
Development of Power-to-X Catalytic Processes for CO2 Valorisation: From the Molecular Level to the Reactor Architecture
 , , , , , and
, , , , , and
Abstract
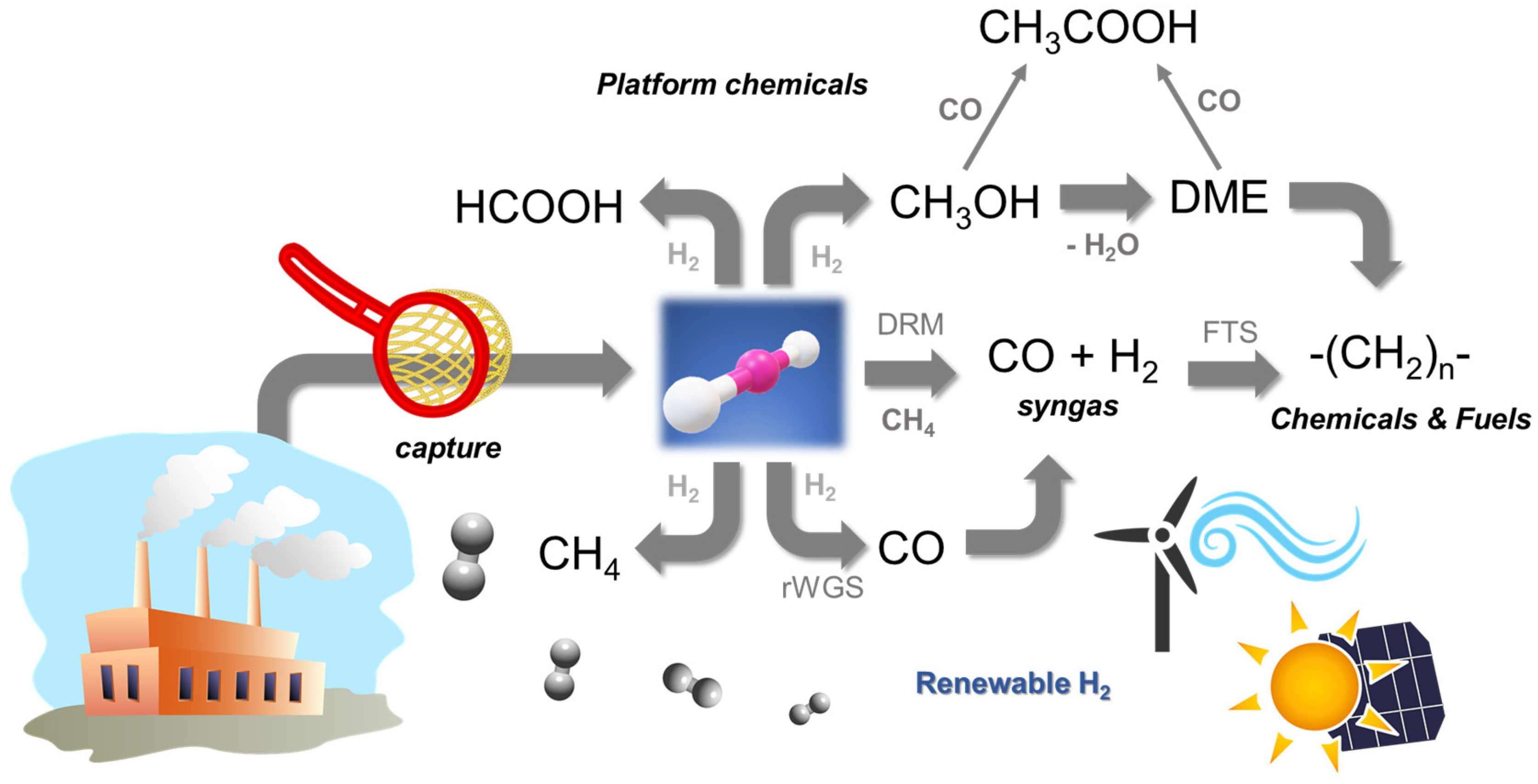
