese, W.M.; Greenstein, M.; Jompa, J.; Tahir, A.; Ireland, C.M.J. Novel Polyketide Metabolites from a Species of Marine Fungi. Nat. Prod. 2000, 63, 142–145. [Google Scholar] [CrossRef]
- Antane, S.; Caufield, C.E.; Hu, W.; Keeney, D.; Labthavikul, P.; Morris, K.; Naughton, S.M.; Petersen, P.J.; Rasmussen, B.A.; Singh, G.; et al. Pulvinones as bacterial cell wall biosynthesis inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 176–180. [Google Scholar] [CrossRef]
- Li, R.-T.; Han, Q.-B.; Zheng, Y.-T.; Wang, R.-R.; Yang, L.-M.; Lu, Y.; Sang, S.-Q.; Zheng, Q.-T.; Zhao, Q.-S.; Sun, H.-D. Structure and anti-HIV activity of micrandilactones B and C, new nortriterpenoids possessing a unique skeleton from Schisandra micrantha. Chem. Commun. 2005, 23, 2936–2938. [Google Scholar] [CrossRef]
- Yu, C.; Ji, P.; Zhang, Y.; Meng, X.; Wang, W. Construction of Enantioenriched γ,γ-Disubstituted Butenolides Enabled by Chiral Amine and Lewis Acid Cascade Cocatalysis. Org. Lett. 2021, 23, 7656–7660. [Google Scholar] [CrossRef]
