
How an Internal Supramolecular Interaction Determines the Stereochemistry of a Metal Center

Abstract
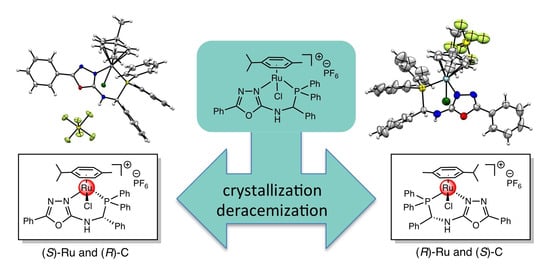
:
1. Introduction
2. Materials and Methods
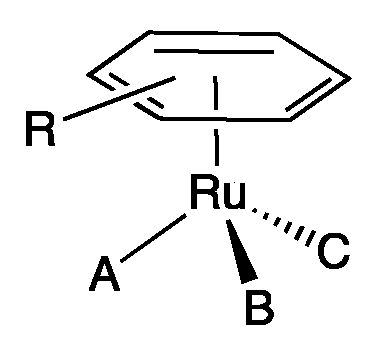
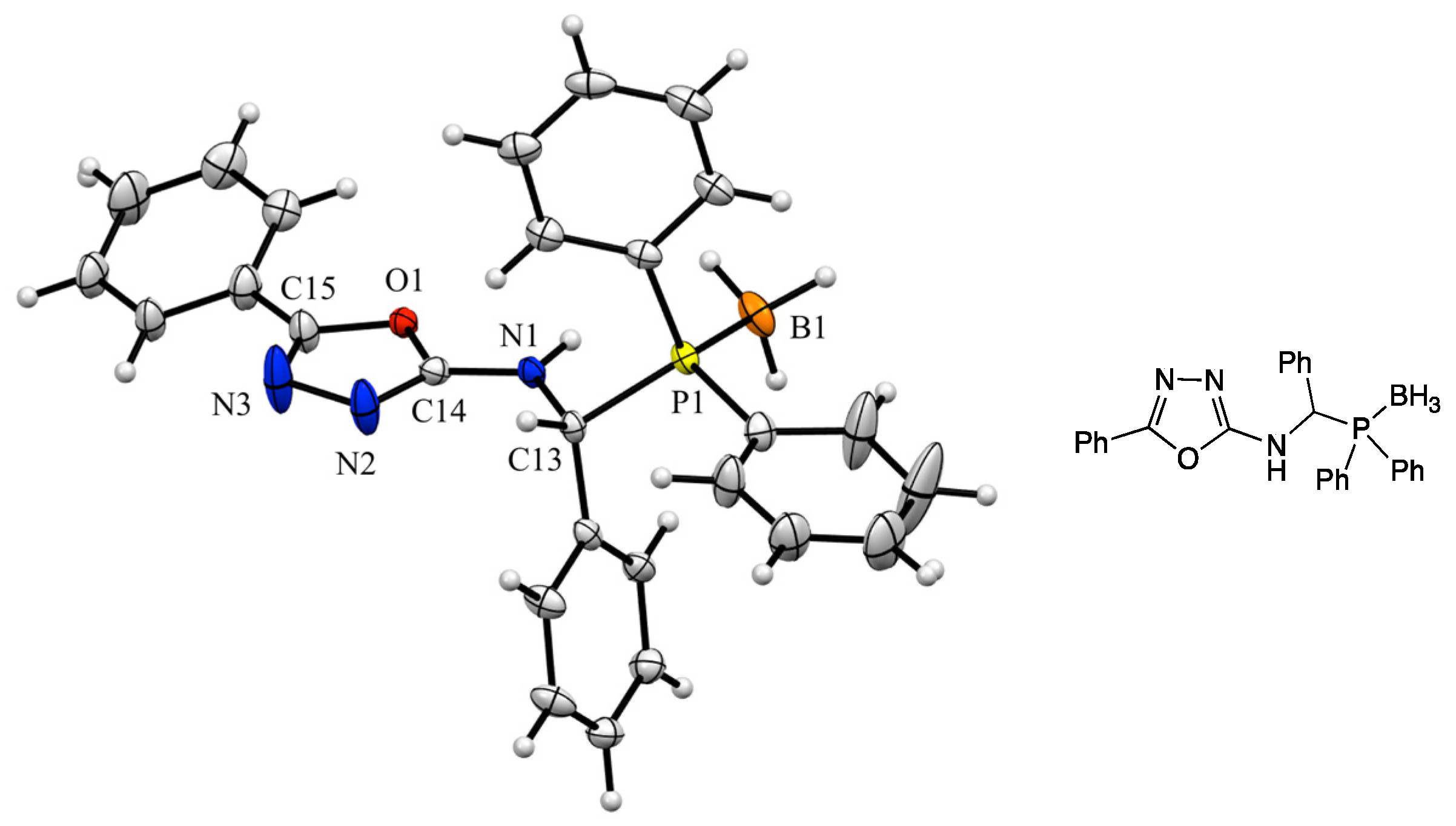
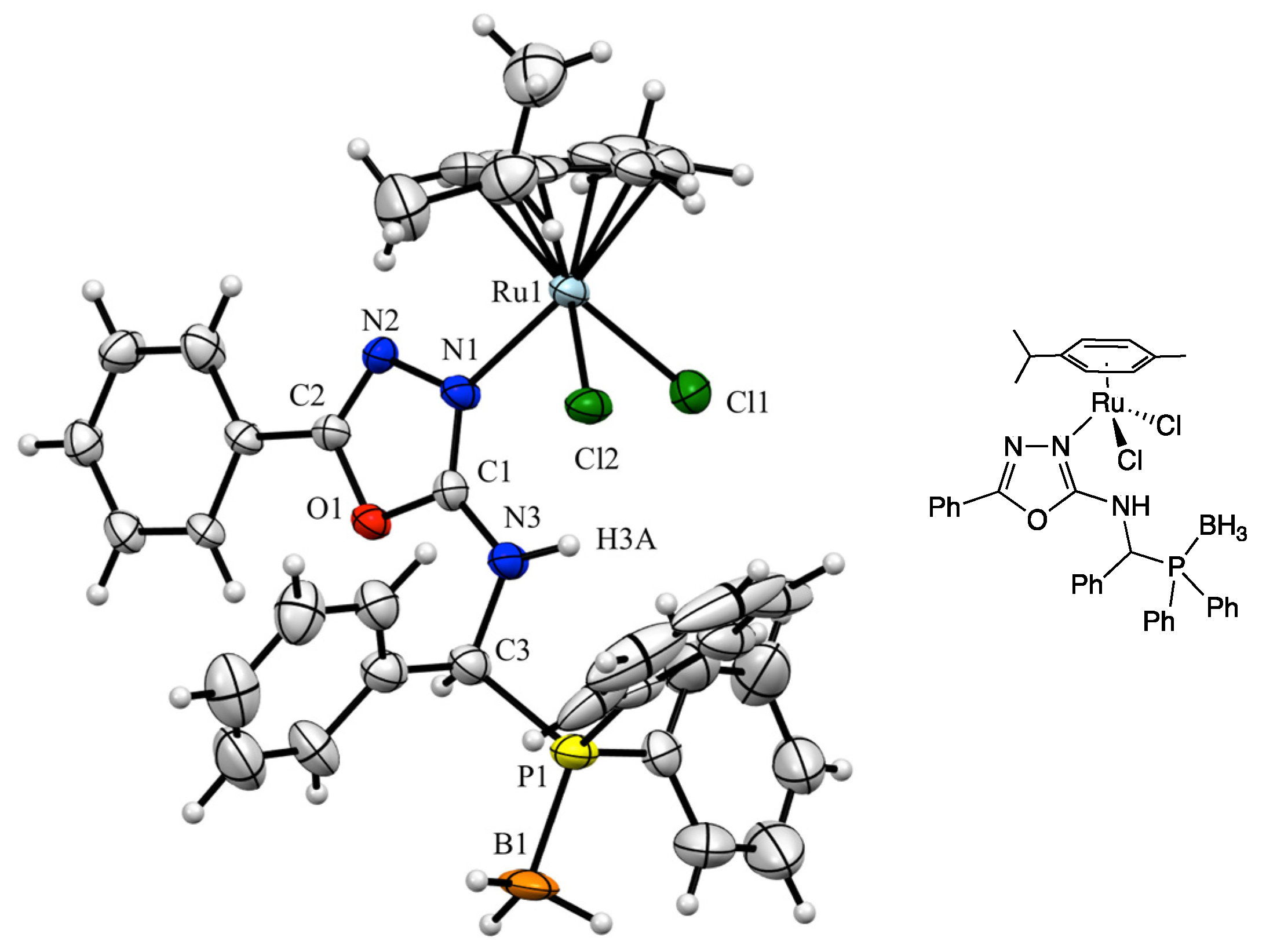
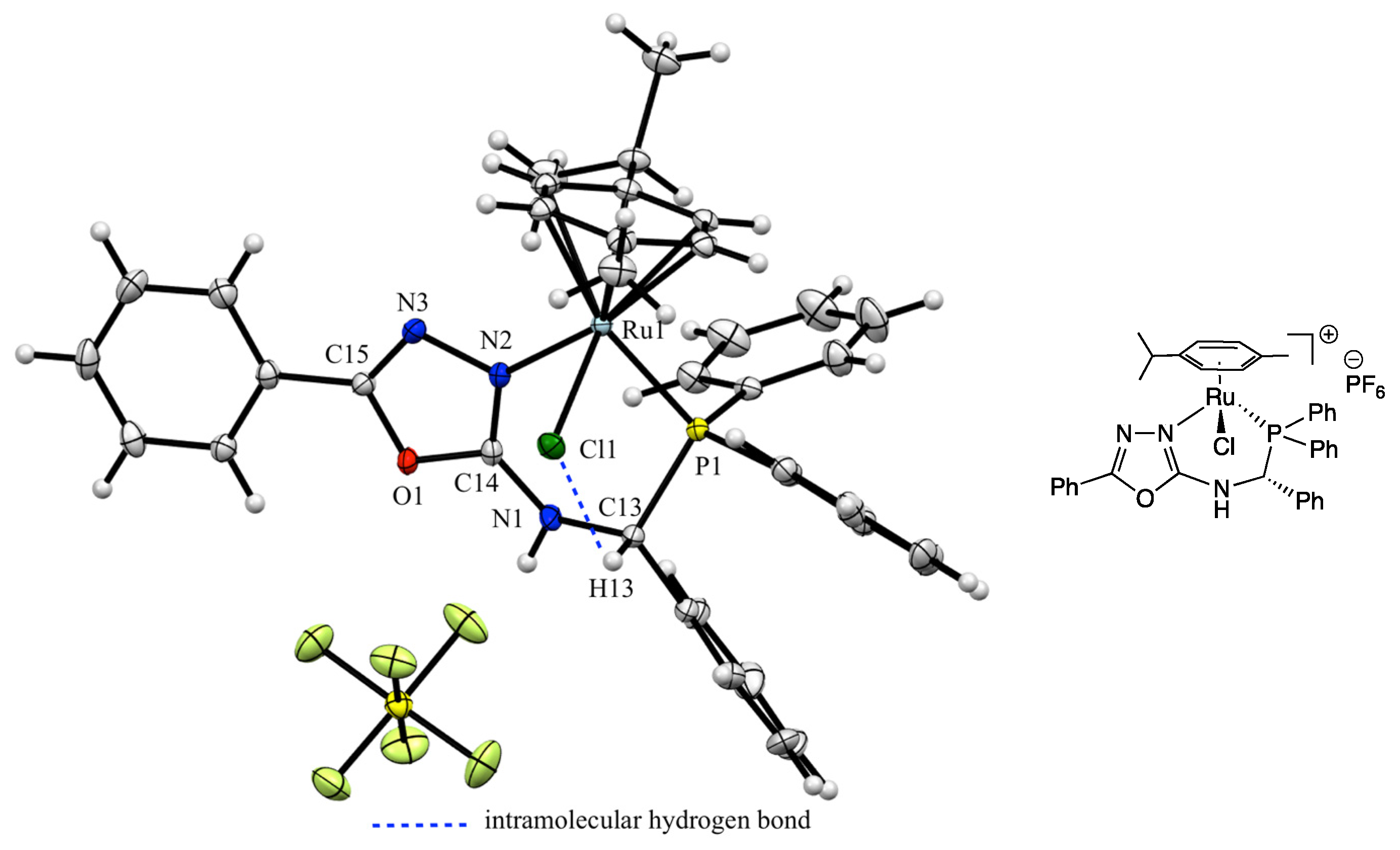
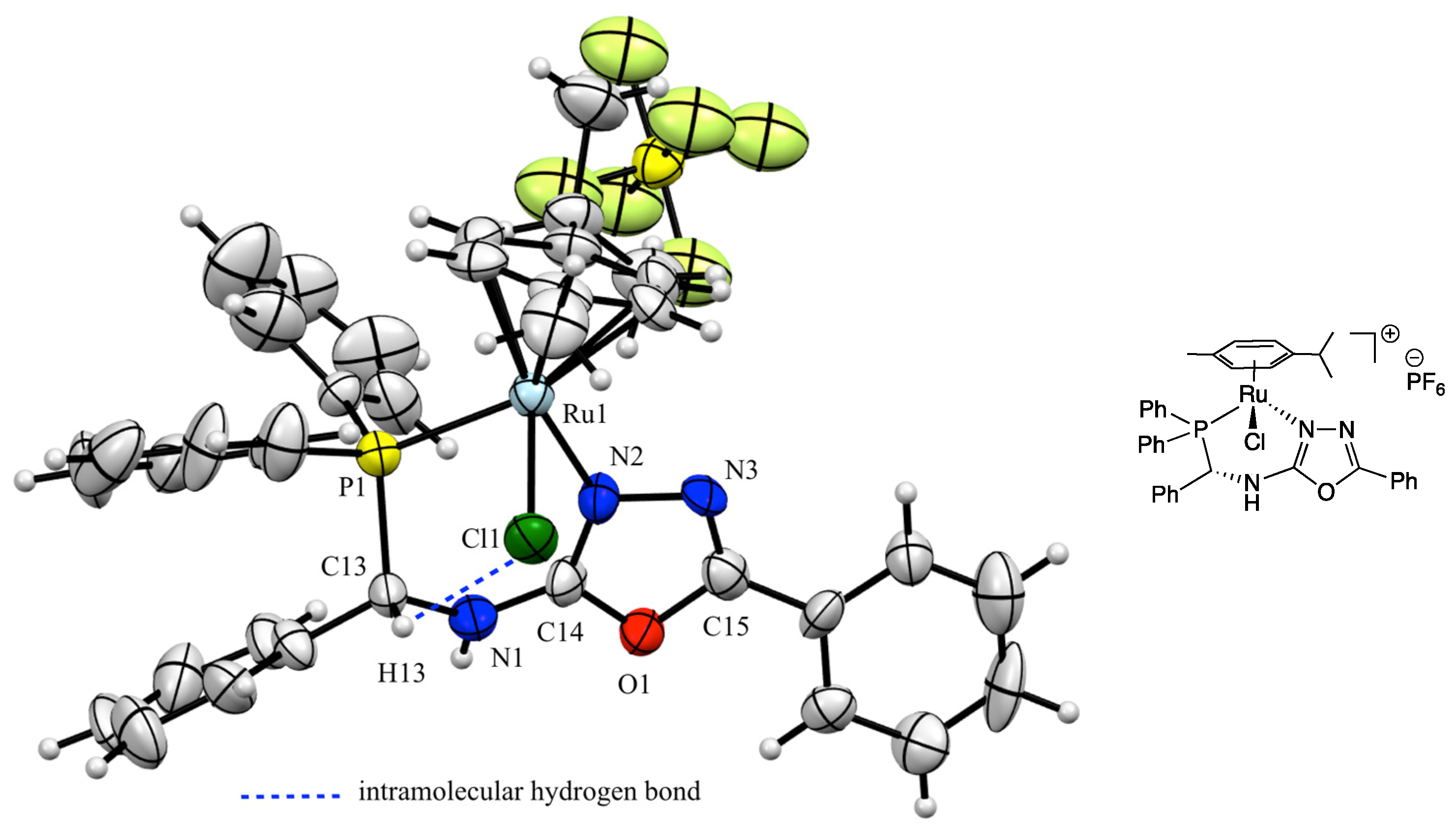
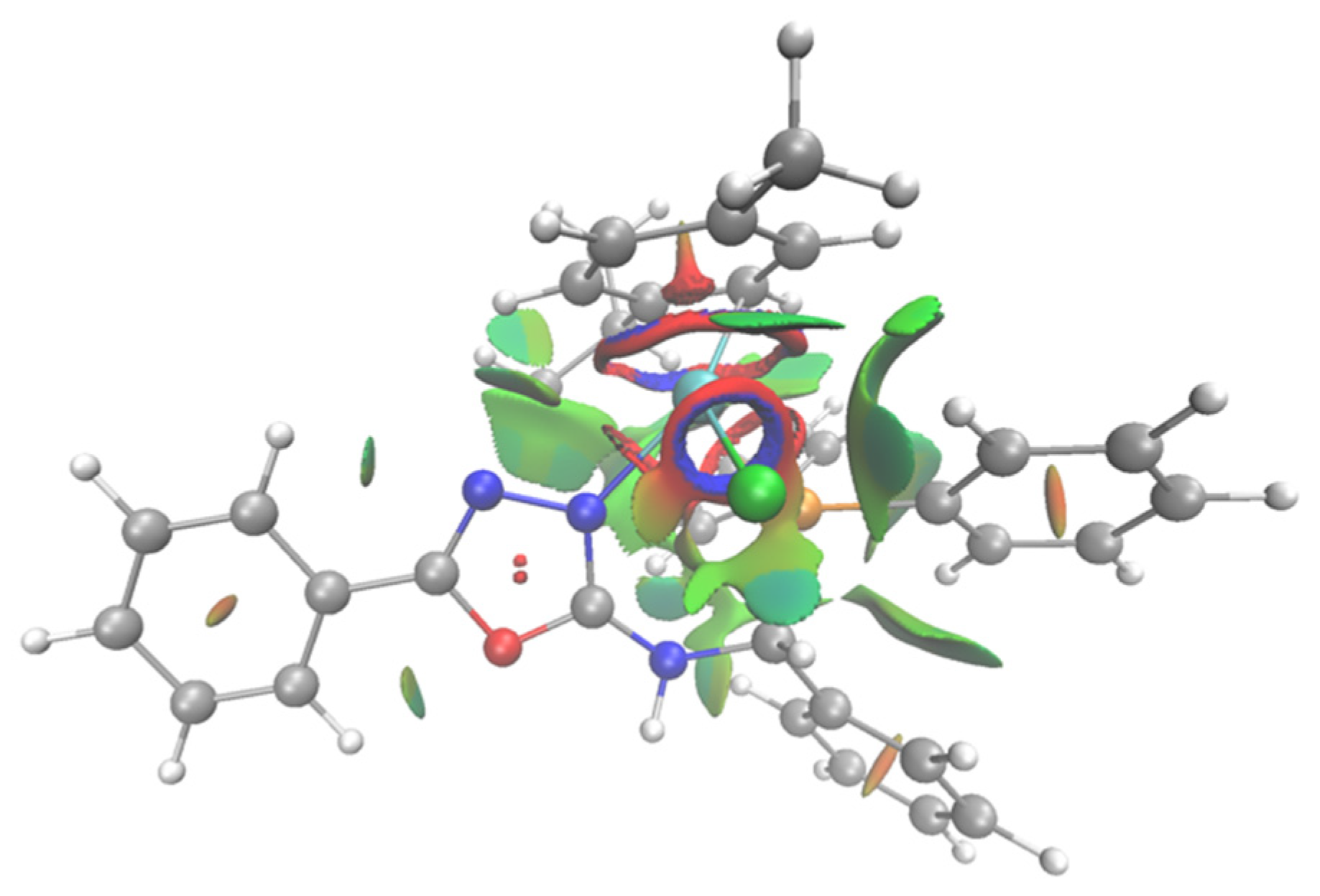
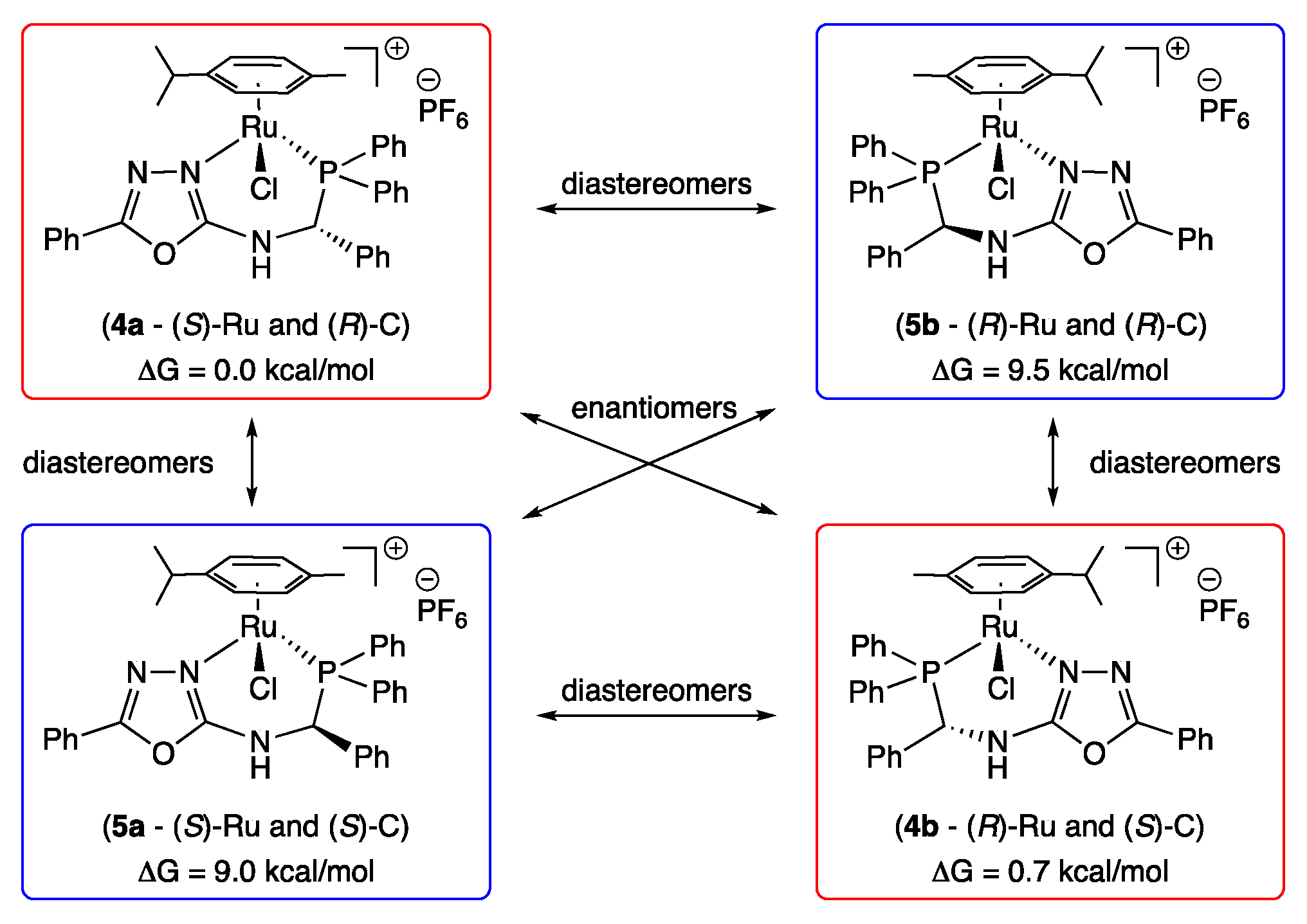
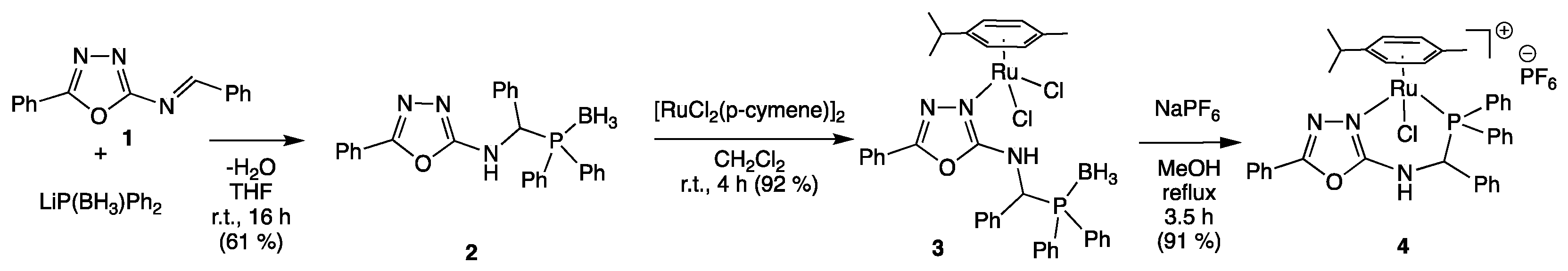
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flack, H.D. Louis Pasteur’s discovery of molecular chirality and spontaneous resolution in 1848, together with a complete review of his crystallographic and chemical work. Acta Crystallogr. Sect. A 2009, A65, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Fogassy, E.; Nógrádi, M.; Kozma, D.; Egri, G.; Pálovics, E.; Kiss, V. Optical resolution methods. Org. Biomol. Chem. 2006, 4, 3011–3030. [Google Scholar] [CrossRef] [PubMed]
- Siedlecka, R. Recent developments in optical resolution. Tetrahedron 2013, 69, 6331–6363. [Google Scholar] [CrossRef]
- Brill, Z.G.; Condakes, M.L.; Ting, C.P.; Maimone, T.J. Navigating the chiral pool in the total synthesis of complex terpene natural products. Chem. Rev. 2017, 117, 11753–11795. [Google Scholar] [CrossRef]
- Santos, R.; Pontes, K.V.; Nogueira, I.B.R. Enantiomers and their resolution. Encyclopedia 2022, 2, 151–188. [Google Scholar] [CrossRef]
- Yoon, T.P.; Jacobsen, E.N. Privileged chiral catalysts. Science 2003, 299, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Gopalaiah, K. Chiral iron catalysts for asymmetric synthesis. Chem. Rev. 2013, 113, 3248–3296. [Google Scholar] [CrossRef]
- Shaw, S.; White, J.D. cis-2,5-Diaminobicyclo [2.2.2]octane, a new chiral scaffold for asymmetric catalysis. Acc. Chem. Res. 2016, 49, 1825–1834. [Google Scholar] [CrossRef]
- Barbaro, P.; Bianchini, C.; Giambastiani, G.; Parisel, S.L. Progress in stereoselective catalysis by metal complexes with chiral ferrocenyl phosphines. Coord. Chem. Rev. 2004, 248, 2131–2150. [Google Scholar] [CrossRef]
- Berthod, M.; Mignani, G.; Woodward, G.; Lemaire, M. Modified BINAP: The how and the why. Chem. Rev. 2005, 105, 1801–1836. [Google Scholar] [CrossRef]
- Li, Y.-M.; Kwong, F.-Y.; Yu, W.-Y.; Chan, A.S.C. Recent advances in developing new axially chiral phosphine ligands for asymmetric catalysis. Coord. Chem. Rev. 2007, 251, 2119–2144. [Google Scholar] [CrossRef]
- Xi, J.-H.; Zhou, Q.-L. Chiral diphosphine and monodentate phosphorus ligands on a spiro scaffold for transition-metal-catalyzed asymmetric reactions. Acc. Chem. Res. 2008, 41, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Tang, W. Chiral monophosphorus ligands for asymmetric catalytic reactions. ACS Catal. 2016, 6, 4814–4858. [Google Scholar] [CrossRef]
- Yanagisawa, A.; Arai, T. Recent advances in chiral phosphine–silver(I) complex-catalyzed asymmetric reactions. Chem. Commun. 2008, 10, 1165–1172. [Google Scholar] [CrossRef]
- Gual, A.; Godard, C.; Castillón, S.; Claver, C. Highlights of the Rh-catalysed asymmetric hydroformylation of alkenes using phosphorus donor ligands. Tetrahedron Asymmetry 2010, 21, 1135–1146. [Google Scholar] [CrossRef]
- Wang, T.; Han, X.; Zhong, F.; Yao, W.; Lu, X. Amino acid-derived bifunctional phosphines for enantioselective transformations. Acc. Chem. Res. 2016, 49, 1369–1378. [Google Scholar] [CrossRef]
- Dutartre, M.; Bayardon, J.; Jugé, S. Applications and stereoselective syntheses of P-chirogenic phosphorus compounds. Chem. Soc. Rev. 2016, 45, 5771–5794. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-S.; Houa, C.-J.; Hu, X.-P. Chiral phosphine-phosphoramidite ligands in asymmetric catalysis. Synth. Commun. 2016, 46, 917–941. [Google Scholar] [CrossRef]
- Misra, A.; Dwivedi, J.; Kishore, D. Role of the transition metal complexes of 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) in asymmetric catalysis. Synth. Commun. 2017, 47, 497–535. [Google Scholar] [CrossRef]
- Budagumpi, S.; Keri, R.S.; Achar, G.; Brinda, K.N. Coinage metal complexes of chiral N-heterocyclic carbine ligands: Syntheses and applications in asymmetric catalysis. Adv. Synth. Catal. 2020, 362, 970–997. [Google Scholar] [CrossRef]
- Kong, L.; Morvan, J.; Pichon, D.; Jean, M.; Albalat, M.; Vives, T.; Colombel-Rouen, S.; Giorgi, M.; Dorcet, V.; Roisnel, T.; et al. From pochiral N-heterocyclic carbenes to optically pure metal complexes: New opportunities in asymmetric catalysis. J. Am. Chem. Soc. 2020, 142, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, A.; Raveedran, A.V.; Latha, A.T.; Priyadarshini, D.; Swamy, P.C.A. Coordination versatility of NHC-metal topologies in asymmetric catalysis: Synthetic insights and recent trends. Coord. Chem. Rev. 2023, 478, 214922. [Google Scholar] [CrossRef]
- Talavera, G.; Fariña, A.S.; Zanotti-Gerosa, A.; Nedden, H.G. Structural diversity in ruthenium-catalyzed asymmetric transfer hydrogenation reactions. In Organometallics in Process Chemistry; Colacot, T.J., Sivakumar, V., Eds.; Topics in Organometallic Chemistry; Springer: Berlin, Germany, 2019; Volume 65, pp. 73–114. [Google Scholar]
- Cotman, A.J. Escaping from flatland: Stereoconvergent synthesis of three-dimensional scaffolds via ruthenium(II)-catalyzed Noyori-Ikariya transfer hydrogenation. Chem. Eur. J. 2021, 27, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, E.; Shezaf, J.Z.; Shen, W.; Krische, M.J. Historical perspective on ruthenium-catalyzed hydrogen transfer and survey of enantioselective hydrogen auto-transfer processes for the conversion of lower alcohols to higher alcohols. Chem. Sci. 2022, 13, 12625–12633. [Google Scholar] [CrossRef]
- Ritleng, V.; Michon, C. Bidentate donor-functionalized N-heterocyclic carbenes: Valuable ligands for ruthenium-catalyzed transfer hydrogenation. Molecules 2022, 27, 4703. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Wang, J. Enantioselective C−H bond functionalization involving arene ruthenium(II) catalysis. Chem. Eur. J. 2023, 29, e202202461. [Google Scholar] [CrossRef] [PubMed]
- Werner, A. Zur Kenntnis des asymmetrischen Kobaltatoms. I. Ber. Dtsch. Chem. Ges. 1911, 44, 1887–1898. [Google Scholar] [CrossRef]
- Ganter, C. Chiral organometallic half-sandwich complexes with defined metal configuration. Chem. Soc. Rev. 2003, 32, 130–138. [Google Scholar] [CrossRef]
- Kumar, P.; Gupta, R.K.; Pandey, D.S. Half-sandwich arene ruthenium complexes: Synthetic strategies and relevance in catalysis. Chem. Soc. Rev. 2014, 43, 707–733. [Google Scholar] [CrossRef]
- Steinlandt, P.S.; Zhang, L.; Meggers, E. Metal stereogenicity in asymmetric transition metal catalysis. Chem. Rev. 2023, 123, 4764–4794. [Google Scholar] [CrossRef]
- Buhse, T.; Cruz, J.-M.; Noble-Terán, M.E.; Hochberg, D.; Ribó, J.M.; Crusats, J.; Micheau, J.-C. Spontaneous deracemizations. Chem. Rev. 2021, 121, 2147–2229. [Google Scholar] [CrossRef] [PubMed]
- Putman, J.I.; Armstrong, D.W. Recent advances in the field of chiral crystallization. Chirality 2022, 34, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Sun, X.; Du, P. Designing single chirality via crystallization method: Spontaneous chiral symmetry breaking and deracemization. Mater. Today Chem. 2023, 32, 101636. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]
- Hkiri, S.; Gourlaouen, C.; Touil, S.; Samarat, A.; Sémeril, D. 1,3,4-Oxadiazole-functionalized α-amino-phosphonates as ligands for the ruthenium-catalyzed reduction of ketones. New J. Chem. 2021, 45, 11327. [Google Scholar] [CrossRef]
- Hkiri, S.; Touil, S.; Samarat, A.; Sémeril, D. Functionalized-1,3,4-oxadiazole ligands for the ruthenium-catalyzed Lemieux-Johnson type oxidation of olefins and alkynes in water. Mol. Catal. 2022, 517, 112014. [Google Scholar] [CrossRef]
- Hkiri, S.; Touil, S.; Samarat, A.; Sémeril, D. Palladium-catalyzed Suzuki–Miyaura cross-coupling with α-aminophosphonates based on 1,3,4-oxadiazole as ligands. C. R. Chim. 2022, 25, 53–65. [Google Scholar] [CrossRef]
- Hkiri, S.; Coşkun, K.A.; Üstün, E.; Samarat, A.; Tutar, Y.; Şahin, N.; Sémeril, D. Silver(I) complexes based on oxadiazole-functionalized α-aminophosphonate: Synthesis, structural study, and biological activities. Molecules 2022, 27, 8131. [Google Scholar] [CrossRef]
- Van Overschelde, M.; Verveckena, E.; Modha, S.G.; Cogen, S.; Van der Eycken, E. Catalyst-free alcoholysis of phosphane-boranes: A smooth, cheap, and efficient deprotection procedure. Tetrahedron 2009, 65, 6410–6415. [Google Scholar] [CrossRef]
- Fecher, G.-H.; Kübler, J.; Felser, C. Chirality in the solid state: Chiral crystal structures in chiral and achiral space groups. Materials 2022, 15, 5812. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B 2013, B69, 249–259. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 2 | 3 | 4a | 4b | |
|---|---|---|---|---|---|
| CCDC depository | 2296142 | 2296144 | 2296150 | 2296149 | |
| Color | Colorless | Colorless | Orange | Colorless | |
| Shape | Block | Block | Prism | Plate | |
| Chemical formula | C27H25BN3OP | C38H41BCl4N3OPRu | C39H40Cl5F6N3OP2Ru | C75H74Cl4F12N6O2P4Ru2 | |
| Formula weight (g mol−1) | 449.28 | 840.39 | 1021.00 | 1787.22 | |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | |
| Space group | P-1 | P-1 | P1 | P1 | |
| Unit cell | a (Å) | 9.9471(5) | 11.7923(13) | 10.2633(4) | 10.430(8) |
| b (Å) | 9.9734(5) | 12.8299(13) | 13.0695(4) | 13.135(9) | |
| c (Å) | 13.1802(7) | 14.0024(16) | 16.6645(6) | 16.931(12) | |
| Parameters | α (°) | 84.889(2) | 81.052(4) | 104.9230(10) | 104.27(2) |
| β (°) | 69.461(2) | 83.206(4) | 97.9710(10) | 98.05(3) | |
| γ (°) | 70.877(2) | 65.607(4) | 90.0220(10) | 90.16(3) | |
| Volume (Å3) | 1156.38(10) | 1902.4(4) | 2137.53(13) | 2224(3) | |
| Z | 2 | 2 | 2 | 1 | |
| D (g cm−3) | 1.290 | 1.467 | 1.586 | 1.334 | |
| μ (mm−1) | 0.144 | 0.770 | 0.816 | 0.600 | |
| Tmin, Tmax | 0.7046, 0.7456 | 0.887, 0.927 | 0.867, 0.908 | 0.931, 0.965 | |
| F(000) | 472 | 860 | 1032 | 906 | |
| Crystal size (mm) | 0.22 × 0.14 × 0.12 | 0.16 × 0.12 × 0.10 | 0.18 × 0.14 × 0.12 | 0.12 × 0.08 × 0.06 | |
| Index ranges | −13 ≤ h ≤ 13 | −13 ≤ h ≤ 15 | −14 ≤ h ≤ 14 | −13 ≤ h ≤ 11 | |
| −13 ≤ k ≤ 13 | −16 ≤ k ≤ 16 | −18 ≤ k ≤ 17 | −17 ≤ k ≤ 17 | ||
| −17 ≤ l ≤ 17 | −17 ≤ l ≤ 18 | −23 ≤ l ≤ 23 | −22 ≤ l ≤ 22 | ||
| θ range for data collection (°) | 2.162 ≤ θ ≤ 28.012 | 2.002 ≤ θ ≤ 28.087 | 2.218 ≤ θ ≤ 30.050 | 1.254 ≤ θ ≤ 28.463 | |
| Reflections collected | 40,699 | 26,123 | 107,287 | 44,157 | |
| Independent/observed | 5538/5255 | 8925/6192 | 22,909/20,812 | 17,986/9156 | |
| Rint | 0.0350 | 0.0692 | 0.0544 | 0.1330 | |
| Data/restraints/parameters | 5,538/27/368 | 8,925/9/455 | 22,909/3/1041 | 17,986/10/862 | |
| Goodness-of-fit on F2 | 1.082 | 1.053 | 1.044 | 0.946 | |
| Final R indices (I > 2.0 σ(I)) | R1 = 0.0804 | R1 = 0.1136 | R1 = 0.0313 | R1 = 0.0765 | |
| wR2 = 0.2000 | wR2 = 0.2540 | wR2 = 0.0605 | wR2 = 0.1501 | ||
| R indices (all data) | R1 = 0.0826 | R1 = 0.1583 | R1 = 0.0408 | R1 = 0.1625 | |
| wR2 = 0.2010 | wR2 = 0.2851 | wR2 = 0.0651 | wR2 = 0.1875 | ||
| Δρmax, Δρmin (eÅ−3) | 0.718, −0.592 | 2.766, −2.183 | 0.964, −0.646 | 0.918, −0.822 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinmetz, M.; Gourlaouen, C.; Sémeril, D. How an Internal Supramolecular Interaction Determines the Stereochemistry of a Metal Center. Organics 2024, 5, 1-11. https://doi.org/10.3390/org5010001
Steinmetz M, Gourlaouen C, Sémeril D. How an Internal Supramolecular Interaction Determines the Stereochemistry of a Metal Center. Organics. 2024; 5(1):1-11. https://doi.org/10.3390/org5010001
Chicago/Turabian StyleSteinmetz, Maxime, Christophe Gourlaouen, and David Sémeril. 2024. "How an Internal Supramolecular Interaction Determines the Stereochemistry of a Metal Center" Organics 5, no. 1: 1-11. https://doi.org/10.3390/org5010001