:1. Introduction
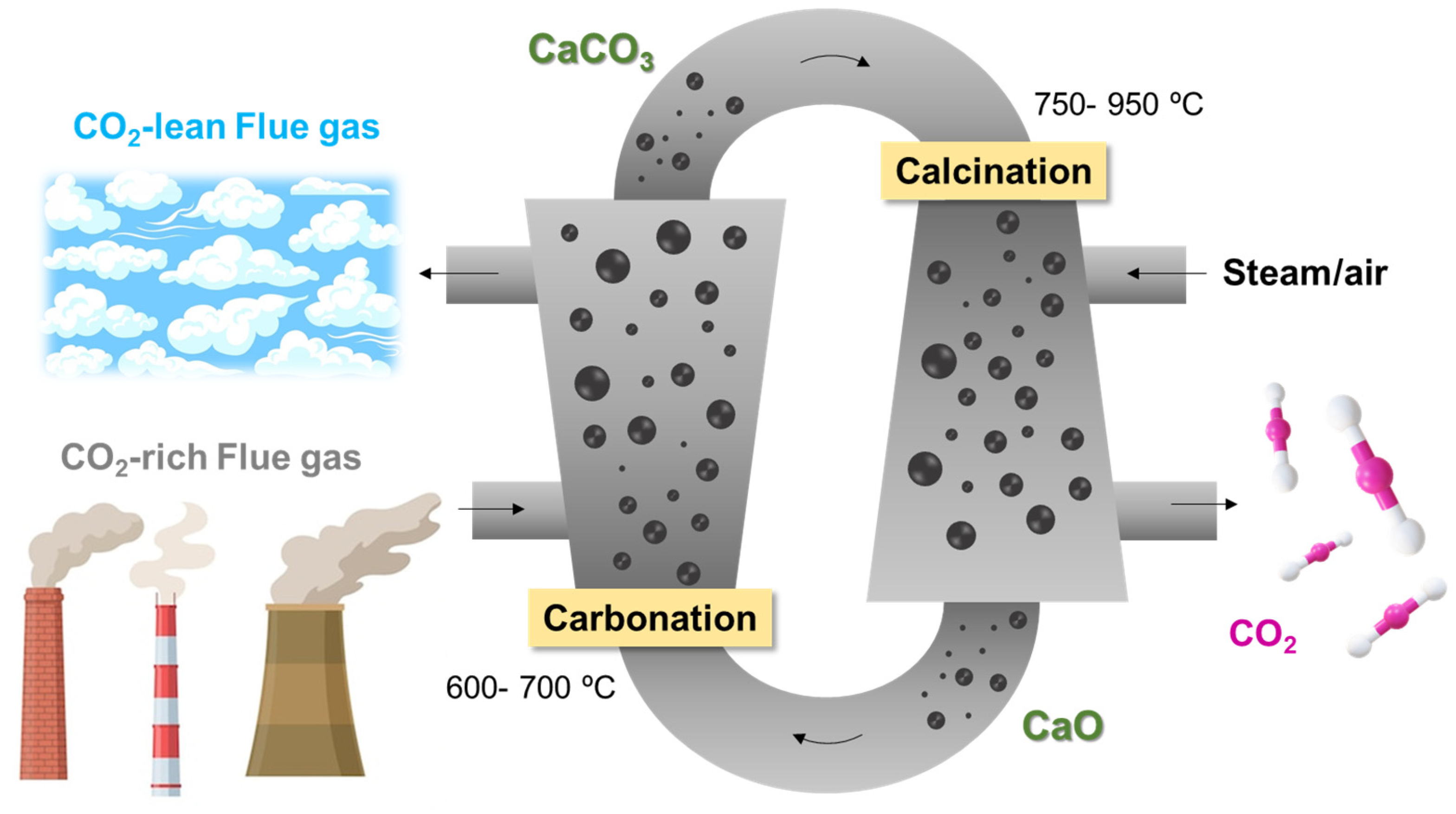
2. Carbon Capture and Storage (CCS)
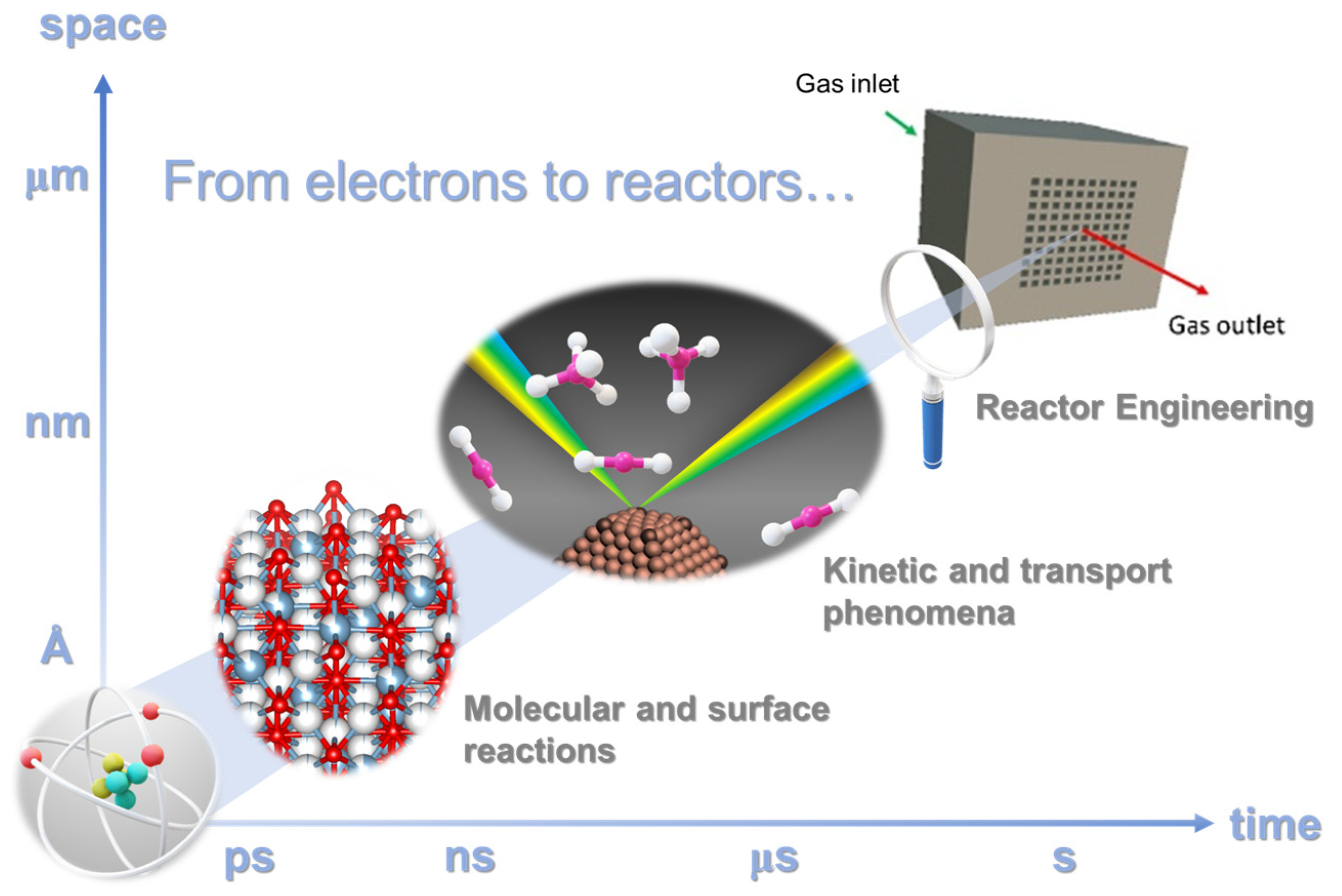
3. Catalytic CO2 Conversion: From Active Sites to Reactor Architecture
3.1. Design of Nanoscale Catalysts with Atomic-Scale Precision
- ▪
- To generate multifunctional catalysts formed by nanoscale particles of metals and bimetallic alloys by fine-tuning of the interfaces.
- ▪
- To understand the nature and composition of the active surface, its evolution with time, and the role of the adsorbed species along the reaction time that may modify the catalytic activity, as well as the selectivity of the reactions.
- ▪
- To selectively activate the very strong C=O bonds of the CO2 molecule at moderate temperatures.
- ▪
- To understand the reaction pathways that generate the desired products.
3.2. Structured Catalysts and Microreactors
3.3. Modelling and Simulation of Structured Catalytic Reactors
4. Chemical Routes for CO2 Conversion
4.1. Reverse Water—Gas Shift (r-WGS) Reaction
4.2. CO2 Methanation: Sabatier Reaction
4.3. Synthesis of Formic Acid (FA) as Energy Vector
4.4. Direct Synthesis of Dimethyl Ether (DME)
4.4.1. One-Step Process
4.4.2. Catalysts for the One-Step Process
4.4.3. Dimethyl Ether from CO2
4.4.4. Production of DME in Compact Systems
4.5. The Synthesis of Acetic Acid Using CO2-Rich Feedstocks
5. Concluding Remarks and Future Trends
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IEA. Energy Technology Perspectives 2016. Towards Sustainable Urban Energy Systems; OECD/IEA: Paris, France, 2016. [Google Scholar]
- Smith, P.; Davis, S.J.; Creutzig, F.; Fuss, S.; Minx, J.; Gabrielle, B.; Kato, E.; Jackson, R.B.; Cowie, A.; Kriegler, E.; et al. Biophysical and economic limits to negative CO2 emissions. Nat. Clim. Chang. 2016, 6, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, A.; Rajab Khalilpour, K.; Milani, D.; Panahi, M. Trends in CO2 conversion and utilization: A review from process systems perspective. J. Environ. Chem. Eng. 2018, 6, 5771–5794. [Google Scholar] [CrossRef]
- Dairanieh, I.; Bernard, D.; Mason, F.; Stokes, G.; de Brun, S. Carbon Dioxide Utilization—ICEF Roadmap 1.0; Innovation for Cool Earth Forum (ICEF): Tokyo, Japan, 2016. [Google Scholar]
- Onarheim, K.; Hannula, A.A.I.; Karki, J.; Lehtonen, J.; Vainikka, P. Carbon Capture and Utilization; A VTT Discussion Paper; VTT: Espoo, Finland, 2017. [Google Scholar]
- Arasto, A.; Onarheim, K.; Tsupari, E.; Kärki, J. Bio-CCS: Feasibility comparison of large scale carbon-negative solutions. Energy Procedia 2014, 63, 6756–6769. [Google Scholar] [CrossRef] [Green Version]
- Otto, A.; Grube, T.; Schiebahn, S.; Stolten, D. Closing the loop: Captured CO2 as a feedstock in the chemical industry. Energy Environ. Sci. 2015, 8, 3283–3297. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, H.; Roser, M.; Rosado, P. CO2 and Greenhouse Gas Emissions. Published Online at OurWorldInData.org. 2020. Available online: https://ourworldindata.org/co2-and-other-greenhouse-gas-emissions (accessed on 7 July 2022).