- Hug, J.J.; Kjaerulff, L.; Garcia, R.; Müller, R. New Deoxyenhygrolides from Plesiocystis pacifica Provide Insights into Butenolide Core Biosynthesis. Mar. Drugs 2022, 20, 72. [Google Scholar] [CrossRef]
- Pelter, A.; Al-Bayati, R.I.H.; Ayoub, M.T.; Lewis, W.; Pardasani, P.; Hänsel, R. Synthetic routes to the piperolides, fadyenolides, epoxypiperolides, and related compounds. J. Chem. Soc. Perkin Trans. 1987, 1, 717–742. [Google Scholar] [CrossRef]
- Boukouvalas, J.; Maltais, F. An efficient total synthesis of the antibiotic patulin. Tetrahedron Lett. 1995, 36, 7175–7176. [Google Scholar] [CrossRef]
- Brown, S.P.; Goodwin, N.C.; MacMillan, D.W.C.J. The First Enantioselective Organocatalytic Mukaiyama−Michael Reaction: A Direct Method for the Synthesis of Enantioenriched γ-Butenolide Architecture. J. Am. Chem. Soc. 2003, 125, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Foxman, B.M.; Deng, L. Asymmetric Vinylogous Aldol Reaction of Silyloxy Furans with a Chiral Organic Salt. J. Am. Chem. Soc. 2010, 132, 9558–9560. [Google Scholar] [CrossRef] [PubMed]
- Pansare, S.V.; Paul, E.K. The Organocatalytic Vinylogous Aldol Reaction: Recent Advances. Chem. Eur. J. 2011, 17, 8770–8779. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Yu, X.; Zu, L.; Wang, W. Chiral Binaphthyl-Derived Amine-Thiourea Organocatalyst-Promoted Asymmetric Morita−Baylis−Hillman Reaction. Org. Lett. 2005, 7, 4293–4296. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Duan, W.; Zu, L.; Wang, W. Organocatalytic Asymmetric Michael Addition of 2,4-Pentandione to Nitroolefins. Org. Lett. 2005, 7, 4713–4716. [Google Scholar] [CrossRef]
- Fleming, E.M.; McCabe, T.; Connon, S.J. Novel axially chiral bis-arylthiourea-based organocatalysts for asymmetric Friedel–Crafts type reactions. Tetrahedron Lett. 2006, 47, 7037–7042. [Google Scholar] [CrossRef]
- Ollevier, T.; Bouchard, J.-E.; Desyroy, V. Diastereoselective Mukaiyama Aldol Reaction of 2-(Trimethylsilyloxy)Furan Catalyzed by Bismuth Triflate. J. Org. Chem. 2008, 73, 331–334. [Google Scholar] [CrossRef]
- Della Sala, P.; Del Regno, R.; Capobianco, A.; Iuliano, V.; Talotta, C.; Geremia, S.; Hickey, N.; Neri, P.; Gaeta, C. Confused-Prism[5]arene: A Conformationally Adaptive Host by Stereoselective Opening of the 1,4-Bridged Naphthalene Flap. Chem. Eur. J. 2023, 29, e2022030. [Google Scholar] [CrossRef]
- Zhu, J.-L.; Zhang, Y.; Liu, C.; Zheng, A.-M.; Wang, W.J. Insights into the Dual Activation Mechanism Involving Bifunctional Cinchona Alkaloid Thiourea Organocatalysts: An NMR and DFT Study. Org. Chem. 2012, 77, 9813–9825. [Google Scholar] [CrossRef] [PubMed]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, M.K.; Schreiner, P.R. (Thio)urea Organocatalyst Equilibrium Acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, 1st ed.; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Lan-Hua, W.; Yong-Bing, H.; Kuo-Xi, X.; Shun-Ying, L.; Ling-Zhi, M. Chiral Fluorescent Receptors Based on (R)-1,1′-Binaphthylene-2,2′-bisthiourea: Synthesis and Chiral Recognition. Chin. J. Chem. 2005, 23, 757–761. [Google Scholar] [CrossRef]
- Sohtome, Y.; Tanatani, A.; Hashimoto, Y.; Nagasawa, K. Development of bis-thiourea-type organocatalyst for asymmetric Baylis–Hillman reaction. Tetrahedron Lett. 2004, 45, 5589–5592. [Google Scholar] [CrossRef]
- Berkessel, A.; Roland, K.; Neudörfl, J.M. Asymmetric Morita−Baylis−Hillman Reaction Catalyzed by Isophoronediamine-Derived Bis(thio)urea Organocatalysts. Org. Lett. 2006, 8, 4195–4198. [Google Scholar] [CrossRef]
- Szlosek, M.; Figadére, B. Highly Enantioselective Aldol Reaction with 2-Trimethylsilyloxyfuran: The First Catalytic Asymmetric Autoinductive Aldol Reaction. Angew. Chem. Int. Ed. 2000, 39, 1799–1801. [Google Scholar] [CrossRef]
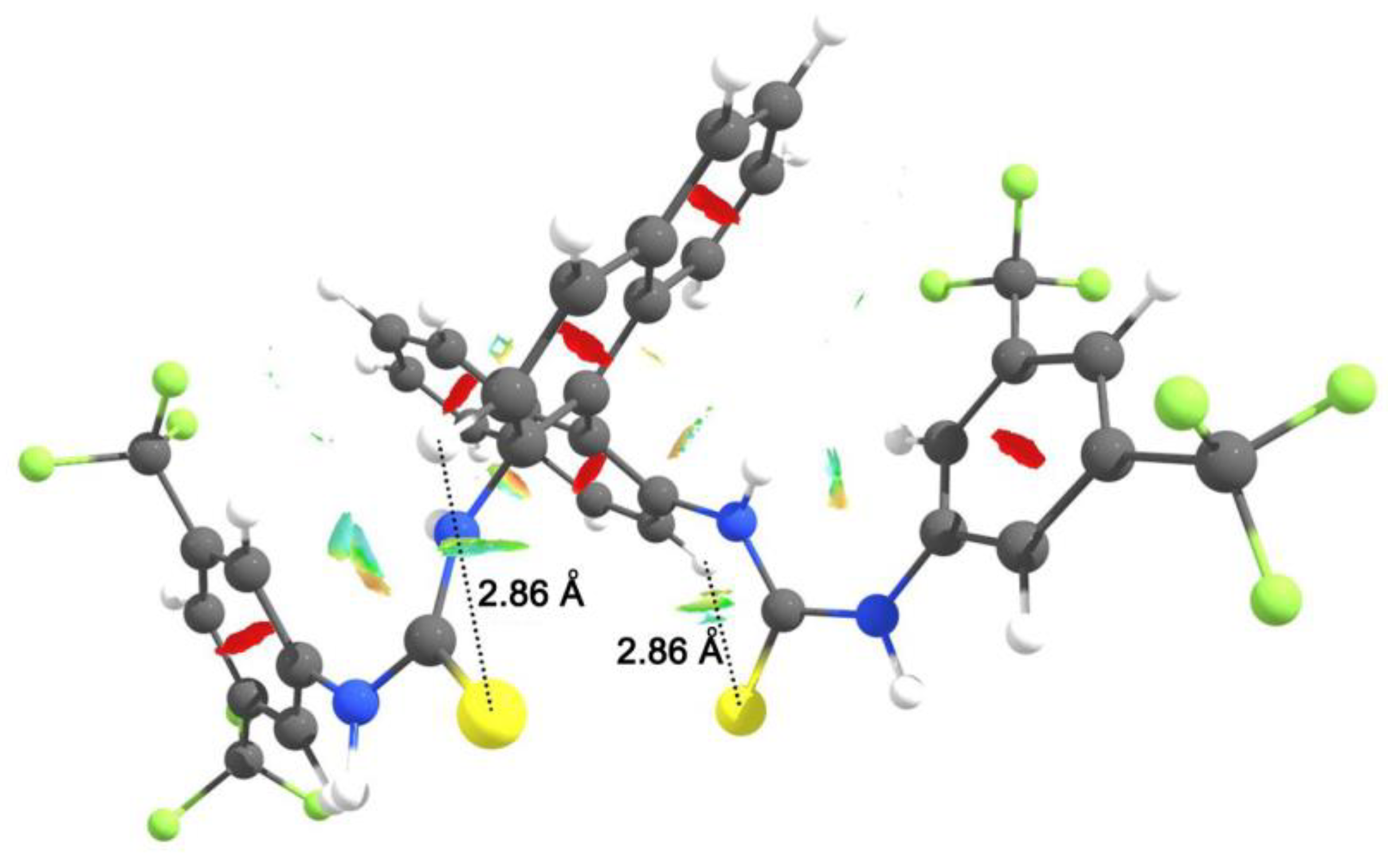
- Ghosh, S.; Chopra, P.; Wategaonkar, S. C–H⋯S interaction exhibits all the characteristics of conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2020, 22, 17482–17493. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
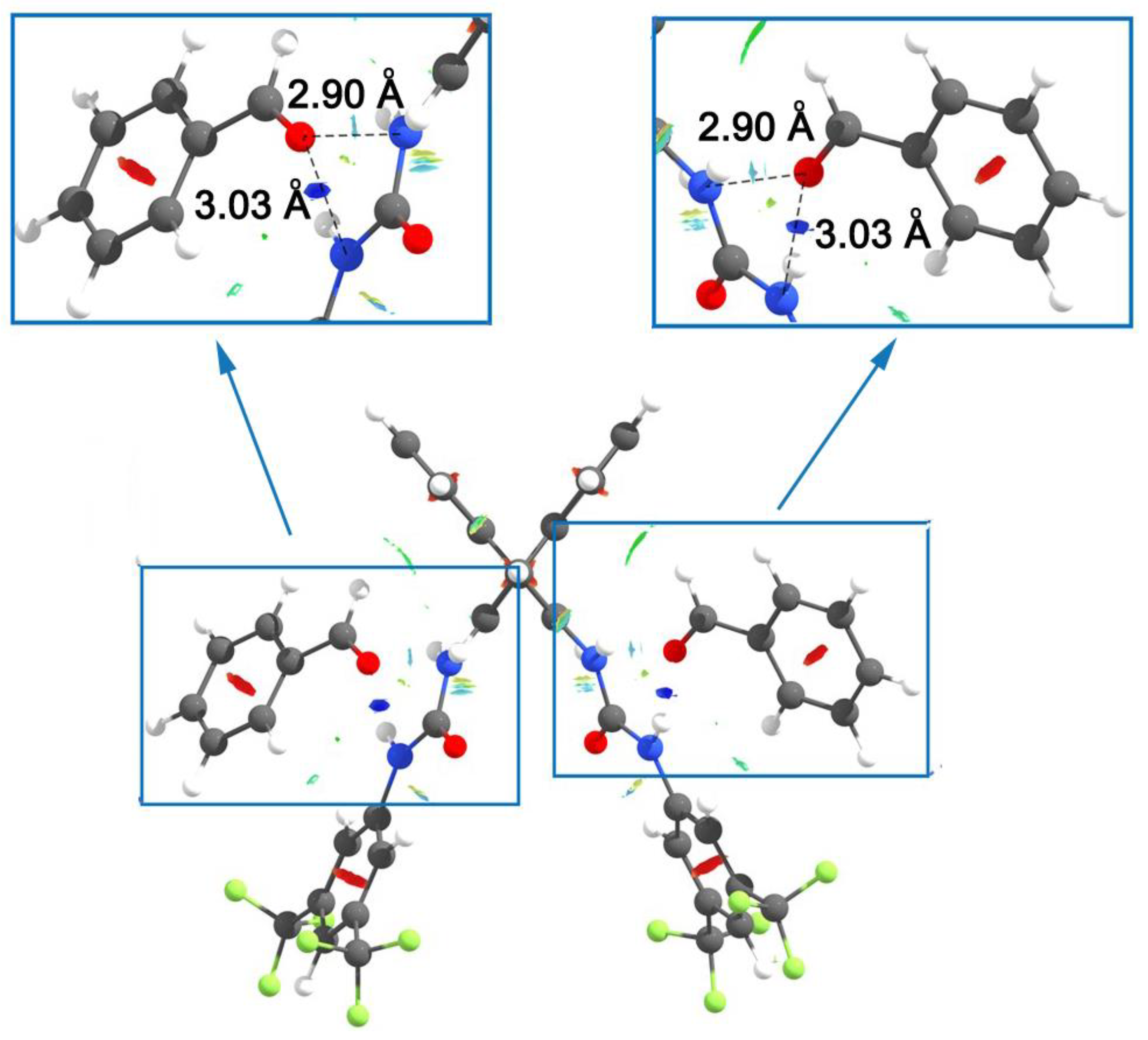{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Equivalents of Aldehyde | T/t (°C/h) | Yield (%) 2 | d.r. Erythro/Threo 3 | e.e. 4 Erythro (%) | e.e. 4 Threo (%) |
| 1 | 2 | 5 | Rt/24 | 29 | 70/30 | 9 | 21 |
| 2 | 3 | 5 | Rt/24 | 56 | 70/30 | 20 | 26 |
| Rotamers | ΔEisomer 1 (kcal/mol) | Ecoord (kcal/mol) | Yield (%) |
|---|---|---|---|
| 2-trans-A | 0.0 | - | - |
| 2-trans-B | 3.0 | - | - |
| 2-cis-A | 2.1 | - | - |
| 2-cis-B | 2.5 | - | - |
| 3-trans-A | 0.0 | - | - |
| 3-trans-B | 3.4 | - | - |
| 3-cis-A | 0.7 | - | - |
| 3-cis-B | 8.9 | - | - |
| 5@2-cis-A | 1.5 | −5.4 | 29 |
| 5@2-trans-A | 0.0 | −4.7 | - |
| 5@3-cis-A | 0.0 | −7.8 | 56 |
| 5@3-trans-A | 1.4 | −5.7 | - |
 | |||||||
|---|---|---|---|---|---|---|---|
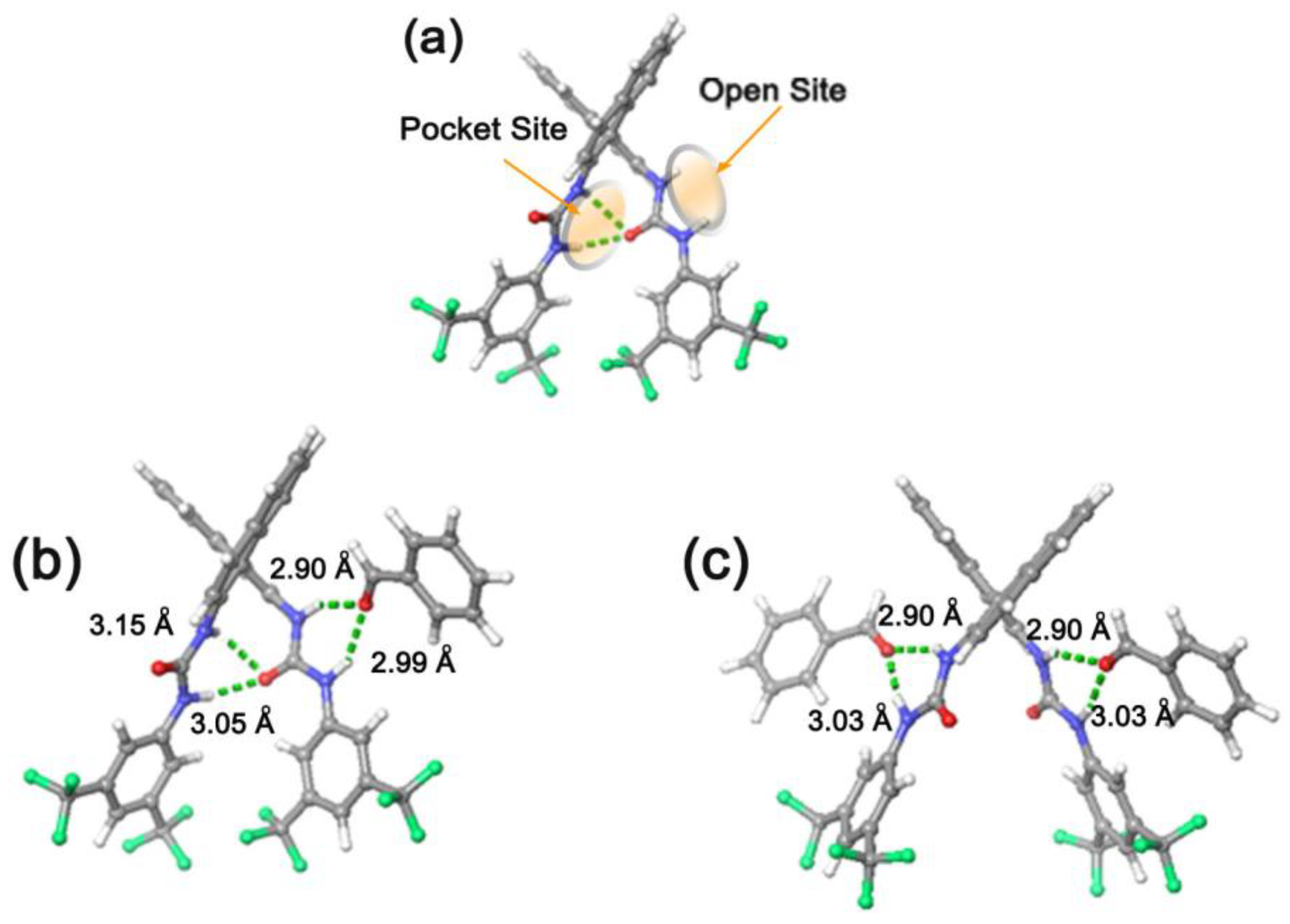
| Entry | Catalyst | Equivalents of Aldehyde | T/t (°C/h) | Yield (%) 2 | d.r. Erythro/Threo 3 | e.e. 4 Erythro (%) | e.e. 4 Threo (%) |
| 1 | 7 | 5 | Rt/24 | 45 | 30/70 | 32 | 93 |
| 2 | 8 | 5 | Rt/24 | 53 | 30/70 | 45 | 90 |
| 3 | 7 | 5 | Rt/48 | 45 | 30/70 | 20 | 91 |
| 4 | 8 | 5 | Rt/48 | 53 | 37/63 | 39 | 89 |
| 5 | 7 | 5 | Rt/72 | 50 | 30/70 | 15 | 91 |
| 6 | 8 | 5 | Rt/72 | 45 | 34/66 | 36 | 85 |
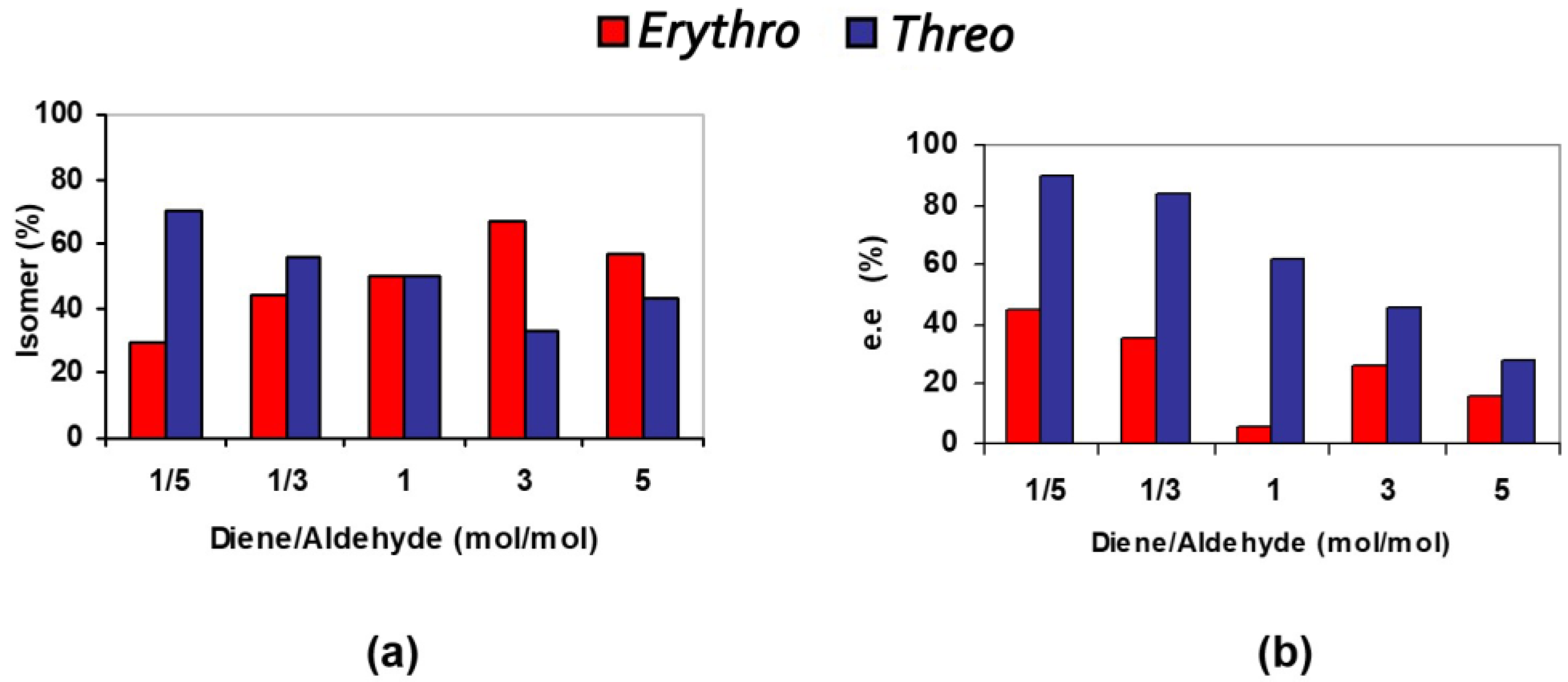
| Entry | TMSOF/ Aldehyde | T/t (°C/h) | Solvent | Yield (%) 2 | d.r. (Erythro/Threo) 3 | e.e. (%) Erythro 4 | e.e. (%) Threo 4 |
|---|---|---|---|---|---|---|---|
| 1 | 1/5 | Rt/24 | - | 53 | 30/70 | 45 | 90 |
| 2 | 1/3 | Rt/24 | - | 70 | 44/56 | 36 | 84 |
| 3 | 1/1 | Rt/24 | - | 45 | 50/50 | 6 | 62 |
| 4 | 1/1 | Rt/24 | Toluene (0.2 mL) | 62 | 48/52 | 2 | 24 |
| 5 | 3/1 | Rt/24 | 43 | 67/33 | 26 | 46 | |
| 6 | 5/1 | Rt/24 | 51 | 57/43 | 16 | 28 |
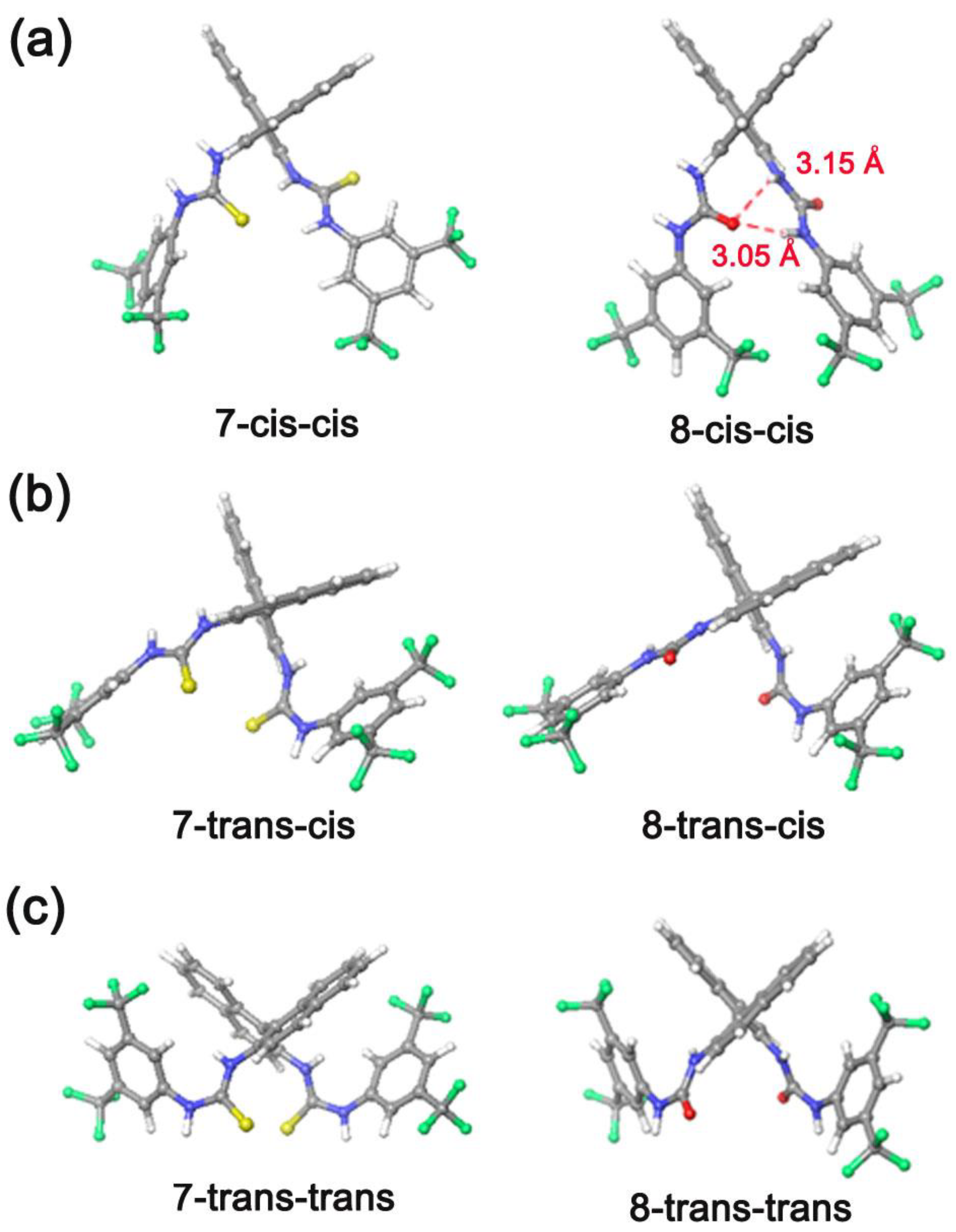
| Rotamers | ΔEisomer 1 (kcal/mol) | Ecoord (kcal/mol) |
|---|---|---|
| 7-cis-cis | 1.77 | - |
| 7-trans-cis | 2.41 | - |
| 7-trans-trans | 0.00 | - |
| 8-cis-cis | 0.00 | - |
| 8-trans-cis | 5.33 | - |
| 8-trans-trans | 8.07 | - |
| 5@8-cis-cis | - | −8.9 |
| (5)2@8-cis-cis | - | −12.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iuliano, V.; Della Sala, P.; Talotta, C.; De Rosa, M.; Gaeta, C.; Neri, P.; Soriente, A. Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives. Organics 2024, 5, 32-45. https://doi.org/10.3390/org5020003
Iuliano V, Della Sala P, Talotta C, De Rosa M, Gaeta C, Neri P, Soriente A. Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives. Organics. 2024; 5(2):32-45. https://doi.org/10.3390/org5020003
Chicago/Turabian StyleIuliano, Veronica, Paolo Della Sala, Carmen Talotta, Margherita De Rosa, Carmine Gaeta, Placido Neri, and Annunziata Soriente. 2024. "Supramolecular Catalysis with Chiral Mono- and Bis-(Thio)Urea-Derivatives" Organics 5, no. 2: 32-45. https://doi.org/10.3390/org5020003