
A New Rapid and Specific Iodination Reagent for Phenolic Compounds

Abstract
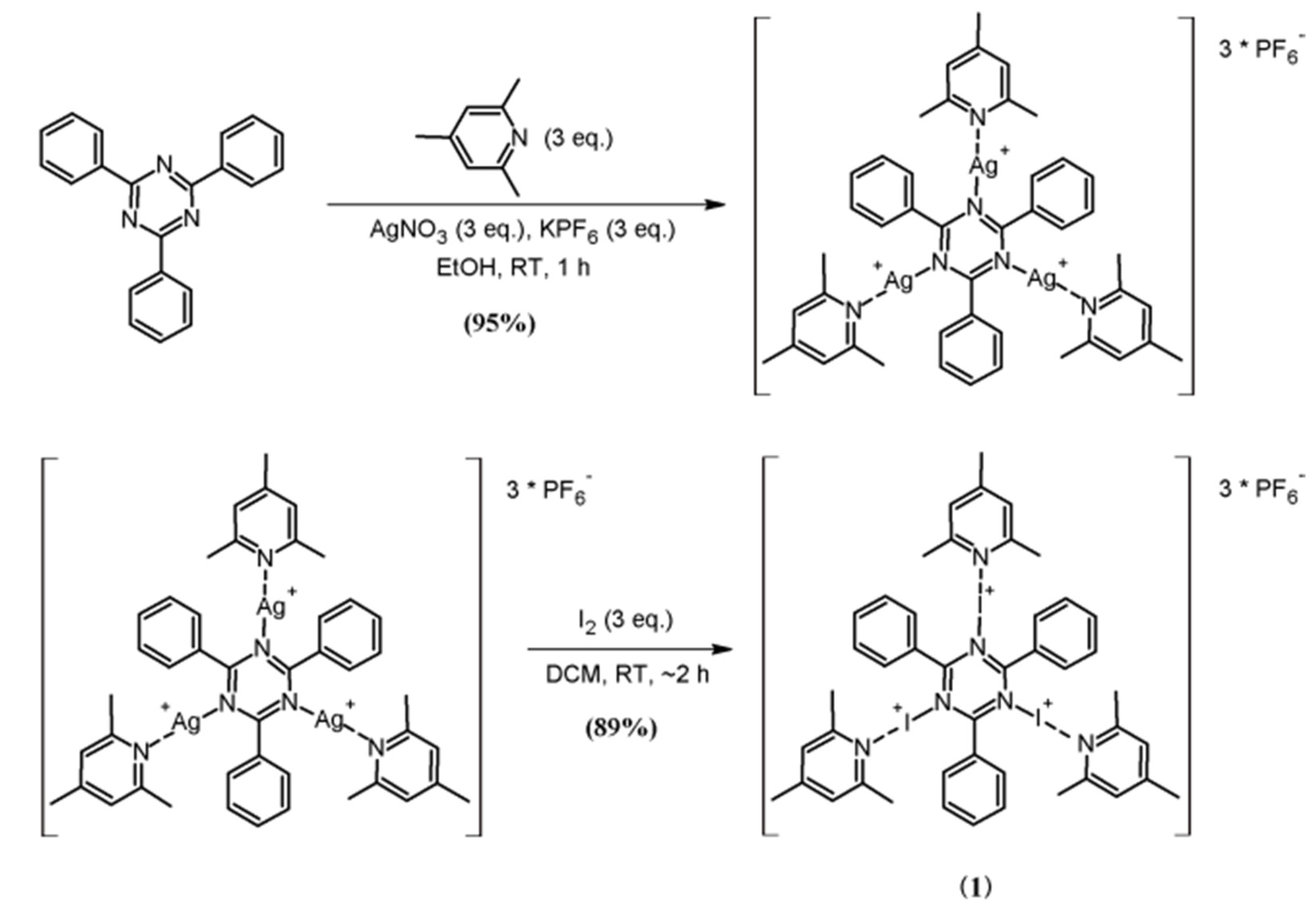
:1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. A New, One-Step, Effective Protocol for the Iodination of Aromatic and Heterocyclic Compounds via Aprotic Diazotization of Amines. Synthesis 2007, 1, 81–84. [Google Scholar] [CrossRef]
- Hubbard, A.; Okazaki, T.; Laali, K.K. Halo- and Azidodediazoniation of Arenediazonium Tetrafluoroborates with Trimethylsilyl Halides and Trimethylsilyl Azide and Sandmeyer-Type Bromodediazoniation with Cu(I)Br in [BMIM][PF6] Ionic Liquid. J. Org. Chem. 2008, 73, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, V.D.; Trusova, M.; Postnikov, P.; Krasnokutskaya, E.A.; Lee, Y.M.; Hwang, H.Y.; Kim, H.; Chi, K.-W. Unusually Stable, Versatile, and Pure Arenediazonium Tosylates: Their Preparation, Structures, and Synthetic Applicability. Org. Lett. 2008, 10, 3961–3964. [Google Scholar] [CrossRef]
- Filimonov, V.D.; Semenischeva, N.I.; Krasnokutskaya, E.A.; Tretyakov, A.N.; Hwang, H.Y.; Chi, K.-W. Sulfonic Acid Based Cation-Exchange Resin: A Novel Proton Source for One-Pot Diazotization-Iodination of Aromatic Amines in Water. Synthesis 2008, 2, 185–187. [Google Scholar] [CrossRef]
- Leas, D.A.; Dong, Y.; Vennerstrom, J.L.; Stack, D.E. One-Pot, Metal-Free Conversion of Anilines to Aryl Bromides and Iodides. Org. Lett. 2017, 19, 2518–2521. [Google Scholar] [CrossRef] [Green Version]
- Hauenschild, T.; Hinderberger, D. A Platform of Phenol-Based Nitroxide Radicals as an “EPR Toolbox” in Supramolecular and Click Chemistry. ChemPlusChem 2019, 84, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Thiebes, C.; Surya Prakash, G.K.; Petasis, N.A.; Olah, G.A. Mild Preparation of Haloarenes by Ipso-Substitution of Arylboronic Acids with N-Halosuccinimides. Synlett 1998, 2, 141–142. [Google Scholar] [CrossRef]
- MacNeil, S.L.; Familoni, O.B.; Snieckus, V. Selective Ortho and Benzylic Functionalization of Secondary and Tertiary p-Tolylsulfonamides. Ipso-Bromo Desilylation and Suzuki Cross-Coupling Reactions. J. Org. Chem. 2001, 66, 3662–3670. [Google Scholar] [CrossRef]
- Klapars, A.; Buchwald, S.L. Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction. J. Am. Chem. Soc. 2002, 124, 14844–14845. [Google Scholar] [CrossRef]
- Stavber, S.; Kralj, P.; Zupan, M. Progressive Direct Iodination of Sterically Hindered Alkyl Substituted Benzenes. Synthesis 2002, 11, 1513–1518. [Google Scholar] [CrossRef]
- Castanet, A.-S.; Colobert, F.; Broutin, P.-E. Mild and regioselective iodination of electron-rich aromatics with N-iodosuccinimide and catalytic trifluoroacetic acid. Tetrahedron Lett. 2002, 43, 5047–5048. [Google Scholar] [CrossRef]
- Lulinski, P.; Kryska, A.; Sosnowski, M.; Skulski, L. Eco-friendly Oxidative Iodination of Various Arenes with a Urea-Hydrogen Peroxide Adduct (UHP) as the Oxidant [1]. Synthesis 2004, 3, 441–445. [Google Scholar] [CrossRef]
- Iskra, J.; Stavber, S.; Zupan, M. Nonmetal-Catalyzed Iodination of Arenes with Iodide and Hydrogen Peroxide. Synthesis 2004, 11, 1869–1873. [Google Scholar] [CrossRef] [Green Version]
- Prakash, G.K.S.; Mathew, T.; Hoole, D.; Esteves, P.M.; Wang, Q.; Rasul, G.; Olah, G.A. N-Halosuccinimide/BF3−H2O, Efficient Electrophilic Halogenating Systems for Aromatics. J. Am. Chem. Soc. 2004, 126, 15770–15776. [Google Scholar] [CrossRef]
- Sedelmeier, J.; Bolm, C. Efficient Copper-Catalyzed N-Arylation of Sulfoximines with Aryl Iodides and Aryl Bromides. J. Org. Chem. 2005, 70, 6904–6906. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, D.; Dick, A.R.; Anani, W.Q.; Sanford, M.S. A Simple Catalytic Method for the Regioselective Halogenation of Arenes. Org. Lett. 2006, 8, 2523–2526. [Google Scholar] [CrossRef]
- Kraszkiewicz, L.; Sosnowski, M.; Skulski, L. Oxidative Iodination of Deactivated Arenes in Concentrated Sulfuric Acid with I2/NaIO4 and KI/NaIO4 Iodinating Systems. Synthesis 2006, 7, 1195–1199. [Google Scholar]
- Ganguly, N.C.; Barik, S.K.; Dutta, S. Ecofriendly Iodination of Activated Aromatics and Coumarins Using Potassium Iodide and Ammonium Peroxodisulfate. Synthesis 2010, 9, 1467–1472. [Google Scholar] [CrossRef]
- Qiu, D.; Mo, F.; Zheng, Z.; Zhang, Y.; Wang, J. Gold(III)-Catalyzed Halogenation of Aromatic Boronates with N-Halosuccinimides. Org. Lett. 2010, 12, 5474–5477. [Google Scholar] [CrossRef]
- Gallo, R.D.C.; Gebara, K.S.; Muzzi, R.M.; Raminelli, C. Efficient and selective iodination of phenols promoted by iodine and hydrogen peroxide in water. Braz. J. Chem. Soc. 2010, 21, 770–774. [Google Scholar] [CrossRef]
- Rodríguez-Lojo, D.; Cobas, A.; Peña, D.; Pérez, D.; Guitián, E. Aryne Insertion into I–I σ-Bonds. Org. Lett. 2012, 14, 1363–1365. [Google Scholar] [CrossRef]
- Jakab, G.; Hosseini, A.; Hausmann, H.; Schreiner, P.R. Mild and Selective Organocatalytic Iodination of Activated Aromatic Compounds. Synthesis 2013, 45, 1635–1640. [Google Scholar]
- Du, B.; Jiang, X.; Sun, P. Palladium-Catalyzed Highly Selective ortho-Halogenation (I, Br, Cl) of Arylnitriles via sp2 C–H Bond Activation Using Cyano as Directing Group. J. Org. Chem. 2013, 78, 2786–2791. [Google Scholar] [CrossRef]
- Partridge, B.M.; Hartwig, J.F. Sterically Controlled Iodination of Arenes via Iridium-Catalyzed C–H Borylation. Org. Lett. 2013, 15, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Zhang, H.; Yang, H.; Fu, H. Metal-Free Iodination of Arylboronic Acids and the Synthesis of Biaryl Derivatives. Synlett 2014, 25, 995–1000. [Google Scholar]
- Leboeuf, D.; Ciesielsk, J.; Frontier, A.J. Gold(I)-Catalyzed Iodination of Arenes. Synlett 2014, 25, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Sun, X.; Li, X.; Yuan, Y.; Jiao, N. Efficient and Practical Oxidative Bromination and Iodination of Arenes and Heteroarenes with DMSO and Hydrogen Halide: A Mild Protocol for Late-Stage Functionalization. Org. Lett. 2015, 17, 2886–2889. [Google Scholar] [CrossRef]
- Chen, J.; Xiong, X.; Chen, Z.; Huang, J. Imidazolium Salt Catalyzed para-Selective Halogenation of Electron-Rich Arenes. Synlett 2015, 26, 2831–2834. [Google Scholar]
- Li, L.; Liu, W.; Zeng, H.; Mu, X.; Cosa, G.; Mi, Z.; Li, C.-J. Photo-induced Metal-Catalyst-Free Aromatic Finkelstein Reaction. J. Am. Chem. Soc. 2015, 137, 8328–8331. [Google Scholar] [CrossRef] [Green Version]
- Racys, D.T.; Warrilow, C.E.; Pimlott, S.L.; Sutherland, A. Highly Regioselective Iodination of Arenes via Iron(III)-Catalyzed Activation of N-Iodosuccinimide. Org. Lett. 2015, 17, 4782–4785. [Google Scholar]
- Racys, D.T.; Sharif, S.A.I.; Pimlott, S.L.; Sutherland, A. Silver(I)-Catalyzed Iodination of Arenes: Tuning the Lewis Acidity of N-Iodosuccinimide Activation. J. Org. Chem. 2016, 81, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Li, Z.; Song, Y.; Yang, R.; Liu, Y.; Cai, H. Decarboxylative Halogenation and Cyanation of Electron-Deficient Aryl Carboxylic Acids via Cu Mediator as Well as Electron-Rich Ones through Pd Catalyst under Aerobic Conditions. J. Org. Chem. 2016, 81, 2794–2803. [Google Scholar] [CrossRef]
- Ajvazi, N.; Stavber, S. Electrophilic Iodination of Organic Compounds Using Elemental Iodine or Iodides: Recent Advances 2008–2021: Part I. Compounds 2022, 2, 3–24. [Google Scholar] [CrossRef]
- Brunei, Y.; Rousseau, G. Iodination of phenols and anilines with bis(sym-collidine)iodine(I) hexafluorophosphate. Tetrahedron Lett. 1995, 36, 8217–8220. [Google Scholar] [CrossRef]
- Homsi, F.; Robin, S.; Rousseau, G.; Patterson, S.; Hart, D.J. Preparation of Bis(2,4,6-Trimethylpyridine)Iodine(I) Hexafluorophosphate and Bis(2,4,6-Trimethylpyridine) Bromine(I) Hexafluorophosphate. Org. Synth. 2000, 77, 206–211. [Google Scholar]
- Rosenthaler, L.; Capuano, L. Process for preparing and therapeutical applications of the 2,4,6-triiodophenol. Pharm. Acta Helv. 1946, 21, 225–228. [Google Scholar] [PubMed]
- Emmanuvel, L.; Shukla, R.K.; Sudalai, A.; Gurunath, S.; Sivaram, S. NaIO4/KI/NaCl: A new reagent system for iodination of activated aromatics through in situ generation of iodine monochloride. Tetrahedron Lett. 2006, 47, 4793–4796. [Google Scholar] [CrossRef]
- Hauenschild, T.; Reichenwallner, J.; Enkelmann, V.; Hinderberger, D. Characterizing Active Pharmaceutical Ingredient Binding to Human Serum Albumin by Spin-Labeling and EPR Spectroscopy. Chem. Eur. J. 2016, 22, 12825–12838. [Google Scholar] [CrossRef]
- Sultani, H.N.; Haeri, H.H.; Hinderberger, D.; Westermann, B. Spin-labelled diketopiperazines and peptide–peptoid chimera by Ugi-multi-component-reactions. Org. Biomol. Chem. 2016, 14, 11336–11341. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Entry | Substrate | Reaction Conditions a (Reaction Time (min); eq. of FIC*17*) | Product | Yield b (%) |
|---|---|---|---|---|
| 1 |  | (5; 1) |  | 98 |
| 2 |  | (10; 1) |  | 99 |
| 3 |  | (5; 1) |  | 95 |
| 4 |  | (10; 1) |  | 96 |
| 5 |  | (5; 1) |  | 95 |
| 6 |  | (10; 1) |  | 95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hauenschild, T.; Hinderberger, D. A New Rapid and Specific Iodination Reagent for Phenolic Compounds. Organics 2023, 4, 137-145. https://doi.org/10.3390/org4020011
Hauenschild T, Hinderberger D. A New Rapid and Specific Iodination Reagent for Phenolic Compounds. Organics. 2023; 4(2):137-145. https://doi.org/10.3390/org4020011
Chicago/Turabian StyleHauenschild, Till, and Dariush Hinderberger. 2023. "A New Rapid and Specific Iodination Reagent for Phenolic Compounds" Organics 4, no. 2: 137-145. https://doi.org/10.3390/org4020011