- Ursua, A.; Gandia, L.M.; Sanchis, P. Hydrogen production from water electrolysis: Current status and future trends. Proc. IEEE Inst. Electr. Electron. Eng. 2012, 100, 410–426. [Google Scholar] [CrossRef]
- Gandía, L.M.; Oroz, R.; Ursúa, A.; Sanchis, P.; Diéguez, P.M. Renewable hydrogen production: Performance of an alkaline water electrolyzer working under emulated wind conditions. Energy Fuels 2007, 21, 1699–1706. [Google Scholar] [CrossRef]
- Vázquez, F.V.; Koponen, J.; Ruuskanen, V.; Bajamundi, C.; Kosonen, A.; Simell, P.; Ahola, J.; Frilund, C.; Elfving, J.; Reinikainen, M.; et al. Power-to-X technology using renewable electricity and carbon dioxide from ambient air: SOLETAIR proof-of-concept and improved process concept. J. CO2 Util. 2018, 28, 235–246. [Google Scholar] [CrossRef]
- Aicher, T. Renewable Hydrogen Technologies. Production, Purification, Storage, Applications, and Safety. Edited by Luis M. Gandía, Gurutze Arzamendi, and Pedro M. Diéguez. Energy Technol. 2015, 3, 90–91. [Google Scholar] [CrossRef]
- Fennell, P.; Anthony, B. (Eds.) Calcium and Chemical Looping Technology for Power Generation and Carbon Dioxide (CO2) Capture; Woodhead Publishing: Boca Raton, FL, USA, 2015; pp. 439–446. [Google Scholar]
- Shimizu, T.; Hirama, T.; Hosoda, H.; Kitano, K.; Inagaki, M.; Tejima, K. A twin fluid-bed reactor for removal of CO2 from combustion processes. Chem. Eng. Res. Des. 1999, 77, 62–68. [Google Scholar] [CrossRef]
- Stanmore, B.R.; Gilot, P. Review—Calcination and carbonation of limestone during thermal cycling for CO2 sequestration. Fuel Process. Technol. 2005, 86, 1707–1743. [Google Scholar] [CrossRef]
- Han, R.; Wang, Y.; Xing, S.; Pang, C.; Hao, Y.; Song, C.; Liu, Q. Progress in reducing calcination reaction temperature of Calcium-Looping CO2 capture technology: A critical review. Chem. Eng. J. 2022, 450, 137952. [Google Scholar] [CrossRef]
- Hornberger, M.; Spörl, R.; Scheffknecht, G. Calcium looping for CO2 capture in cement plants—Pilot scale test. Energy Procedia 2017, 114, 6171–6174. [Google Scholar] [CrossRef]
- Dieter, H.; Bidwe, A.R.; Varela-Duelli, G.; Charitos, A.; Hawthorne, C.; Scheffknecht, G. Development of the calcium looping CO2 capture technology from lab to pilot scale at IFK, University of Stuttgart. Fuel 2014, 127, 23–37. [Google Scholar] [CrossRef]
- Sanchez-Biezma, A.; Paniagua, J.G.; Díaz, L.Á.V.; Lorenzo, M.; Álvarez, J.F.G.; Martinez, D.; Arias, B.; Diego, M.E.; Abanades, J.C. Testing postcombustion CO2 capture with CaO in a 1.7 MWt pilot facility. Energy Procedia 2013, 37, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Adánez, J.; Gayán, P.; García-Labiano, F. Comparison of mechanistic models for the sulfation reaction in a broad range of particle sizes of sorbents. Ind. Eng. Chem. Res. 1996, 35, 2190–2197. [Google Scholar] [CrossRef]
- Agnew, J.; Hampartsoumian, E.; Jones, J.M.; Nimmo, W. The simultaneous calcination and sintering of calcium based sorbents under a combustion atmosphere. Fuel 2000, 79, 1515–1523. [Google Scholar] [CrossRef]
- Abanades, J.C.; Álvarez, D. Conversion limits in the reaction of CO2 with lime. Energy Fuels 2003, 17, 308–315. [Google Scholar] [CrossRef]
- Salvador, C.; Lu, D.; Anthony, E.J.; Abanades, J.C. Enhancement of CaO for CO2 capture in an FBC environment. Chem. Eng. J. 2003, 96, 187–195. [Google Scholar] [CrossRef]
- Fennell, P.S.; Pacciani, R.; Dennis, J.S.; Davidson, J.F.; Hayhurst, A.N. The effects of repeated cycles of calcination and carbonation on a variety of different limestones, as measured in a hot fluidized bed of sand. Energy Fuels 2007, 21, 2072–2081. [Google Scholar] [CrossRef]
- Al-Jeboori, M.J.; Nguyen, M.; Dean, C.; Fennell, P.S. Improvement of limestone-based CO2 sorbents for Ca-Looping by HBr and other mineral acids. Ind. Eng. Chem. Res. 2013, 52, 1426–1433. [Google Scholar] [CrossRef]
- Dean, C.C.; Dugwell, D.; Fennell, P.S. Investigation into potential synergy between power generation, cement manufacture and CO2 abatement using the calcium looping cycle. Energy Environ. Sci. 2011, 4, 2050–2053. [Google Scholar] [CrossRef]
- Telesca, A.; Calabrese, D.; Marroccoli, M.; Tomasulo, M.; Valenti, G.L.; Duelli, G.; Montagnaro, F. Spent limestone sorbent from calcium looping cycle as a raw material for the cement industry. Fuel 2014, 118, 202–205. [Google Scholar] [CrossRef]
- Hanak, D.P.; Anthony, E.J.; Manovic, V. A review of developments in pilot-plant testing and modelling of calcium looping process for CO2 capture from power generation systems. Energy Environ. Sci. 2015, 8, 2199–2249. [Google Scholar] [CrossRef] [Green Version]
- Müller, K.; Fleige, M.; Rachow, F.; Schmeißer, D. Sabatier based CO2-methanation of flue gas emitted by conventional power plants. Energy Procedia 2013, 40, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Huijgen, W.J.J.; Comans, R.N.J.; Witkamp, G.-J. Cost evaluation of CO2 sequestration by aqueous mineral carbonation. Energy Conv. Manag. 2007, 48, 1923–1935. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, M.I.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Physicochemical characterization and use of wastes from stainless steel mill. Environ. Prog. Sustain. Energy 2010, 29, 471–480. [Google Scholar] [CrossRef]
- Yi, H.; Xu, G.; Cheng, H.; Wang, J.; Wan, Y.; Chen, H. An Overview of Utilization of Steel Slag. Procedia Environ Sci. 2012, 16, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Doucet, F.J. Effective CO2-specific sequestration capacity of steel slags and variability in their leaching behaviour in view of industrial mineral carbonation. Miner. Eng. 2010, 23, 262–269. [Google Scholar] [CrossRef]
- Bobicki, E.R.; Liu, Q.; Xu, Z.; Zeng, H. Carbon capture and storage using alkaline industrial wastes. Prog. Energy Combust. Sci. 2012, 38, 302–320. [Google Scholar] [CrossRef]
- Sanna, A.; Dri, M.; Hall, M.R.; Maroto-Valer, M. Waste materials for carbon capture and storage by mineralisation (CCSM)—A UK perspective. Appl. Energy 2012, 99, 545–554. [Google Scholar] [CrossRef]
- Pan, S.-Y.; Chang, E.E.; Chiang, P.-C. CO2 capture by accelerated carbonation of alkaline wastes: A review on its principles and applications. Aerosol Air Qual. Res. 2012, 12, 770–791. [Google Scholar] [CrossRef]
- Yu, J.; Wang, K. Study on characteristics of steel slag for CO2 capture. Energy Fuels 2011, 25, 5483–5492. [Google Scholar] [CrossRef]
- Miranda-Pizarro, J.; Perejón, A.; Valverde, J.M.; Sánchez-Jiménez, P.E.; Pérez-Maqueda, L.A. Use of steel slag for CO2 capture under realistic calcium-looping conditions. RSC Adv. 2016, 6, 37656–37663. [Google Scholar] [CrossRef] [Green Version]
- Valverde, J.M.; Miranda-Pizarro, J.; Perejón, A.; Sánchez-Jiménez, P.E.; Pérez-Maqueda, L.A. Calcium-Looping performance of steel and blast furnace slags for thermochemical energy storage in concentrated solar power plants. J. CO2 Utili. 2017, 22, 143–154. [Google Scholar] [CrossRef]
- Romero-Hermida, I.; Santos, A.; Pérez-López, R.; García-Tenorio, R.; Esquivias, L.; Morales-Flórez, V. New method for carbon dioxide mineralization based on phosphogypsum and aluminium-rich industrial wastes resulting in valuable carbonated by-products. J. CO2 Util. 2017, 18, 15–22. [Google Scholar] [CrossRef] [Green Version]
- El-Naas, M.H.; El Gamal, M.; Hameedi, S.; Mohamed, A.-M.O. CO2 sequestration using accelerated gas-solid carbonation of pre-treated EAF steel-making bag house dust. J. Environ. Manag. 2015, 156, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Aalto University. Available online: http://eng.aalto.fi/en/current/current_archive/news/2014-09-16/ (accessed on 12 July 2022).
- Torres-Sempere, G.; Pastor-Pérez, L.; Odriozola, J.A.; Yu, J.; Durán-Olivencia, F.J.; Bobadilla, L.F.; Reina, T.R. Recent advances on gas-phase CO2 conversion: Catalysis design and chemical processes to close the carbon cycle. Curr. Opin. Green Sustain. Chem. 2022, 36, 100647. [Google Scholar] [CrossRef]
- Zhao, G.; Huang, X.; Wang, X.; Wang, X. Progress in catalyst exploration for heterogeneous CO2 reduction and utilization: A critical review. J. Mater. Chem. A 2017, 5, 21625–21649. [Google Scholar] [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, A.; Borges, M.; Corral-Pérez, J.J.; Olcina, J.G.; Hu, L.; Cornu, D.; Huang, R.; Stoian, D.; Urakawa, A. CO2 Activation over catalytic surfaces. ChemPhysChem 2017, 18, 3135–3141. [Google Scholar] [CrossRef]
- Sharma, D.; Sharma, R.; Chand, D.; Chaudhary, A. Nanocatalysts as potential candidates in transforming CO2 into valuable fuels and chemicals: A review. Environ. Nanotechnol. Monit. Manag. 2022, 18, 100671. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, C.; Sun, K.; Jia, X.; Ye, J.; Liu, C.-J. Advances in studies of the structural effects of supported Ni catalysts for CO2 hydrogenation: From nanoparticle to single atom catalyst. J. Mater. Chem. A 2022, 10, 5792–5812. [Google Scholar] [CrossRef]
- Heidary, N.; Ly, K.H.; Kornienko, N. Probing CO2 conversion chemistry on nanostructured surfaces with operando vibrational spectroscopy. Nano Lett. 2019, 19, 4817–4826. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Wang, Y.; Guo, M.; Zhang, J.; Li, Z.; Deng, T.; Zhang, Z.; Yan, B. In-situ/operando techniques to identify active sites for thermochemical conversion of CO2 over heterogeneous catalysts. J. Energy Chem. 2021, 62, 153–171. [Google Scholar] [CrossRef]
- van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Lou, Y.; Xu, J.; Zhang, Y.; Pan, C.; Dong, Y.; Zhu, Y. Metal-support interaction for heterogeneous catalysis: From nanoparticles to single atoms. Mater. Today Nano 2020, 12, 100093. [Google Scholar] [CrossRef]
- Fujiwara, K.; Okuyama, K.; Pratsinis, S.E. Metal-support interactions in catalysts for environmental remediation. Environ. Sci. Nano 2017, 4, 2076–2092. [Google Scholar] [CrossRef]
- Hernández, W.Y.; Centeno, M.A.; Romero-Sarria, F.; Odriozola, J.A. Synthesis and characterization of Ce1−xEuxO2−x/2 mixed oxides and their catalytic activities for CO oxidation. J. Phys. Chem. C 2009, 113, 5629–5635. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Santos, J.L.; Ivanova, S.; Odriozola, J.A.; Urakawa, A. Unravelling the role of oxygen vacancies in the mechanism of the reverse water–gas Shift reaction by operando DRIFTS and Ultraviolet–Visible spectroscopy. ACS Catal. 2018, 8, 7455–7467. [Google Scholar] [CrossRef]
- Laguna, O.H.; Romero Sarria, F.; Centeno, M.A.; Odriozola, J.A. Gold supported on metal-doped ceria catalysts (M = Zr, Zn and Fe) for the preferential oxidation of CO (PROX). J. Catal. 2010, 276, 360–370. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef] [Green Version]
- Azancot, L.; Bobadilla, L.F.; Centeno, M.A.; Odriozola, J.A. Effect of potassium loading on basic properties of Ni/MgAl2O4 catalyst for CO2 reforming of methane. J. CO2 Util. 2021, 52, 101681. [Google Scholar] [CrossRef]
- Azancot, L.; Bobadilla, L.F.; Centeno, M.A.; Odriozola, J.A. IR spectroscopic insights into the coking-resistance effect of potassium on nickel-based catalyst during dry reforming of methane. Appl. Catal. B Environ. 2021, 285, 119822. [Google Scholar] [CrossRef]
- Jia, J.; Qian, C.; Dong, Y.; Li, Y.F.; Wang, H.; Ghoussoub, M.; Butler, K.T.; Walsh, A.; Ozin, G.A. Heterogeneous catalytic hydrogenation of CO2 by metal oxides: Defect engineering—perfecting imperfection. Chem. Soc. Rev. 2017, 46, 4631–4644. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.; Moulijn, J.A. Process Intensification. Ind. Eng. Chem. Res. 2002, 41, 1920–1924. [Google Scholar] [CrossRef]
- Giani, L.; Groppi, G.; Tronconi, E. Heat transfer characterization of metallic foams. Ind. Eng. Chem. Res. 2005, 44, 9078–9085. [Google Scholar] [CrossRef]
- Tonkovich, A.Y.; Zilka, J.L.; Lamont, M.; Wang, Y.; Wegeng, R.S. Microchannel reactors for fuel processing applications: Water gas shift reactor. Chem. Eng. Sci. 1999, 54, 2947–2951. [Google Scholar] [CrossRef]
- Domínguez, M.I.; Centeno, M.A.; Martínez, T.M.; Bobadilla, L.F.; Laguna, Ó.H.; Odriozola, J.A. Current scenario and prospects in manufacture strategies for glass, quartz, polymers and metallic microreactors: A comprehensive review. Chem. Eng. Res. Des. 2021, 171, 13–35. [Google Scholar] [CrossRef]
- Laguna, O.H.; Domínguez, M.I.; Centeno, M.A.; Odriozola, J.A. Chapter 4—Catalysts on Metallic Surfaces: Monoliths and Microreactors. In New Materials for Catalytic Applications; Parvulescu, V.I., Kemnitz, E., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 81–120. [Google Scholar]
- Ivanova, S.; Laguna, O.H.; Centeno, M.Á.; Eleta, A.; Montes, M.; Odriozola, J.A. Chapter 10—Microprocess Technology for Hydrogen Purification. In Renewable Hydrogen Technologies; Gandía, L.M., Arzamendi, G., Diéguez, P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 225–243. [Google Scholar]
- Lämmermann, M.; Schwieger, W.; Freund, H. