The Regio- and Stereoselective Synthesis of 1,4-Diarylbut-1-en-3-ynes Having Aryl Groups at the Mutual Syn Positions


Abstract
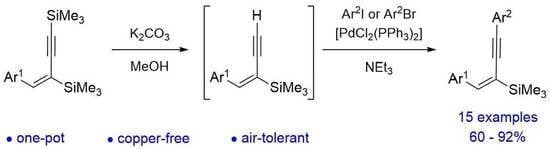
:
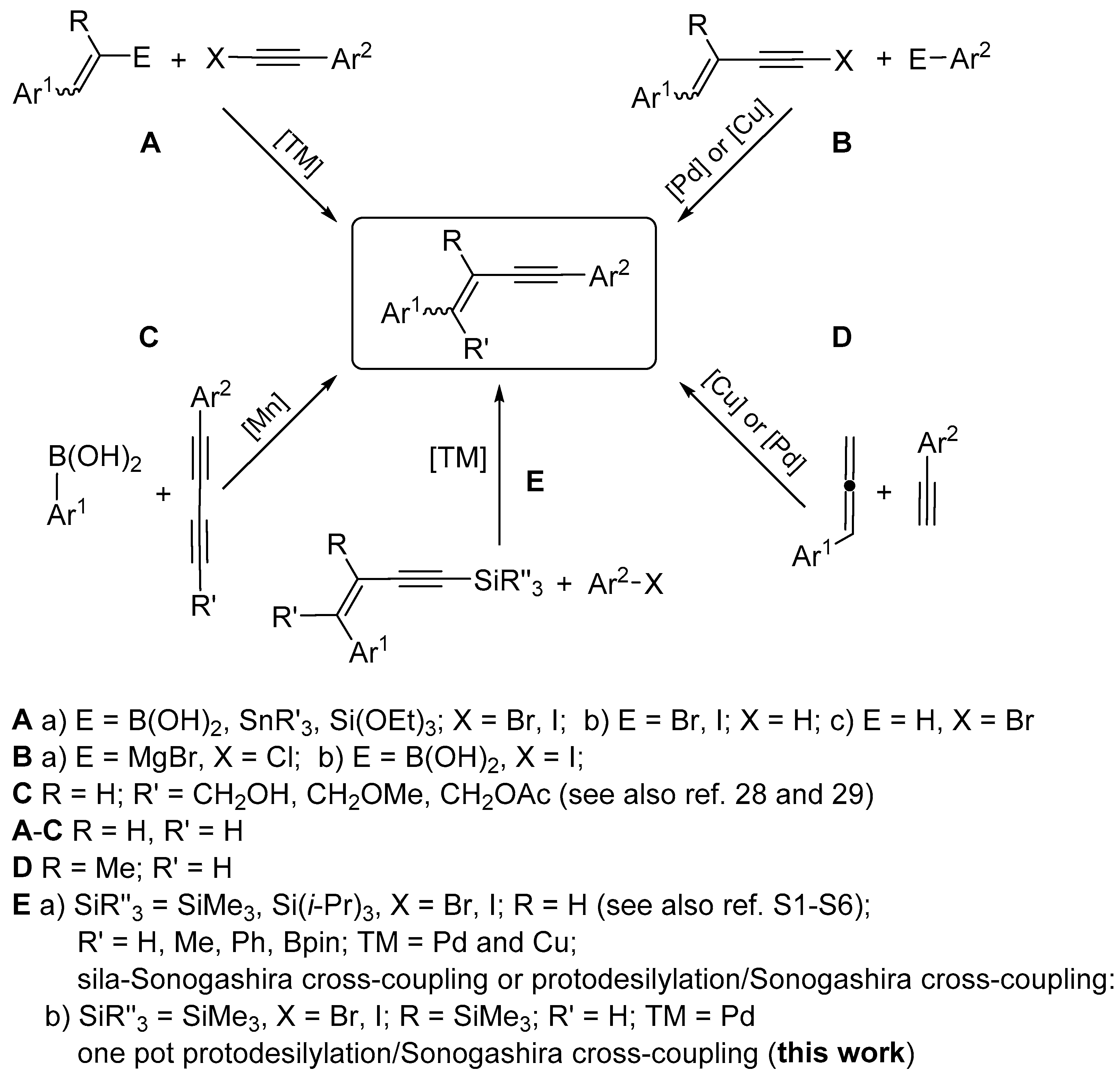
1. Introduction
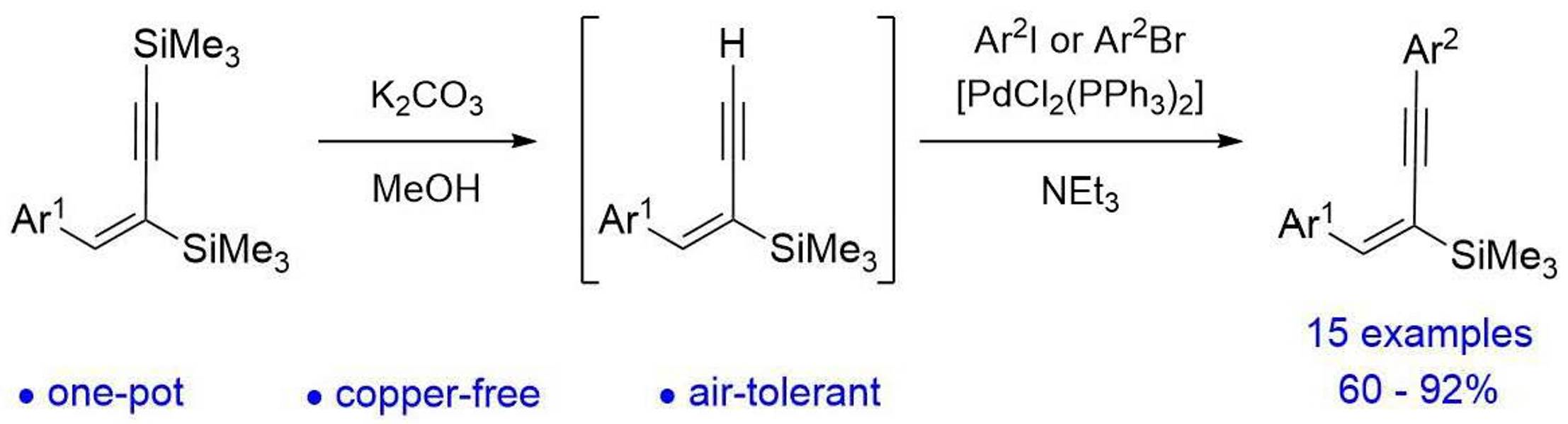
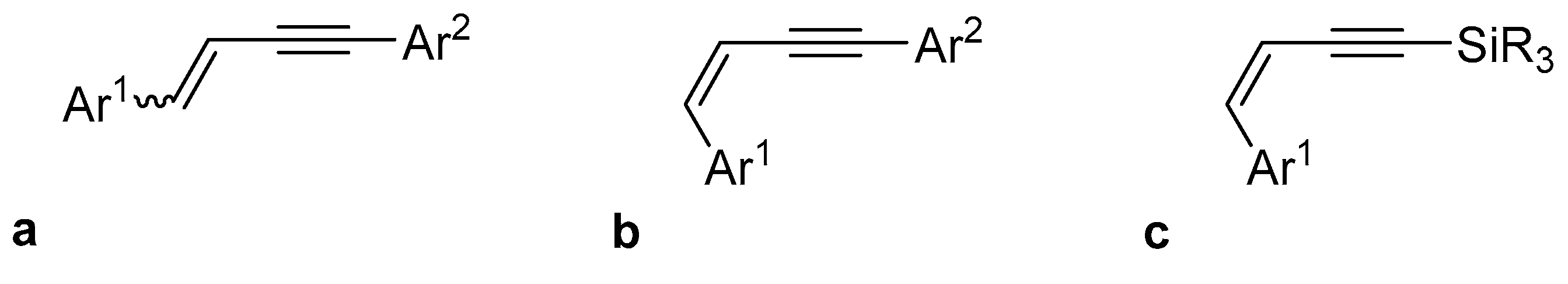
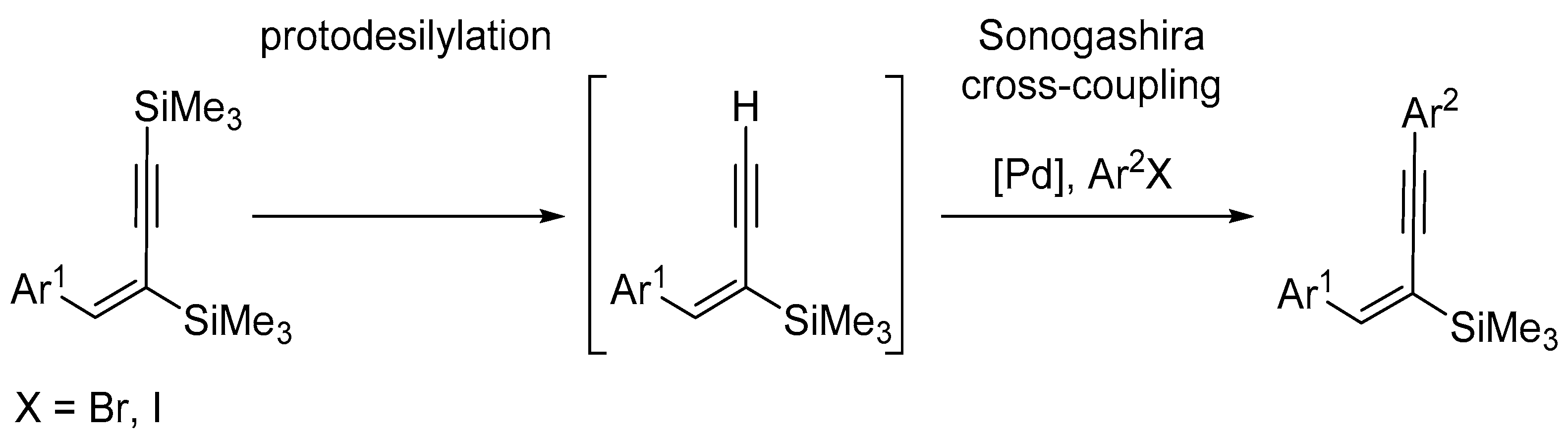
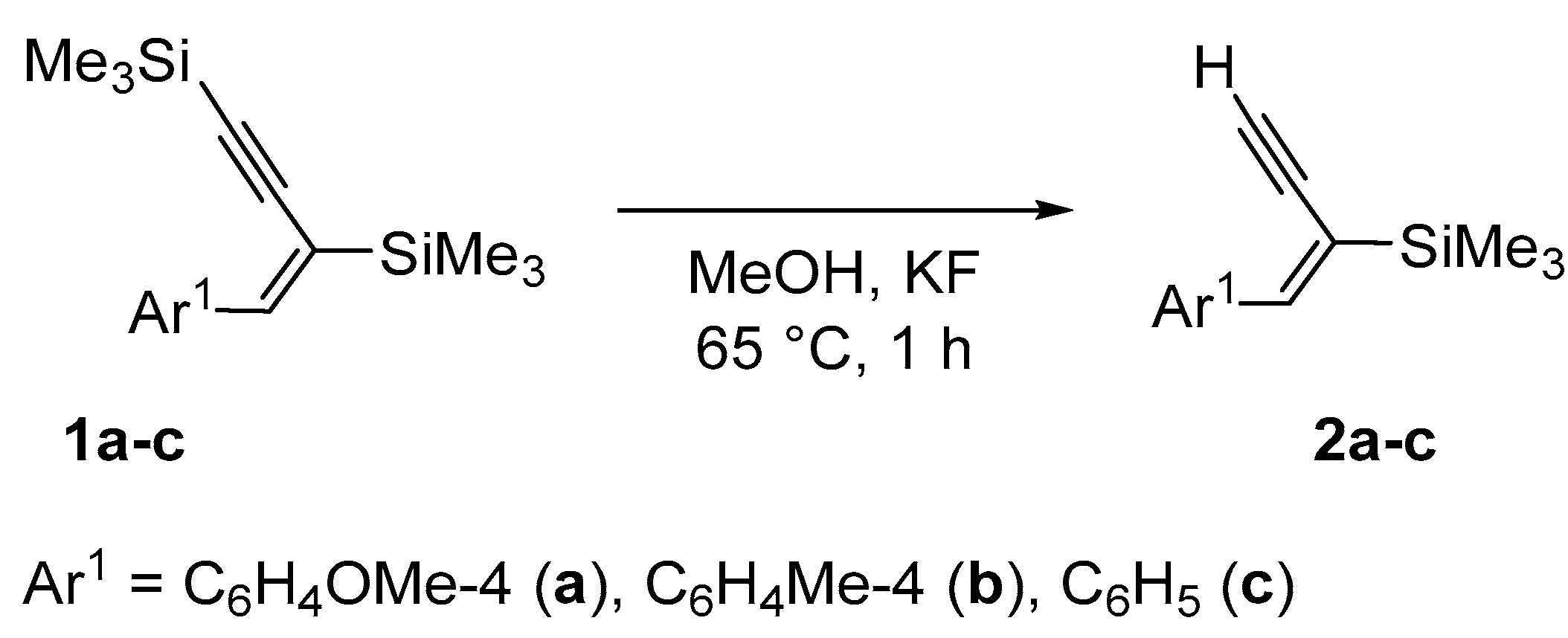
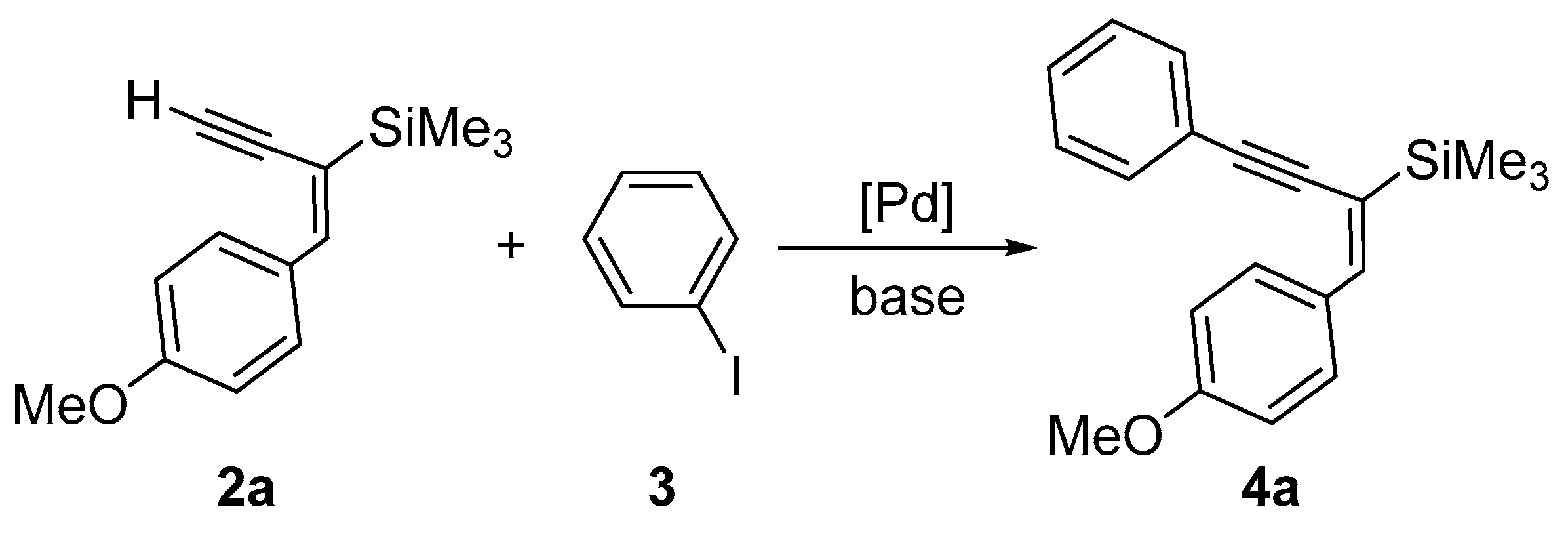
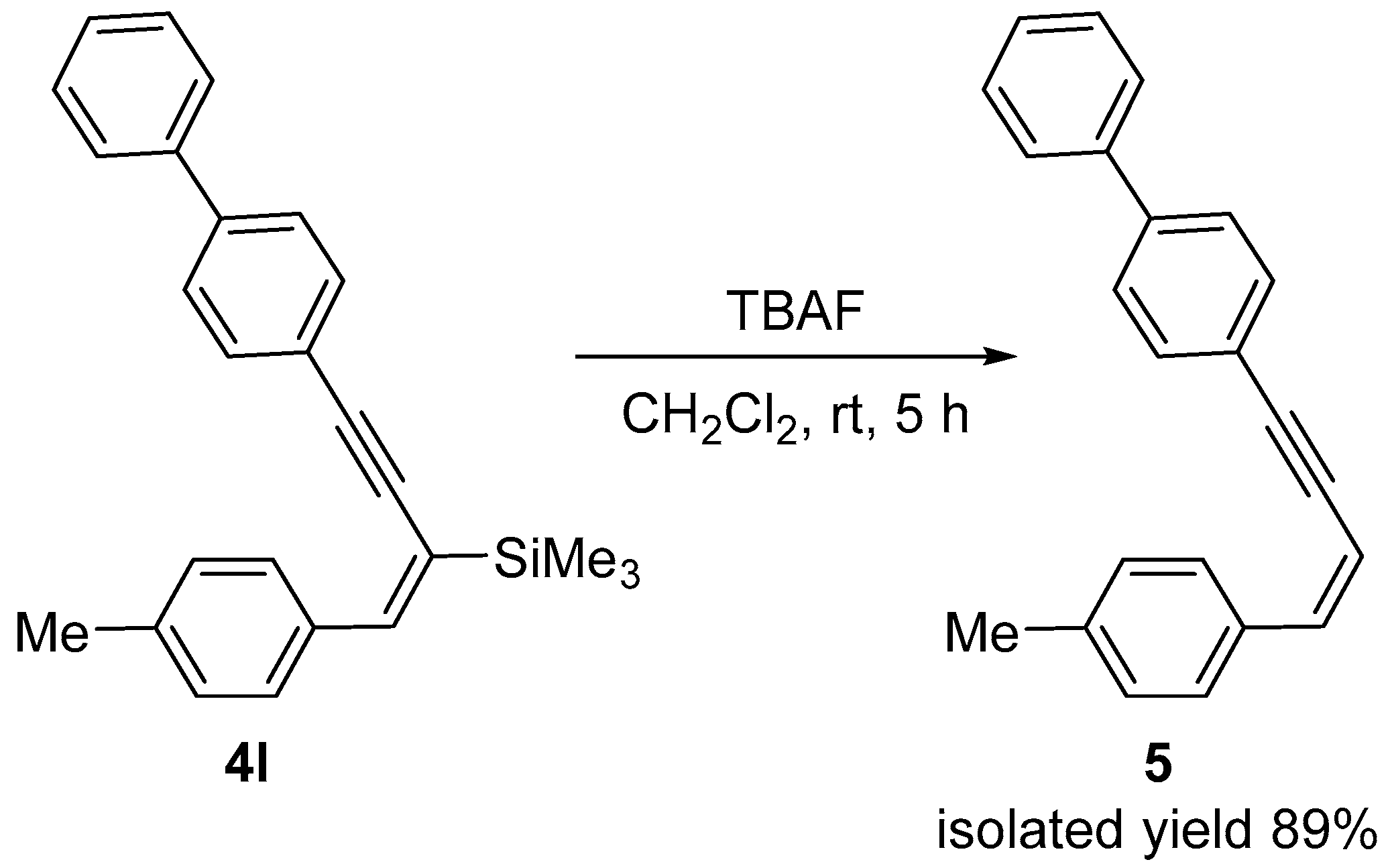
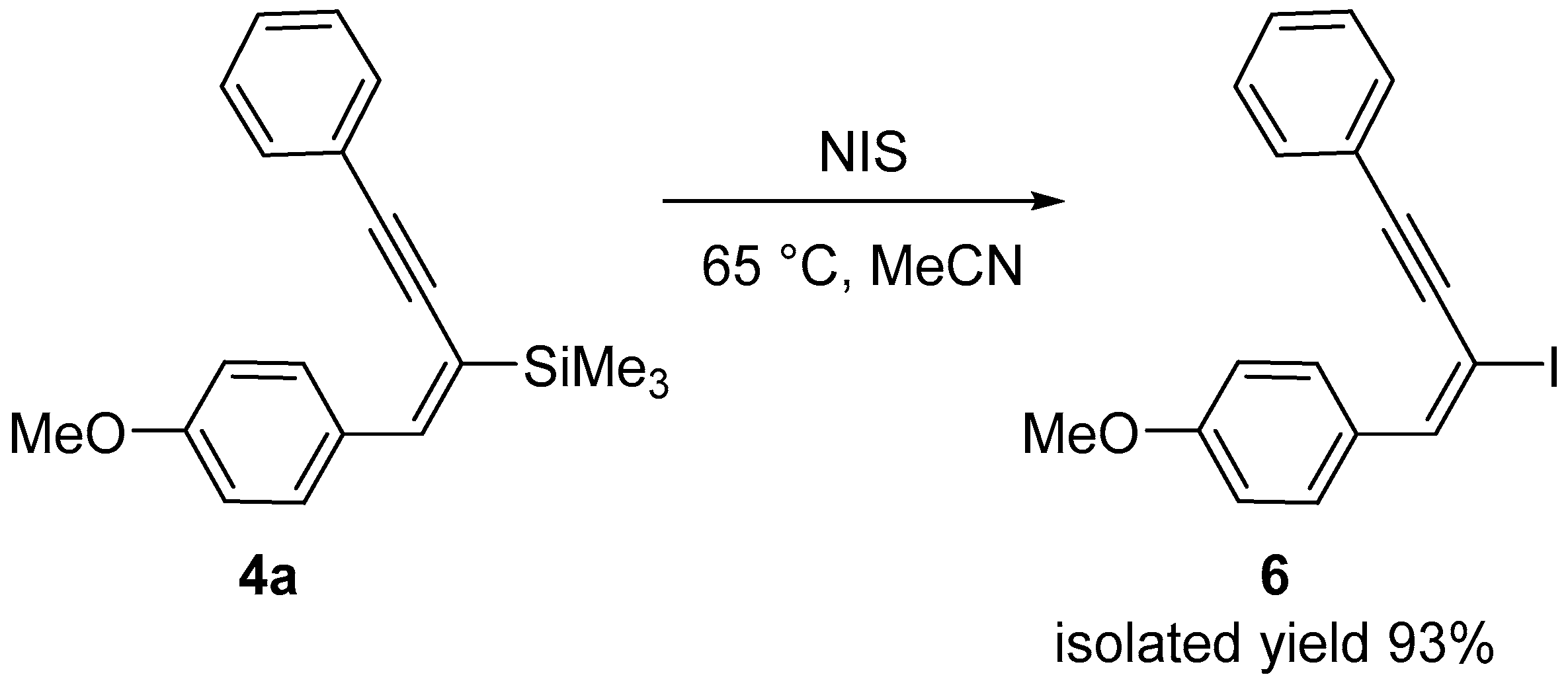
2. Results and Discussion
3. Conclusions
4. Experimental
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Masters, J.T. Transition metal-catalyzed couplings of alkynes to 1,3-enynes: Modern methods and synthetic applications. Chem. Soc. Rev. 2016, 45, 2212–2238. [Google Scholar] [CrossRef] [PubMed]
- Dherbassy, Q.; Manna, S.; Talbot, F.J.T.; Prasitwatcharakorn, W.; Perry, G.J.P.; Procter, D.J. Copper-catalyzed functionalization of enynes. Chem. Sci. 2020, 11, 11380–11393. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Ren, J.; Yang, Y.; Ye, X.; Wang, B.; Wang, H. 2-Activated 1,3-enynes in enantioselective synthesis. Org. Biomol. Chem. 2020, 18, 7977–7986. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, P.; Fu, Y.; Hao, T.; Liu, X.; Ding, Q.; Peng, Y. Recent advances in the tandem annulation of 1,3-enynes to functionalized pyridine and pyrrole derivatives. Beilstein J. Org. Chem. 2021, 17, 2462–2476. [Google Scholar] [CrossRef]
- Gevorgyan, V.; Yamamoto, Y. Palladium-catalyzed enyne–yne [4+2] benzannulation as a new and general approach to polysubstituted benzenes. J. Organomet. Chem. 1999, 576, 232–247. [Google Scholar] [CrossRef]
- Iverson, S.L.; Uetrecht, J.P. Identification of a reactive metabolite of terbinafine: Insights into terbinafine-induced hepatotoxicity. Chem. Res. Toxicol. 2001, 14, 175–181. [Google Scholar] [CrossRef]
- Shun, A.L.K.S.; Tykwinski, R.R. Synthesis of naturally occurring polyynes. Angew. Chem. Int. Ed. 2006, 45, 1034–1057. [Google Scholar] [CrossRef]
- Wang, D.; Gao, S. Sonogashira coupling in natural product synthesis. Org. Chem. Front. 2014, 1, 556–566. [Google Scholar] [CrossRef]
- El-Shazly, M.; Barve, B.D.; Korinek, M.; Liou, J.-R.; Chuang, D.-W.; Cheng, Y.-B.; Hou, M.-F.; Wang, J.-J.; Wu, Y.-C.; Chang, F.-R. Insights on the Isolation, biological activity and synthetic protocols of enyne derivatives. Curr. Top. Med. Chem. 2014, 14, 1076–1093. [Google Scholar] [CrossRef]
- Liu, Y.; Nishiura, M.; Wang, Y.; Hou, Z. π-Conjugated aromatic enynes as a single-emitting component for white electroluminescence. J. Am. Chem. Soc. 2006, 128, 5592–5593. [Google Scholar] [CrossRef]
- Peng, T.; Li, G.; Liu, Y.; Wua, Y.; Ye, K.; Yao, D.; Yuan, Y.; Hou, Z.; Wang, Y. High-efficiency and deep-blue fluorescent organic light-emitting diodes with the easily controlled doping concentrations. Org. Electron. 2011, 12, 1068–1072. [Google Scholar] [CrossRef]
- Doucet, H.; Hierso, J.-C. Palladium-based catalytic systems for the synthesis of conjugated enynes by Sonogashira reactions and related alkynylations. Angew. Chem. Int. Ed. 2007, 46, 834–871. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Wang, J. Recent advances in transition-metal-catalyzed synthesis of conjugated enynes. Org. Biomol. Chem. 2016, 14, 6638–6650. [Google Scholar]
- Fu, L.; Greßies, S.; Chen, P.; Liu, G. Recent advances and perspectives in transition metal-catalyzed 1,4-Functionalizations of unactivated 1,3-enynes for the synthesis of allenes. Chin. J. Chem. 2020, 38, 91–100. [Google Scholar] [CrossRef]
- Mao, L.; Bose, S.K. Hydroboration of enynes and mechanistic insights. Adv. Synth. Catal. 2020, 362, 4174–4188. [Google Scholar] [CrossRef]
- Ahammed, S.; Kundu, D.; Ranu, B.C. Cu-Catalyzed Fe-driven Csp−Csp and Csp−Csp2 cross-coupling: An access to 1,3-diynes and 1,3-enynes. J. Org. Chem. 2014, 79, 7391–7398. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, E.; Yoshida, H.; Kurahashi, T.; Nakao, Y.; Hiyama, T. Carbostannylation of alkynes catalyzed by an iminophosphine-palladium complex. J. Am. Chem. Soc. 1998, 120, 2975–2976. [Google Scholar] [CrossRef]
- Cornelissen, L.; Lefrancq, M.; Riant, O. Copper-catalyzed cross-coupling of vinylsiloxanes with bromoalkynes: Synthesis of enynes. Org. Lett. 2014, 16, 3024–3027. [Google Scholar] [CrossRef]
- Xie, X.; Xu, X.; Li, H.; Xu, X.; Yang, J.; Li, Y. Iron-catalyzed cross-coupling reactions of terminal alkynes with vinyl iodides. Adv. Synth. Catal. 2009, 351, 1263–1267. [Google Scholar] [CrossRef]
- Huang, M.; Feng, Y.; Wu, Y. Enyne synthesis through a modified Sonogashira cross-coupling reaction catalyzed by cyclopalladated complexes. Tetrahedron 2012, 68, 376–381. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Wang, Y.-J.; Cheng, J.-H.; Lee, C.-F. Copper-catalyzed coupling of alkynes with alkenyl halides. Synlett 2012, 23, 930–934. [Google Scholar]
- Mi, X.; Huang, M.; Feng, Y.; Wu, Y. Discovery of a novel palladium catalyst for the preparation of enynes with a copper- and ligand-free Sonogashira reaction. Synlett 2012, 23, 1257–1261. [Google Scholar] [CrossRef]
- Mukherjee, N.; Kundu, D.; Ranu, B.C. A co-operative Ni/Cu system for Csp-Csp and Csp-Csp2 cross-coupling providing a direct access to unsymmetrical 1,3-diynes and en-ynes. Chem. Commun. 2014, 50, 15784–15787. [Google Scholar] [CrossRef]
- Wena, Y.; Wang, A.; Jiang, H.; Zhu, S.; Huang, L. Highly regio- and stereoselective synthesis of 1,3-enynes from unactivated ethylenes via palladium-catalyzed cross-coupling. Tetrahedron Lett. 2011, 52, 5736–5739. [Google Scholar] [CrossRef]
- Provot, O.; Giraud, A.; Peyrat, J.-F.; Alami, M.; Brion, J.-D. Synthetic approach to enyne and enediyne analogues of anticancer agents. Tetrahedron Lett. 2005, 46, 8547–8550. [Google Scholar] [CrossRef]
- Tikad, A.; Hamze, A.; Provot, O.; Brion, J.-D.; Alami, M. Suzuki coupling reactions of (E)- and (Z)-chloroenynes with boronic acids:versatile access to functionalized 1,3-enynes. Eur. J. Org. Chem. 2010, 2010, 725–731. [Google Scholar] [CrossRef]
- Yan, Z.; Zhu, C.; Xie, J. Manganese(I)-catalyzed selective functionalization of alkynes. Synlett 2018, 29, 124–128. [Google Scholar]
- Yan, Z.; Yuan, X.-A.; Zhao, Y.; Zhu, C.; Xie, J. Selective hydroarylation of 1,3-diynes using a dimeric manganese catalyst: Modular synthesis of Z-enynes. Angew. Chem. Int. Ed. 2018, 57, 12906–12910. [Google Scholar] [CrossRef]
- Zhu, L.; Guo, H.; Feng, X.; Yamamoto, Y.; Bao, M. Copper-catalyzed one-pot synthesis of 1,3-enynes from 2-chloro-N-(quinolin-8-yl)acetamides and terminal alkynes. J. Org. Chem. 2020, 85, 8740–8748. [Google Scholar] [CrossRef]
- Liu, Z.-K.; Yang, Y.; Zhan, Z.-P. Preparation of (E)-1,3-enyne derivatives through palladium catalyzed hydroalkynylation of allenes. J. Org. Chem. 2022, 87, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Lee, S. Selective mono- and dialkynylation of 1-fluoro-2,2-diiodovinylarenes using Pd-catalyzed decarboxylative coupling reactions. Org. Lett. 2019, 21, 7923–7927. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Komatsu, R.; Uchida, N.; Ikeda, R.; Konakahara, T. A single-step synthesis of enynes: Pd-catalyzed arylalkynylation of aryl iodides, internal alkynes, and alkynylsilanes. Org. Lett. 2010, 12, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, J.A.; Poveda, R.R.; Brenes, J.A. One-pot conversion of aldehydes and ketones into 1-substituted and 1,4-disubstituted 1,3-enynes. Synthesis 2018, 50, 3307–3321. [Google Scholar] [CrossRef]
- Liu, Y.-W.; Li, L.-J.; Xu, H.; Dai, H.-X. Palladium-catalyzed alkynylation of enones with alkynylsilanes via C−C bond activation. J. Org. Chem. 2022, 87, 6807–6811. [Google Scholar] [CrossRef]
- Shao, L.-X.; Shi, M. Copper-catalyzed coupling reactions of alkenyl halides with alkynes in the absence of palladium and ligand. Tetrahedron 2007, 63, 11938–11942. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, T.; Qu, X.; Sun, P.; Yang, H.; Mao, J. Copper(I)-catalyzed synthesis of 1,3-enynes via coupling between vinyl halides and alkynes or domino coupling of vinyl halides. Org. Biomol. Chem. 2011, 9, 7309–7312. [Google Scholar] [CrossRef]
- Kumar, R.; Zajc, B. Stereoselective synthesis of conjugated fluoro enynes. J. Org. Chem. 2012, 77, 8417–8427. [Google Scholar] [CrossRef]
- Wada, T.; Iwasaki, M.; Kondoh, A.; Yorimitsu, H.; Oshima, K. Palladium-catalyzed addition of silyl-substituted chloroalkynes to terminal alkynes. Chem. Eur. J. 2010, 16, 10671–10674. [Google Scholar] [CrossRef]
- Finkbeiner, P.; Kloeckner, U.; Nachtsheim, B.J. OH-Directed alkynylation of 2-vinylphenols with ethynyl benziodoxolones: A fast access to terminal 1,3-enynes. Angew. Chem. Int. Ed. 2015, 54, 4949–4952. [Google Scholar] [CrossRef]
- Caspers, L.D.; Finkbeiner, P.; Nachtsheim, B.J. Direct electrophilic C–H alkynylation of unprotected 2-vinylanilines. Chem. Eur. J. 2017, 23, 2748–2752. [Google Scholar] [CrossRef]
- Sang, H.L.; Hu, Y.; Ge, S. Cobalt-catalyzed regio- and stereoselective hydrosilylation of 1,3-diynes to access silyl-functionalized 1,3-enynes. Org. Lett. 2019, 21, 5234–5237. [Google Scholar] [CrossRef]
- Cembellín, S.; Dalton, T.; Pinkert, T.; Scha, F.; Glorius, F. Highly selective synthesis of 1,3-enynes, pyrroles, and furans by manganese(I)-catalyzed C−H activation. ACS Catal. 2020, 10, 197–202. [Google Scholar] [CrossRef]
- Guo, L.-Y.; Li, Q.; Liu, Y.-T.; Li, L.; Ni, Y.-Q.; Li, Y.; Pan, F. Palladium-catalyzed alkynylation of alkenes via C–H Activation for the preparation of conjugated 1,3-enynes. Adv. Synth. Catal. 2022, 364, 1109–1116. [Google Scholar] [CrossRef]
- Sang, H.L.; Wu, C.; Phua, G.G.D.; Ge, S. Cobalt-catalyzed regiodivergent stereoselective hydroboration of 1,3-diynes to access boryl-functionalized Enynes. ACS Catal. 2019, 9, 10109–10114. [Google Scholar] [CrossRef]
- Rogalski, S.; Kubicki, M.; Pietraszuk, C. Palladium catalysed regio- and stereoselective synthesis of (E)-4-aryl-1,3-bis(trimethylsilyl)but-3-en-1-ynes. Tetrahedron 2018, 74, 6192–6198. [Google Scholar] [CrossRef]
- Pawluć, P.; Hreczycho, G.; Szudkowska, J.; Kubicki, M.; Marciniec, B. New one-pot synthesis of (E)-β-aryl vinyl halides from styrenes. Org. Lett. 2009, 11, 3390–3393. [Google Scholar] [CrossRef]
- Wang, E.; Fu, X.; Xie, X.; Chen, J.; Gao, H.; Liu, Y. Gold and Bronsted acid catalyzed isomerization of [3]cumulenols and [3]cumulenones: Efficient syntheses of 1,5-dien-3-ynes and furan derivatives. Tetrahedron Lett. 2011, 52, 1968–1972. [Google Scholar] [CrossRef]
- Komiya, S. (Ed.) Synthesis of Organometallic Compounds; John Wiley and Sons: Hoboken, NJ, USA, 1997. [Google Scholar]
- Heck, R.F. Palladium Reagents in Organic Synthesis; Academic: New York, NY, USA, 1985. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
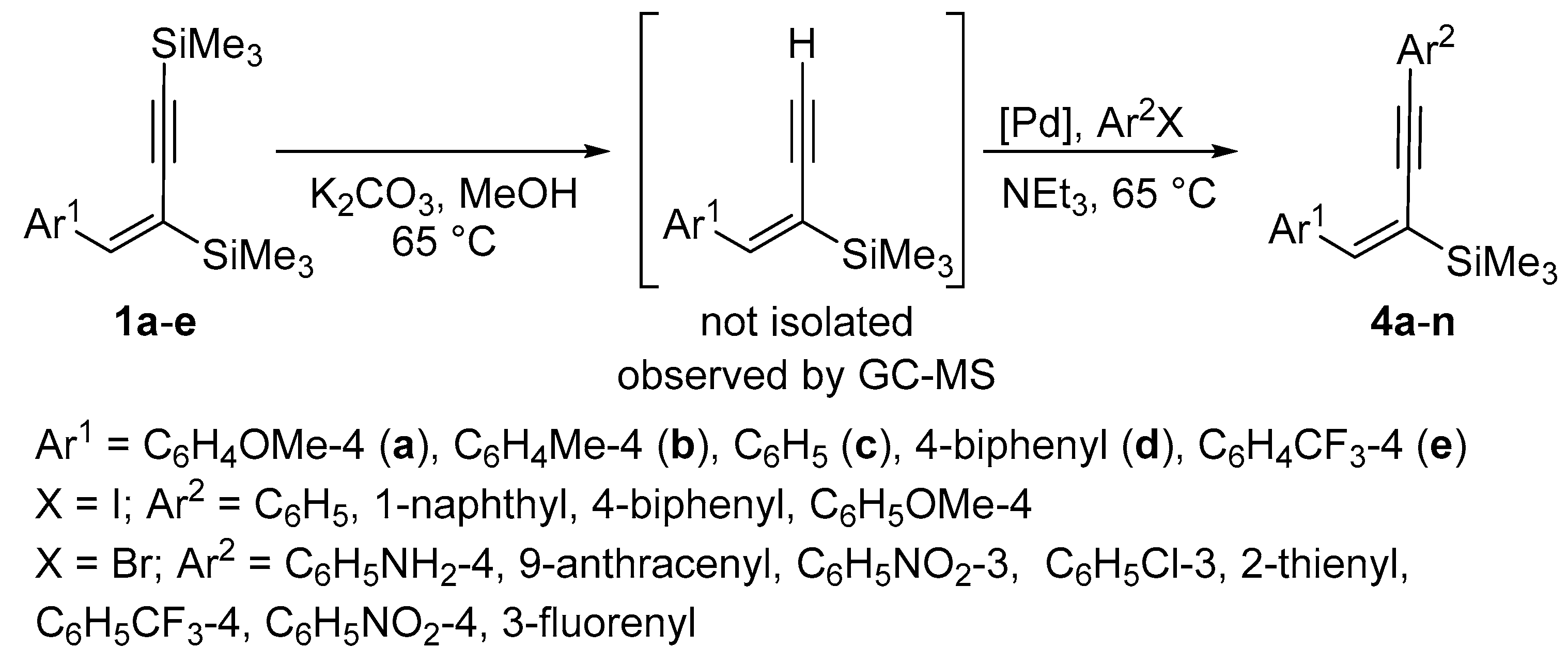{kind=link}
{kind=link}
{kind=link}
| Enyne | Base | Time [h] | Solvent | Conv. | Yield [%] |
|---|---|---|---|---|---|
| 1a | KF | 3 | toluene | 0 | 0 |
| 1a | K2CO3 | 3 | toluene | 0 | 0 |
| 1a | KF | 3 | DMF | 27 | 22 |
| 1a | KF | 3 | THF | 5 | 5 |
| 1a | KF | 3 | MeOH | 100 a | 99 a |
| 1a | CsF | 2.5 | MeOH | 100 | 99 |
| 1a | NaF | 3 | MeOH | 65 | 65 |
| 1a | K2CO3 | 3 | MeOH | 100 a | 99 a |
| 1a | KF | 1 | MeOH | 100 | 99 |
| 1a | TBAF | 1 | MeOH | 100 | 5 |
| 1a | KOt-Bu | 3 | MeOH | 100 | 30 |
| 1a | KOH | 3 | MeOH | 100 | 22 |
| 1a | K2CO3 | 1 | MeOH | 100 | 99 |
| 1b | K2CO3 | 1 | MeOH | 100 | 98 |
| 1c | K2CO3 | 1 | MeOH | 100 | 96 |
| Cat. | Additive (Amount) a | Base | Conv. [%] | Yield [%] |
|---|---|---|---|---|
| [Pd(PPh3)4] | - | KF | 100 | 99 |
| [Pd(PPh3)4] | - | NEt3 | 100 | 98 |
| [Pd(PPh3)4] | CuI (see text) | KF | 100 | 98 |
| [PdCl2(PPh3)2] | - | KF | 100 | 98 |
| [PdCl2(PPh3)2] | - | NEt3 | 100 | 99 |
| [PdCl2(PPh3)2] | - | NEt3 | 98 a | 98 a |
| [PdCl2(PPh3)2] | CuI (see text) | KF | 100 | 98 |
| [PdCl2(PPh3)2] | - | KF | 97 b | 96 b |
| [PdCl2(PPh3)2] | - | K2CO3 | 90 | 90 |
| PEPPSI-IPr | - | KF | 99 | 95 |
| PEPPSI-IPr | - | NEt3 | 99 | 94 |
| [Pd2(dba)3] | PPh3 (2 equiv) | NEt3 | 99 | 95 |
| [Pd2(dba)3] | SPhos (2 equiv) | NEt3 | 100 | 96 |
| PdCl2 | dppf (1 equiv) | KF | 75 | 75 |
| [PdCl2(PhCN)2] | PPh3 (2 equiv) | KF | 87 | 86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogalski, S.; Szymaszek, N.; Pietraszuk, C. The Regio- and Stereoselective Synthesis of 1,4-Diarylbut-1-en-3-ynes Having Aryl Groups at the Mutual Syn Positions. Organics 2023, 4, 206-218. https://doi.org/10.3390/org4020017
Rogalski S, Szymaszek N, Pietraszuk C. The Regio- and Stereoselective Synthesis of 1,4-Diarylbut-1-en-3-ynes Having Aryl Groups at the Mutual Syn Positions. Organics. 2023; 4(2):206-218. https://doi.org/10.3390/org4020017
Chicago/Turabian StyleRogalski, Szymon, Natalia Szymaszek, and Cezary Pietraszuk. 2023. "The Regio- and Stereoselective Synthesis of 1,4-Diarylbut-1-en-3-ynes Having Aryl Groups at the Mutual Syn Positions" Organics 4, no. 2: 206-218. https://doi.org/10.3390/org4020017