
Total Syntheses of Chloropupukeananin and Its Related Natural Products

Department of Chemistry, Hokkaido University, Sapporo 060-0810, Hokkaido, Japan
Organics 2022, 3(3), 304-319; https://doi.org/10.3390/org3030023
Submission received: 15 July 2022
/
Revised: 22 August 2022
/
Accepted: 5 September 2022
/
Published: 9 September 2022
(This article belongs to the Special Issue New Reactions and Strategies for Natural Product Synthesis)
Abstract
:Chloropupukeananin is a natural product that inhibits HIV-1 replication and has antitumor activity. Its structure consists of a chlorinated tricyclo[4.3.1.03,7]decane core skeleton with an array of highly oxidized multifunctional groups. In the biosynthesis of chloropupukeananin, (+)-iso-A82775C and (−)-maldoxin are employed as biosynthetic precursors for the intermolecular Diels–Alder and carbonyl–ene reactions, followed by the migration of the p-orcellinate group. Chloropupukeanolides and chloropestolides are intermediates and isomers in biosynthesis; their unique chemical structures and biosynthetic pathways have attracted significant attention from synthetic chemists. In this review, I present the synthetic studies on chloropupukeananin and its related compounds that have been conducted thus far.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Chloropupukeananin (1) was originally isolated in 2008 by Che et al. from Pestalotiopsis fici (an endophytic plant fungus) as an antimicrobial agent and inhibitor of HIV-1 replication (Figure 1) [1]. Simultaneously, iso-A82775C (2) and pestheic acid [2,3,4] (3) were also isolated and proposed as biosynthetic precursors of 1 (Scheme 1). Structurally, chloropupkeananin possesses a highly functionalized pupukeanane skeleton [5,6,7,8,9]. Significantly, both 1 and pupukeanane share a common skeleton despite possessing no biosynthetic relationship, as pupukeananes, which are marine sesquiterpenes from sponges, have exclusively been isolated with a single functional group.
The isolation of chloropestolide A (5) [10] from the same fermentation medium as 1 in 2009 led Suzuki and Kobayashi to propose that the actual biosynthetic precursors of these compounds are 2 and maldoxin (4) [4], which is an oxidized form of 3 [11]. Although 4 had already been isolated without the determination of its optical rotation and stereochemistry, [4] (R)-4 was assumed to be generated by asymmetric oxidative dearomatization in the biosynthesis of 1. The subsequent biosynthetic pathway is as follows: the intermolecular reverse electron-demanding Diels–Alder reaction of 2 and 4 gives cycloadduct 5 and its isomers 6–8, possessing a bicyclo[2.2.2]octane skeleton. Moreover, the normal-electron Diels–Alder reaction, using the vinylallene moiety of 2 as a diene, generates 9 and 10. Intramolecular carbonyl–ene reactions between the C5 position in the allene moiety derived from 2 and the C6 ketone derived from 4 afford 11 and 12 with tricyclo [4.3.1.03,7]decane skeletons from 7 and 8, respectively. Finally, the p-orsellinate moiety of 11 migrates to C18-OH to give 1. In the continuing efforts by Che et al. to elucidate the biosynthetic pathway of 1 [12,13,14,15,16], all the aforementioned biosynthetic intermediates 6–12 (chloropestolides B-F and chloropupukenolides C and D) were isolated along with other degradation products 13–17. Notably, optically active (R)-4 was isolated from the related fungus, P. theae, [15] along with chlorotheolides A (18) and B (19) and 1-undecene-2,3-dicarboxylic acid (20), which is a proposed biosynthetic precursor of 18 and 19 (Scheme 2). To identify a biosynthetic gene cluster in chloropupukeananin-producing bacteria, chloropestolides H-K [16] (21–24), which may be produced from 4 and siccayne 25, were isolated using a prenyltransferase gene disruption strain.
The face selectivity of the intermolecular Diels–Alder reactions in biosynthesis requires further discussion. The isolation of chloropestolides (5–8, 21–24) demonstrates the occurrence of all possible isomers in reverse electron-requested Diels–Alder reaction (Scheme 3). In the formation of these bicyclo [2.2.2]octane-containing cycloadducts, the dienophiles approach the Re- or Si-plane of C4’ of diene 4 in syn- or anti-orientation between the R group of the dienophiles (2 or 25) and the C2 acetal site of diene 4. While the usual enzymatic reaction occurs selectively in nature, the current intermolecular Diels–Alder reaction of 4 displays poor selectivity. Therefore, enzymes may not be involved in the Diels–Alder reaction of 4. However, it is reasonable to assume that enzymes are involved elsewhere in the series of biosynthetic reactions because the reaction sites are well controlled despite the presence of several functional groups. The chemical synthesis of the chloropupukeananin family via the Diels–Alder reactions of 4 and biosynthetic dienophiles helps elucidate the detailed mechanism of biosynthesis, especially the occurrence of an enzymatic Diels–Alder reaction. If the enzymatic Diels–Alder reaction does occur, this enzyme is the first example of an intermolecular Diels–Alderase that constructs a highly functional bicyclo [2.2.2]octane skeleton [17,18,19,20]. Because of the high synthetic convergence and the broad diversity of products in the intermolecular Diels–Alder reaction, artificial Diels–Alderases such as antibody catalysts [21], artificial enzymes [22], and supramolecules [23] have been developed. Recently, enzymes promoting the intermolecular Diels–Alder reactions to produce pseudo-dimeric resveratrols have been identified [24]. The identification of an intermolecular heterodimeric Diels–Alderase is expected to pave the way for the production of a variety of bioactive natural product-like compounds through its genetic modification [25].
Therefore, chloropupukeananin has attracted significant research interest, and various studies have been conducted to elucidate its biosynthesis. In this review, reports on chloropupukeananin in the context of synthetic chemistry, the synthesis of biosynthetic precursors, the Diels–Alder reaction mimicking biosynthesis, and the total synthesis of chloropupukeananin, are presented.
2. Synthesis of the Biosynthetic Precursors of Chloropupukeananin
2.1. Pestheic Acid and Maldoxin
Pestheic acid (3) [2], also known as RES-1214-2 [3] and dihydromaldoxin [4], is a metabolite of chlorinated lichexanthone derivatives, such as chloroisosulochin (26) and chloroisosulochin dehydrate (27) [2], and it consists of p-orsellinate and methyl p-chlorobenzoate moieties (Figure 2). Maldoxin (4) was isolated from Xylaria species, which was collected from a Malaysian rain forest by Edwards et al., along with 3 and maldoxone (28) [4]. Similar to the biosynthesis of nidulin, [26,27] 3 and 28 can be generated by the oxidative dearomatization of 26, followed by the hydrolysis of the resulting spiroketone 29. The re-dearomatization of the methyl p-chlorobenzoate moiety of 3 produces 4. These compounds exhibit biological activities; in particular, 3 is a promising selective endothelin A receptor antagonist.
In 2012, Yu and Snyder reported the first total synthesis of pestheic acid and maldoxin based on a biosynthetic pathway (Scheme 4) [28]. The synthesis was initiated with a known six-step conversion of methyl 3,5-dihydroxybenzoate to salicylaldehyde 30. Site-selective methylation followed by chlorination [29,30,31] using SO2Cl2 and 2,2,6,6-tetramethylpiperidine (TMP) afforded 3-chlorosalicyladehyde 31 (58% yield) and 5-chloride (15% yield). The use of a bulkier amine is essential because an undesired 5-chlorination was preferred when using t-butylamine (3-Cl:5-Cl = 1:2.6). After the MOM protection, the lithiated orcinol derivative 33 [32] was added to aldehyde 32 to produce alcohol 34 in 92% yield. Oxidation with Dess–Martin periodinane (DMP) gave benzophenone 35 in 90% yield. Over-reduction during the reductive removal of the benzyl group caused several issues. Various catalysts and reaction times were investigated, and reduction using the Rosenmund catalyst selectively afforded alcohol 36. The resulting primary alcohol was converted to methyl ester 37 via a conventional three-step transformation. The removal of the MOM groups using TsOH produced chloroisosulochin 26. Oxidation with K3Fe(CN)6 [33,34,35] in H2O followed by continuous acidic hydrolysis furnished maldoxone 28. The basic hydrolysis of 28 achieved a total synthesis of 3. The oxidative dearomatization of 3 with iodobenzene diacetate (PIDA) afforded racemic 4 in 31% yield. The observed melting point of synthetic 4 is 193 °C, while that of natural 4 reported in the literature is 143 °C [4], indicating that the natural maldoxin is not racemic.
The asymmetric synthesis of (R)-4 is essential for the total synthesis of the natural products in the chloropupukeananin family. In addition, an efficient and robust synthetic strategy to supply large quantities of 3 and 4 is required. In 2018, Suzuki et al. reported the asymmetric total synthesis of (−)-(R)-4 via the intramolecular SNAr etherification and asymmetric oxidative dearomatization of 3 using the Ishihara catalyst (Scheme 5) [36]. Herein, the right-hand benzoate moiety 40 was synthesized from 5-methoxysalicylic acid. After a three-step conversion to phenol 38 [37], chlorination with sodium hypochlorite proceeded nonselectively to afford a 1:1 mixture of 4-Cl and 6-Cl. Subsequent acidic esterification produced the 4-chlorobenzoate 39 (40% yield in two steps) [11]. The site-selective removal of the methyl group using AlCl3 achieved the synthesis of the right-hand moiety 40. As shown in Snyder’s synthesis, the nonselectivity of the chlorination of these compounds is a fundamental issue that may necessitate alternative functionalization reactions. The alternative synthesis [38] was initiated with commercial 2-chloro-1,4-dimethoxybenzene, which was converted to methyl 4-chloro-2,5-dimethoxybenzoate 42 by three-step sequence (formylation, Pinnick oxidation, and acidic esterification). The oxidative nucleophilic substitution reaction [39,40] using PIDA in trifluoroacetic acid (TFA)/AcOH mixed solvent occurred selectively at the C3 position (C3/C6 = 6:1) to give 43, which was directly subjected to basic hydrolysis to produce phenol 39 in 68% yield.
The left fragment 44 was prepared using two-step transformation: the lithiation of 3,5-difluorotoluene followed by carboxylation with CO2 (64% yield) [41] and intermolecular SNAr etherification of the resulting benzoic acid using KOBn (quantitative yield). The preparation of acyl chloride from the left-hand fragment 44 and the subsequent regioselective esterification with the right-hand fragment 40 afforded benzoate ester 45 in 79% yield. The intramolecular SNAr reaction [42,43] was initiated by the treatment of benzoate 45 with Cs2CO3 (0.05 M, 80 °C) in DMSO, and the one-pot acid hydrolysis of the resulting 7-membered lactone 46 produced diaryl ether 47 in 51% yield. The synthesis of pestheic acid 3 was achieved by removing the Bn group of diaryl ether 47. The asymmetric oxidative dearomatization [44,45,46] of 3 to maldoxin 4 using Ishihara catalysts has been thoroughly investigated. As a result, the optically pure (−)-(R)-4 was successfully obtained with 93% yield using the (2R,2′R)-Ishihara catalyst (10 mol%), mCPBA (1.5 equiv.) as the co-oxidant, methanol (20 equiv.) as an additive, and chloroform (0.01 M) as the solvent [47,48,49]. The stereochemistry of (−)-(R)-4 was confirmed by X-ray crystallographic analysis. The levorotation of synthetic (R)-4 indicates that natural 4 isolated from P. theae possesses the R-configuration. Moreover, as the melting point of synthetic 4 is 142–143 °C, the originally isolated natural 4 was likely to be optically pure.
2.2. Iso-A82775C
As the name indicates, iso-A82775C is a diastereomer of the known natural product A82775C 48 [50] (Figure 3), isolated from an unknown terrestrial fungus collected in Egypt. Another natural diastereomer is Spartinoxide [51] (49), which is an enantiomer of 48, isolated from a marine-derived fungus and identified as an inhibitor of human leukemia elastase. These compounds belong to a class of naturally occurring cyclohexene epoxides. Typically, these compounds (such as eutypoxides, asperpentyne, harveynone, panepoxydone, and isopanepoxydone) possess one prenyl chain; cyclohexene epoxides with two prenyl side chains are rare. To the best of our knowledge, only two natural products (other than compounds 2, 48, and 49) have been reported: pestalofone A (50) [52] and biscogniene B (51) [53]. Significantly, one of the prenyl units of these compounds is oxidized and rich in sp2 carbon. These oxidized prenyl side chains can undergo dimerization, forming a variety of natural products; for instance, pestalofones B and C have been isolated as dimeric natural products of iso-A82775C. Interest in the biological activity, as well as the biosynthetic pathway of these dimers, prompted investigations into the total synthesis of naturally occurring cyclohexene epoxides with prenyl side chains.
Suzuki et al. reported the enantioselective total synthesis of (+)-iso-A82775C in 2017, [54] which is the first report of the synthesis of naturally occurring cyclohexene epoxides with two prenyl side chains. The synthetic strategy is illustrated in Scheme 6. The installation of the labile axially chiral vinylallene to 52 would be achieved in the final stage of the total synthesis using a 2-propenyl metal reagent via an anti-SN2′ reaction. The stereoselective epoxidation of 53 and Pd-catalyzed prenylation would envision vinyl bromide 54. Further, optically active 54 could be obtained via the base-catalyzed asymmetric Diels–Alder reaction reported by Okamura et al. [55,56,57,58] using pyrone 55 and 2-chloroacrylate 56. First, 55 was prepared on a decagram scale from mucic acid using a modified two-step procedure [59,60]. By investigating the intermolecular Diels–Alder reaction using various cinchona alkaloids, the optimal result was obtained using 0.1 equiv of cinchonine in toluene at 0 °C to produce the desired endo cycloadduct 54 with 67% ee (endo:exo = 3.6:1). Recrystallization of 54 (67% ee) from EtOAc/n-hexane gave enantiomerically pure crystalline (−)-54 (>99% ee, 42% yield from pyrone 55), and the absolute stereochemistry of the product was determined via X-ray crystallographic analysis. Chemoselective reduction of the ester with LiBH4, the protection of the resulting alcohol, and the reduction of the lactone moiety with DIBAL afforded α-hydroxylactol 57. The Criegee oxidation of α-hydroxylactol 57 furnished α-bromoenone 58 in 43% overall yield from 54. After the protection of the secondary alcohol, removal of the TES group, and one-pot hydroxyl group-induced reduction using NaBH(OAc)3, 1,3-diol 59 was obtained as a single diastereomer [61]. Pd-coupling precursor 53 was obtained via the TES protection of 1,3-diol 59 in quantitative yield. Pd coupling reactions with various allyl metal reagents were conducted, but allylation occurred only under standard Stille conditions, quantitatively producing 60. Stille coupling with prenylstannane reagents was unsuccessful, and prenylcyclohexene 61 was obtained only in trace amounts. However, the cross-metathesis of 60 with 2-methylbut-2-ene furnished prenylcyclohexene 61 in 91% yield. Selective deprotection, the Dess–Martin oxidation of the resulting primary alcohol, and subsequent Seyferth–Gilbert homologation gave propargylic chloride 62. After the removal of the silyl-protecting groups of alkyne 62, vanadium-catalyzed hydroxyl-directed epoxidation [62], and re-protection of the resulting diol, epoxide 52 was obtained as a single diastereomer. An anti-SN2′ reaction was first conducted with CuCN and isopropenyl-MgBr to afford the corresponding vinylallene as a single diastereomer; however, the conversion was low (~30%), probably because of the competitive deprotonation of the terminal alkyne. In contrast, the use of an organoindium reagent in the presence of a Pd-catalyst [63] resulted in the consumption of all the starting materials, and subsequent deprotection of the TES groups of the resulting allene gave (+)-2 in 92% yield on a subgram scale.
The concise total synthesis of rac-51, which is a naturally occurring cyclohexene epoxide with two prenyl side chains, was achieved by Han et al. in 2018 (Scheme 7) [64], along with the enantioselective total synthesis of (−)-51 and its dimeric congener via biomimetic heterodimerization. The synthesis started with Mehta’s four-step procedure [65] to convert p-methoxyphenol to tricyclic diketone 63. The stereoselective reduction of the less hindered ketone [66] followed by the retro-Diels–Alder reaction afforded prenylated epoxyenone 64. The following three-step conversion (α-iodination of enone, Luche reduction, and Stille coupling with alkynylstannane 66) completed the synthesis of 51. Similarly, Han et al. reported the total synthesis of (+)-1 and (+)-50 using a common synthetic intermediate, rac-64 [67]. The Mitsunobu reaction of rac-64 with O-methyl-D-mandelic acid furnished ester 67 and its diastereomer, which gave pure 67 in 40% yield on chromatographic separation. The removal of the O-methylmandelate group via methanolysis produced prenylated epoxyenone 68, which is an epimer of 64, with 97.5% ee. The protection of the secondary alcohol with a TBS group and α-iodination of the enone moiety furnished iodide 69. The installation of the other prenyl unit via Stille coupling with alkynylstannane 66 was successful, following a procedure similar to the synthesis of 51. Attempts to reduce enone 69 resulted in a 1,2-reduction instead of the desired 1,4-reduction, owing to conjugation with the alkyne moiety. Therefore, after converting the alkyne to dicobalt complex 71, 1,4-reduction using K-selectride and the oxidative decobaltation of the resulting ketone with CAN successfully afforded β,γ-ynone 72. The tautomerization of 72 was achieved by treatment with a catalytic amount of triethylamine [68], resulting in the desired axially chiral vinylallene 73 as a single diastereomer (40% yield, in three steps). The isomerization reaction proceeded in a 3:1 diastereomeric ratio, but the minor product was labile with an affinity for dimerization reaction at room temperature. The 1,2-reduction of ketone 73 occurred diastereoselectively with LiBHEt3 to produce alcohol 74. Desilylation with TBAF achieved the total synthesis of (+)-iso-A82775C (2) (66% yield, over two steps). The authors investigated the dimerization reactions of the synthetic iso-A82775C and its 16-oxo derivative and found that, in contrast to biscognienyne B, these compounds did not undergo dimerization under any condition.
3. Biomimetic Synthesis of Chloropupukeananin
3.1. Model Studies on the Intermolecular Diels–Alder Reaction
Synthetic studies using model compounds of biosynthetic precursors to elucidate the biosynthetic pathway of chloropupukeananin (Scheme 8) were independently conducted by Suzuki et al. and Yu and Snyder. Suzuki and Kobayashi reported the biomimetic Diels–Alder reaction with the simple model compounds 74 and 75 (both achiral compounds) in 2010 [11]. With these substrates, the Diels–Alder reaction barely occurred under heating and Lewis acid conditions, resulting in low yields of cycloadducts. Regarding selectivity, the ratio of syn- and anti-cycloadducts 76 and 77 (corresponding to chloropupukeananin and chloropestolide A, respectively) was 1:3, and a small amount of normal electron-demand (NED) cycloadduct 78 was obtained. The reaction under high-pressure conditions improved the yield and selectivity and produced a mixture of 76 and 77 (76:77 = 1:1.6) in 70% yield.
Yu and Synder in 2011 reported the Diels–Alder reaction using the synthetic racemic 4 and the same achiral vinylallene 74 under the thermal conditions (75 °C, 24 h) [69]. This cycloaddition reaction displayed a selectivity of nearly 1:1:1 for Si-syn, Si-anti, and NED, giving tricyclic cycloadduct 80 (corresponding to chloropupukeanolide D) in 22% yield. This study reveals that the cyclic p-orsellinate moiety accelerates the intermolecular Diels–Alder reaction and completely controls the facial selectivity from the Si face.
In 2013, Suzuki et al. reported synthetic studies with advanced model compounds 83 and 84, cyclohexane possessing a vinylallene and its adjacent hydroxyl group as a model compound for Iso-A82775C, and a compound simplifying to a salicylate moiety instead of the p-orsellinate moiety of maldoxin, respectively [70]. Initially, using both racemic model compounds, the Diels–Alder reaction under high-pressure conditions (1.0 GPa, 96 h) afforded tricyclic compound 85 in 48% yield, and its structure was unambiguously confirmed via X-ray crystallographic analysis. However, the stereochemistry of the other products 86 and 87 (two anti- and one NED cycloadduct) could not be identified using NMR studies. Using optically pure model compounds (+)-83 and (−)-84 (a natural combination), the intermolecular Diels–Alder reaction furnished tricyclic compound 85 in 70% yield, along with anti-cycloadduct 86 in 20% yield. These results indicate that the hydroxyl group is important for Si-syn selectivity, probably because of the hydrogen bonding with the carbonyl groups of the maldoxin unit.
Furthermore, Suzuki et al. studied the thermal intermolecular Diels–Alder reaction of (−)-4 with typical alkenes 88a–c to acquire the trends of the facial selectivity of 4 [36]. In the case of ethyl vinyl ether 88a and styrene 88b, the reactions occurred at room temperature and favored Si-anti cycloadducts 90. However, in the case of methyl acrylate 88c, the reaction required heating to 80 °C and showed a slightly lower Si-anti selectivity.
3.2. Syntheses of Chloropupukeananin and Its Related Natural Products
The total syntheses of the optically pure biosynthetic precursors (+)-2 and (−)-4 enabled the synthesis of chloropupukeananin via the intermolecular Diels–Alder reaction (Scheme 9) [38]. First, the reaction between (+)-2 and (−)-4 was performed under high-pressure conditions. The intermolecular Diels–Alder reaction and the subsequent carbonyl-ene reaction achieved a near-completion after 64 h, producing the desired Si-syn cycloadduct 7 (5%) and carbonyl-ene product 11 (71%), along with Si-anti cycloadduct 6 (17%). Similar to previous studies using enantiopure model compounds, no other cycloadducts were detected. To complete the intramolecular carbonyl-ene reaction, the products of the high-pressure reaction were heated to 60 °C at the atmospheric pressure, furnishing 11 and 6 in 69% and 21% isolated yields, respectively. The thermal conditions required for the Diels–Alder/carbonyl–ene cascade reactions at atmospheric pressure were also investigated. The intermolecular Diels–Alder reaction between (+)-2 and (−)-4 was performed under neat conditions (25 °C, 120 h). After the completion of the Diels–Alder reaction, the mixture was heated (60 °C, 68 h) to afford the target compounds 11 (57% yield) and 6 (25% yield).
The migration of the p-orsellinate group of 11, which was the final step in the biosynthetic pathway of chloropupukeananin, was conducted under basic conditions (Scheme 10). Migration was accomplished by the nucleophilic attack on C26 in the p-orsellinate group by the secondary alkoxide moiety at the C18 position generated from 11 using a strong base. This was followed by the elimination of the p-orsellinate group from the tetrahedral intermediate. The total synthesis of 1 was achieved by the treatment of 11 with KOt-Bu in DMF.
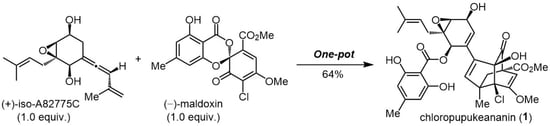
Further, the one-pot biomimetic transformation of (+)-2 and (−)-4 to (+)-1 was accomplished as part of an alternative synthetic approach (Scheme 11); this was easily achieved because the Diels–Alder/carbonyl–ene cascade reaction did not require any reagents or solvents. The cascade reaction at atmospheric pressure (neat, 5 °C, 7 days; thereafter, 60 °C, 9 h) and subsequent migration reaction of the p-orsellinate group (KOt-Bu, DMF) provided (+)-1 in 64% yield, along with 6 in 20% yield.
The synthesis of chloropestolides H-K (21–24) via the intermolecular Diels–Alder reaction of siccayne [71] (25) and (−)-4 was also achieved by Suzuki et al. (Scheme 12) [38]. The intermolecular Diels–Alder reaction using common organic solvents was studied, and the reaction in CH2Cl2 produced a quantitative mixture of the four cycloadducts 21–24 (21:22:23:24 = 14:44:36:6). As expected, owing to the nature of (−)-4, a preference for the Si face was observed, and 22 and 23 were isolated with 39% and 34% yields, respectively. The use of other solvents afforded a mixture predominantly comprising Si-anti 23. Significantly, the ratio of cycloadducts, particularly 22:23, depended on the solvent basicity (SB), [72,73] with the ratio of 23 to 22 increasing as the SB value increased. In a solvent-free reaction, a mixture of 25 and (−)-4 was maintained undisturbed at room temperature for 24 h to complete the Diels–Alder reaction, affording a mixture of 21–24 (21:22:23:24 = 9:32:51:8). A high-pressure reaction in CH2Cl2 at 1.0 GPa for 1 h provided a mixture of the same ratio as that in the reaction under atmospheric pressure conditions (21:22:23:24 = 15:46:33:6). Under high-pressure conditions, the intermolecular Diels–Alder reaction between 25 and (−)-4 was significantly accelerated, but the facial selectivity remained unaffected.
4. Conclusions
This review outlines synthetic studies on the natural product chloropupukeananin and its analogs. Based on synthetic studies using model compounds, the biosynthetic pathway of chloropupukeananin was speculated to involve the intermolecular Diels–Alder reaction between maldoxin and iso-A82775C, carbonyl-ene reaction, and the migration reaction of the p-orsellinate group. Additionally, enantioselective syntheses of both biosynthetic precursors, (−)-maldoxin and (+)-iso-A82775C, were achieved. Combining these findings, the one-pot total synthesis of chloropupukeananin mimicking the biosynthetic pathway was accomplished (overall 4.5% yield, 19 steps from 3-bromo-2-hydroxypyrone). As a further bonus, the total synthesis of chloropestolides B, I, and J, and chloropupukeanolide D was achieved.
However, this synthetic approach preferentially gives Si-syn and Si-anti isomers among the possible cycloadducts in intermolecular Diels–Alder reactions, and it is difficult to synthesize natural products derived from other cycloadducts. Controlling the facial selectivity is possible by using computational chemistry, careful examination of reaction conditions (such as solvents and additives), and modifications to the biosynthetic precursors themselves to create appropriate reaction substrates. Chemical syntheses of natural/non-natural analogs of chloropupukeananin provide a wide variety of bioactive compounds. Additionally, these chemical syntheses are expected to contribute significantly to the identification and elucidation of the function of enzymes involved in chloropupukeananin biosynthesis. I hope that this review will provide new insight into the total synthesis of complex natural products.
Funding
This research was funded by JSPS KAKENHI, grant number JP20K05485. The work was funded by the Naito Foundation, the Uehara Memorial Foundation, the Kurata Memorial Hitachi Science and Technology Foundation, and the Research Foundation for Pharmaceutical Sciences.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to thank Kazutada Ikeuchi (Hokkaido Univ.) for his kind comments on this manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Liu, L.; Liu, S.; Jiang, L.; Chen, X.; Guo, L.; Che, Y. Chloropupukeananin, the first chlorinated pupukeanane derivative, and its precursors from Pestalotiopsis fici. Org. Lett. 2008, 10, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Takahashi, I.; Kawano, T.; Kimura, Y. Chloroisosulochrin, chloroisosulochrin dehydrate, and pestheic acid, plant growth regulators, produced by Pestalotiopsis theae. Z. Naturforsch. B 2001, 56, 797–803. [Google Scholar] [CrossRef]
- Ogawa, T.; Ando, K.; Aotani, Y.; Shinoda, K.; Tanaka, T.; Tsukuda, E.; Yoshida, M.; Matsuda, Y. RES-1214-1 and -2, novel non-peptidic endothelin type A receptor antagonists produced by Pestalotiopsis sp. J. Antibiot. 1995, 48, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Adeboya, M.O.; Edwards, R.L.; Lassøe, T.; Maitland, D.J.; Shields, L.; Whalley, A.J.S. Metabolites of the higher fungi. Part 29. Maldoxin, maldoxone, dihydromaldoxin, isodihydromaldoxin and dechlorodihydromaldoxin. A spirocyclohexadienone, a depsidone and three diphenyl ethers: Keys in the depsidone biosynthetic pathway from a member of the fungus genus Xylaria. J. Chem. Soc. Perkin Trans. 1 1996, 1419–1425. [Google Scholar] [CrossRef]
- Burreson, B.J.; Scheuer, P.J.; Finer, J.S.; Clardy, J. 9-Isocyanopupukeanane, a marine invertebrate allomone with a new sesquiterpene skeleton. J. Am. Chem. Soc. 1975, 97, 4763–4764. [Google Scholar] [CrossRef]
- Hagadone, M.R.; Burreson, B.J.; Scheuer, P.J.; Finer, J.S.; Clardy, J. Defense allomones of the nudibranch Phyllidia varicosa Lamarck 1801. Helv. Chim. Acta. 1979, 62, 2484–2494. [Google Scholar] [CrossRef]
- Fusetani, N.; Wolstenholme, H.J.; Matsunaga, S. Co-occurrence of 9-isocyanopupukeanane and its C-9 epimer in the nudibranch Phyllidia bourguini. Tetrahedron Lett. 1990, 31, 5623–5624. [Google Scholar] [CrossRef]
- He, H.; Salva, J.; Catalos, R.F.; Faulkner, D.J. Sesquiterpene thiocyanates and isothiocyanates from Axinyssa aplysinoides. J. Org. Chem. 1992, 57, 3191–3194. [Google Scholar] [CrossRef]
- Simpson, J.S.; Garson, M.J.; Hooper, J.N.A.; Cline, E.I.; Angerhofer, C.K. Terpene metabolites from the tropical. Marine sponge Axinyssa sp. nov. Aust. J. Chem. 1997, 50, 1123–1127. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Liu, S.; Zheng, Z.; Chen, X.; Zhang, H.; Guo, L.; Che, Y. Chloropestolide A, an antitumor metabolite with an unprecedented spiroketal skeleton from Pestalotiopsis fici. Org. Lett. 2009, 11, 2836–2839. [Google Scholar] [CrossRef]
- Suzuki, T.; Kobayashi, S. Concise approach to pupukeanane skeleton: Synthetic study of chloropupukeananin. Org. Lett. 2010, 12, 2920–2923. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Niu, S.; Lu, X.; Chen, X.; Zhang, H.; Guo, L.; Che, Y. Unique metabolites of Pestalotiopsis fici suggest a biosynthetic hypothesis involving a Diels–Alder reaction and then mechanistic diversification. Chem. Commun. 2010, 46, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Bruhn, T.; Guo, L.; Gotz, D.C.; Brun, R.; Stich, A.; Che, Y.; Bringmann, G. Chloropupukeanolides C-E: Cytotoxic pupukeanane chlorides with a spiroketal skeleton from Pestalotiopsis fici. Chem. Eur. J. 2011, 17, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, L.; Cao, Y.; Guo, L.; Liu, G.; Che, Y. Spiroketals of Pestalotiopsis fici provide evidence for a biosynthetic hypothesis involving diversified Diels-Alder reaction cascades. J. Org. Chem. 2013, 78, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Han, Y.; Xiao, J.; Li, L.; Guo, L.; Jiang, X.; Kong, L.; Che, Y. Chlorotheolides A and B, spiroketals generated via diels-alder reactions in the endophytic fungus Pestalotiopsis theae. J. Nat. Prod. 2016, 79, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, L.; Guan, F.; Li, E.; Jin, J.; Li, J.; Che, Y.; Liu, G. Characterization of a prenyltransferase for iso-A82775C biosynthesis and generation of new congeners of chloropestolides. ACS Chem. Biol. 2018, 13, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Klas, K.; Tsukamoto, S.; Sherman, D.H.; Williams, R.M. Natural Diels-Alderases: Elusive and irresistable. J. Org. Chem. 2015, 80, 11672–11685. [Google Scholar] [CrossRef]
- Minami, A.; Oikawa, H. Recent advances of Diels-Alderases involved in natural product biosynthesis. J. Antibiot. 2016, 69, 500–506. [Google Scholar] [CrossRef]
- Jeon, B.S.; Wang, S.A.; Ruszczycky, M.W.; Liu, H.W. Natural [4+2]-Cyclases. Chem. Rev. 2017, 117, 5367–5388. [Google Scholar] [CrossRef]
- Jamieson, C.S.; Ohashi, M.; Liu, F.; Tang, Y.; Houk, K.N. The expanding world of biosynthetic pericyclases: Cooperation of experiment and theory for discovery. Nat. Prod. Rep. 2019, 36, 698–713. [Google Scholar] [CrossRef]
- Gouverneur, V.E.; Houk, K.N.; de Pascual-Teresa, B.; Beno, B.; Janda, K.D.; Lerner, R.A. Control of the exo and endo pathways of the Diels-Alder reaction by antibody catalysis. Science 1993, 262, 204–208. [Google Scholar] [CrossRef]
- Siegel, J.B.; Zanghellini, A.; Lovick, H.M.; Kiss, G.; Lambert, A.R.; St Clair, J.L.; Gallaher, J.L.; Hilvert, D.; Gelb, M.H.; Stoddard, B.L.; et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 2010, 329, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.; Artelsmair, M.; Wu, G.; Lu, X.; Barrow, S.J.; Uddin, N.; Rosta, E.; Masson, E.; Scherman, O.A. Cucurbit[7]uril as a supra-molecular artificial enzyme for Diels-Alder reactions. Angew. Chem. Int. Ed. 2017, 56, 15688–15692. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Su, C.; Du, X.; Wang, R.; Chen, S.; Zhou, Y.; Liu, C.; Liu, X.; Tian, R.; Zhang, L.; et al. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat. Chem. 2020, 12, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Basler, S.; Studer, S.; Zou, Y.; Mori, T.; Ota, Y.; Camus, A.; Bunzel, H.A.; Helgeson, R.C.; Houk, K.N.; Jimenez-Oses, G.; et al. Efficient Lewis acid catalysis of an abiological reaction in a de novo protein scaffold. Nat. Chem. 2021, 13, 231–235. [Google Scholar] [CrossRef]
- Beach, W.F.; Richards, J.H. The structure and biosynthesis of nidulin. J. Org. Chem. 1963, 28, 2746–2751. [Google Scholar] [CrossRef]
- Hendrickson, J.B.; Ramsay, M.V.J.; Kelly, T.R. New synthesis of depsidones. Diploicin and gangaleoidin. J. Am. Chem. Soc. 1972, 94, 6834–6843. [Google Scholar] [CrossRef]
- Yu, M.; Snider, B.B. Syntheses of chloroisosulochrin and isosulochrin and biomimetic elaboration to maldoxin, maldoxone, dihydromaldoxin, and dechlorodihydromaldoxin. Org. Lett. 2011, 13, 4224–4227. [Google Scholar] [CrossRef]
- Smith, K.; Butters, M.; Nay, B. High ortho-selectivity in the chlorination of phenols with N-chlorodialkylamines in the presence of silica. Tetrahedron Lett. 1988, 29, 1319–1322. [Google Scholar] [CrossRef]
- Gnaim, J.M.; Sheldon, R.A. Highly regioselective ortho-chlorination of phenol with sulfuryl chloride in the presence of amines. Tetrahedron Lett. 1995, 36, 3893–3896. [Google Scholar] [CrossRef]
- Gnaim, J.M.; Sheldon, R.A. Selective ortho-chlorination of phenol using sulfuryl chloride in the presence of t-butylaminomethyl polystyrene as a heterogeneous amine catalyst. Tetrahedron Lett. 2004, 45, 8471–8473. [Google Scholar] [CrossRef]
- Lambert, G.J.; Duffley, R.P.; Dalzell, H.C.; Razdan, R.K. Regioselective aromatic hydroxylation. An oxidative reaction of arylcopper(I) and lithium diarylcopper(I) ate complexes. J. Org. Chem. 1982, 47, 3350–3353. [Google Scholar] [CrossRef]
- Sala, T.; Sargent, M.V. Depsidone synthesis. Part 16. Benzophenone–grisa-3′,5′-diene-2′,3-dione–depsidone interconversion: A new theory of depsidone biosynthesis. J. Chem. Soc. Perkin Trans. 1 1981, 855–869. [Google Scholar] [CrossRef]
- Coomber, M.F.; Sargent, M.V.; Skelton, B.W.; White, A.H. Depsidone synthesis. Part 24. The synthesis of epiphorellic acid 2. A pseudodepsidone and X-ray crystal structure of a grisadienedione epoxide. J. Chem. Soc. Perkin Trans. 1 1989, 441–448. [Google Scholar] [CrossRef]
- Pulgarin, C.; Tabbachi, R. Synthèse du virensate de méthyle. Helv. Chim. Acta 1989, 72, 1061–1065. [Google Scholar] [CrossRef]
- Suzuki, T.; Watanabe, S.; Uyanik, M.; Ishihara, K.; Kobayashi, S.; Tanino, K. Asymmetric total synthesis of (−)-maldoxin, a common biosynthetic ancestor of the chloropupukeananin family. Org. Lett. 2018, 20, 3919–3922. [Google Scholar] [CrossRef]
- Churcher, I.; Hallett, D.; Magnus, P. Synthesis of the antitumor agent aglycon (±)-calicheamicinone using an o-quinone monoketal strategy. J. Am. Chem. Soc. 1998, 120, 10350–10358. [Google Scholar] [CrossRef]
- Suzuki, T.; Watanabe, S.; Ikeda, W.; Kobayashi, S.; Tanino, K. Biomimetic total syntheses of (+)-chloropupukeananin, (−)-chloropupukeanolide D, and chloropestolides. J. Org. Chem. 2021, 86, 15597–15605. [Google Scholar] [CrossRef]
- Kita, Y.; Tohma, H.; Hatanaka, K.; Takada, T.; Fujita, S.; Mitoh, S.; Sakurai, H.; Oka, S. Hypervalent iodine-induced nucleophilic substitution of para-substituted phenol ethers. Generation of cation radicals as reactive intermediates. J. Am. Chem. Soc. 1994, 116, 3684–3691. [Google Scholar] [CrossRef]
- Viswanadh, N.; Ghotekar, G.S.; Thoke, M.B.; Velayudham, R.; Shaikh, A.C.; Karthikeyan, M.; Muthukrishnan, M. Transition metal free regio-selective C–H hydroxylation of chromanones towards the synthesis of hydroxyl-chromanones using PhI(OAc)2 as the oxidant. Chem. Commun. 2018, 54, 2252–2255. [Google Scholar] [CrossRef]
- Koyama, H.; Sahoo, S.P.; Yang, G.X.-Q.; Miller, D.J. P38 Kinase Inhibiting Agents. WIPO (PCT) Patent WO2010129208A1, 11 November 2010. [Google Scholar]
- Ito, Y.; Ohmori, K.; Suzuki, K. Annulation approach to doubly linked (A-type) oligocatechins: Syntheses of (+)-procyanidin A2 and (+)-cinnamtannin B1. Angew. Chem. Int. Ed. 2014, 53, 10129–10133. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Ohmori, K.; Suzuki, K. Stereocontrolled total syntheses of (−)-rotenone and (−)-dalpanol by 1,2-rearrangement and SNAr oxycyclizations. Angew. Chem. Int. Ed. 2017, 56, 182–187. [Google Scholar] [CrossRef]
- Uyanik, M.; Ishihara, K. Asymmetric oxidative dearomatization reaction. In Asymmetric Dearomatization Reactions; You, S.-L., Ed.; John Wiley & Sons: Weinheim, Germany, 2016; pp. 129–151. [Google Scholar]
- Wu, W.T.; Zhang, L.; You, S.-L. Asymmetric dearomatization of phenols. Chem. Soc. Rev. 2016, 45, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, G.; Hong, L.; Wang, R. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Org. Biomol. Chem. 2016, 14, 2164–2176. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Yasui, T.; Ishihara, K. Hydrogen bonding and alcohol effects in asymmetric hypervalent iodine catalysis: Enantioselective oxidative dearomatization of phenols. Angew. Chem. Int. Ed. 2013, 52, 9215–9218. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Sasakura, N.; Mizuno, M.; Ishihara, K. Enantioselective synthesis of masked benzoquinones using designer chiral hypervalent organoiodine(III) catalysis. ACS Catal. 2017, 7, 872–876. [Google Scholar] [CrossRef]
- Uyanik, M.; Yasui, T.; Ishihara, K. Chiral hypervalent organoiodine-catalyzed enantioselective oxidative spirolactonization of naphthol derivatives. J. Org. Chem. 2017, 82, 11946–11953. [Google Scholar] [CrossRef]
- Sanson, D.R.; Gracz, H.; Tempesta, M.S.; Fukuda, D.S.; Nakatsukasa, W.M.; Sands, T.H.; Baker, P.J.; Mynderse, J.S. A82775B and A82775C, novel metabolites of an unknown fungus of the order sphaeropsidales. Tetrahedron 1991, 47, 3633–3644. [Google Scholar] [CrossRef]
- Elsebai, M.F.; Kehraus, S.; Gütschow, M.; König, G.M. Spartinoxide, a new enantiomer of A82775C with inhibitory activity toward HLE from the marine-derived fungus Phaeosphaeria spartinae. Nat. Prod. Commun. 2010, 5, 1071–1076. [Google Scholar] [CrossRef]
- Liu, L.; Liu, S.; Chen, X.; Guo, L.; Che, Y. Pestalofones A–E, bioactive cyclohexanone derivatives from the plant endophytic fungus Pestalotiopsis Fici. Bioorg. Med. Chem. 2009, 17, 606–613. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, G.-D.; Zou, J.; He, R.-R.; Qin, S.-Y.; Hu, D.; Li, G.-Q.; Guo, L.-D.; Yao, X.-S.; Gao, H. Dimericbiscognienyne A: A meroterpenoid dimer from Biscogniauxia sp. with new skeleton and its activity. Org. Lett. 2017, 19, 38–41. [Google Scholar] [CrossRef]
- Suzuki, T.; Watanabe, S.; Kobayashi, S.; Tanino, K. Enantioselective total synthesis of (+)-iso-A82775C, a proposed biosynthetic precursor of chloropupukeananin. Org. Lett. 2017, 19, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Iwagawa, T.; Nakatani, M. A base catalyzed Diels-Alder reaction of 3-hydroxy-2-pyrone. Tetrahedron Lett. 1995, 36, 5939–5942. [Google Scholar] [CrossRef]
- Okamura, H.; Nakamura, Y.; Iwagawa, T.; Nakatani, M. Asymmetric base-catalyzed Diels-Alder reaction of 3-hydroxy-2-pyrone with N-methylmaleimide. Chem. Lett. 1996, 25, 193–194. [Google Scholar] [CrossRef]
- Shimizu, H.; Okamura, H.; Iwagawa, T.; Nakatani, M. Asymmetric synthesis of (−)- and (+)-eutipoxide B using a base-catalyzed Diels–Alder reaction. Tetrahedron 2001, 57, 1903–1908. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Wang, Y.-Q.; Liu, Y.; Foxman, B.M.; Deng, L. Asymmetric Diels−Alder reactions of 2-pyrones with a bifunctional organic catalyst. J. Am. Chem. Soc. 2007, 129, 6364–6365. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, F.; Lewis, J.; Heroux, K.; Cohen, S. Characterization and evaluation of pyrone and tropolone chelators for use in metalloprotein inhibitors. Inorg. Chim. Acta 2007, 360, 264–272. [Google Scholar] [CrossRef]
- Mayer, R. Pseudo-α-Tropolone vom typ des 3-hydroxy-α-pyrons, 3-hydroxy-γ-pyrons und der 3-hydroxy-thia-pyrone. Chem. Ber. 1957, 90, 2369–2372. [Google Scholar] [CrossRef]
- Saksena, A.K.; Mangiaracina, P. Recent studies on veratrum alkaloids: A new reaction of sodium triacetoxyborohydride [NaBH(OAc)3]. Tetrahedron Lett. 1983, 24, 273–276. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Verhoeven, T.R. Metal-catalyzed, highly selective oxygenations of olefins and acetylenes with tert-butyl hydroperoxide. Practical considerations and mechanisms. Aldrichimica Acta 1979, 12, 63–74. [Google Scholar]
- Riveiros, R.; Rodríguez, D.; Pérez Sestelo, J.; Sarandeses, L.A. Palladium-catalyzed cross-coupling reaction of triorganoindium reagents with propargylic esters. Org. Lett. 2006, 8, 1403–1406. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, M.J.; Chung, G.; Lee, H.-Y.; Han, S. (+)-Dimericbiscognienyne A: Total synthesis and mechanistic investigations of the key heterodimerization. Org. Lett. 2018, 20, 6886–6890. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Pan, S.C. Total synthesis of the novel antifungal agent (±)-jesterone. Org. Lett. 2004, 6, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Kumar, Y.S.; Das, M. A de novo Diels–Alder strategy toward the novel pentacyclic natural product fluostatin C: A concise synthesis of 6-deoxyfluostatin C. Tetrahedron Lett. 2011, 52, 3505–3508. [Google Scholar] [CrossRef]
- Kim, G.; Kim, T.; Han, S. Total synthesis of (+)-pestalofone A and (+)-iso-A82775C. J. Org. Chem. 2020, 85, 6815–6821. [Google Scholar] [CrossRef]
- Line, N.J.; Witherspoon, B.P.; Hancock, E.N.; Brown, M.K. Synthesis of ent-[3]-ladderanol: Development and application of intramolecular chirality transfer [2+2] cycloadditions of allenic ketones and alkenes. J. Am. Chem. Soc. 2017, 139, 14392–14395. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Snider, B.B. Diels–Alder reaction of maldoxin with an isopropenylallene. Tetrahedron 2011, 67, 9473–9478. [Google Scholar] [CrossRef]
- Suzuki, T.; Miyajima, Y.; Suzuki, K.; Iwakiri, K.; Koshimizu, M.; Hirai, G.; Sodeoka, M.; Kobayashi, S. Unexpected Diels-Alder/carbonyl-ene cascade toward the biomimetic synthesis of chloropupukeananin. Org. Lett. 2013, 15, 1748–1751. [Google Scholar] [CrossRef]
- Pinault, M.; Frangin, Y.; Genet, J.P.; Zamarlik, H. Total synthesis of siccayne. Synthesis 1990, 935–937. [Google Scholar] [CrossRef]
- Catalán, J.; Palomar, J.; Díaz, C.; de Paz, J.L.G. On solvent basicity: Analysis of the SB scale. J. Phys. Chem. A 1997, 101, 5183–5189. [Google Scholar] [CrossRef]
- Catalán, J.; Díaz, C.; López, V.; Pérez, P.; De Paz, J.-L.; Rodríguez, J.G. A Generalized solvent basicity scale: The solvatochromism of 5-nitroindoline and its homomorph 1-methyl-5-nitroindoline. Liebigs Ann. Chem. 1996, 1996, 1785–1794. [Google Scholar] [CrossRef]
Figure 1.
The chemical structure of chloropupukeananin and pupukeanane.

Scheme 1.
The biosynthetic pathway of chloropupukeananin and its related natural products.

Scheme 2.
Natural products derived by the intermolecular Diels–Alder reaction of maldoxin.

Scheme 3.
The facial selectivity of the intermolecular Diels–Alder reaction of maldoxin.

Figure 2.
The chemical structure of pestheic acid, maldoxin, and these related natural compounds.

Scheme 4.
Total synthesis of pestheic acid and rac-maldoxin by Yu and Snyder. MOM—methoxymethyl, DIPEA—diisopropylethylamine, DMP—Dess-Martin periodinane, PIDA—(diacetoxyiodo)benzene.
Scheme 4.
Total synthesis of pestheic acid and rac-maldoxin by Yu and Snyder. MOM—methoxymethyl, DIPEA—diisopropylethylamine, DMP—Dess-Martin periodinane, PIDA—(diacetoxyiodo)benzene.

Scheme 5.
Enantioselective total synthesis of (−)-maldoxin by Suzuki et al. TFA—trifluoroacetic acid, DMSO—dimethylsulfoxide, mCPBA—3-chloroperbenzoic acid.
Scheme 5.
Enantioselective total synthesis of (−)-maldoxin by Suzuki et al. TFA—trifluoroacetic acid, DMSO—dimethylsulfoxide, mCPBA—3-chloroperbenzoic acid.

Figure 3.
The chemical structure of iso-A82775C and its related natural products.

Scheme 6.
Enantioselective total synthesis of (+)-iso-A82775C by Suzuki et al. TES—triethylsilyl, DIBAL—diisobutylaluminium hydride, TBS—tert-butyldimethylsilyl, TBAF—tetra-n-butylammonium fluoride.
Scheme 6.
Enantioselective total synthesis of (+)-iso-A82775C by Suzuki et al. TES—triethylsilyl, DIBAL—diisobutylaluminium hydride, TBS—tert-butyldimethylsilyl, TBAF—tetra-n-butylammonium fluoride.

Scheme 7.
Total syntheses of rac-biscognienyne B and (+)-iso-A82775C by Han et al. DMAP—4-dimethylaminopyridine, DEAD—diethyl azodicarboxylate, CAN—cerium ammonium nitrate.
Scheme 7.
Total syntheses of rac-biscognienyne B and (+)-iso-A82775C by Han et al. DMAP—4-dimethylaminopyridine, DEAD—diethyl azodicarboxylate, CAN—cerium ammonium nitrate.

Scheme 8.
Model studies on the intermolecular Diels–Alder reaction toward the biomimetic synthesis of chloropupukeananin. All yields in italics are calculated by 1H NMR. a carbonyl-ene isomer 79 derived from 76 was obtained (11% yield). b the whole structure could not be identified.
Scheme 8.
Model studies on the intermolecular Diels–Alder reaction toward the biomimetic synthesis of chloropupukeananin. All yields in italics are calculated by 1H NMR. a carbonyl-ene isomer 79 derived from 76 was obtained (11% yield). b the whole structure could not be identified.

Scheme 9.
The intermolecular Diels–Alder/carbonyl–ene cascade reaction using (+)-iso-A82775C and (−)-maldoxin. All yields in italics are calculated by 1H NMR.
Scheme 9.
The intermolecular Diels–Alder/carbonyl–ene cascade reaction using (+)-iso-A82775C and (−)-maldoxin. All yields in italics are calculated by 1H NMR.

Scheme 10.
Migration reaction of p-orsellinate group of chloropupukeanolide D. DMF—N,N-dimethylformaminde.
Scheme 10.
Migration reaction of p-orsellinate group of chloropupukeanolide D. DMF—N,N-dimethylformaminde.

Scheme 11.
One-pot biomimetic synthesis of chloropupukeananin.

Scheme 12.
The intermolecular Diels–Alder reaction using siccayne and (−)-maldoxin. Isolated yield in parentheses. SB—solvent basicity.
Scheme 12.
The intermolecular Diels–Alder reaction using siccayne and (−)-maldoxin. Isolated yield in parentheses. SB—solvent basicity.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suzuki, T. Total Syntheses of Chloropupukeananin and Its Related Natural Products. Organics 2022, 3, 304-319. https://doi.org/10.3390/org3030023
AMA Style
Suzuki T. Total Syntheses of Chloropupukeananin and Its Related Natural Products. Organics. 2022; 3(3):304-319. https://doi.org/10.3390/org3030023
Chicago/Turabian StyleSuzuki, Takahiro. 2022. "Total Syntheses of Chloropupukeananin and Its Related Natural Products" Organics 3, no. 3: 304-319. https://doi.org/10.3390/org3030023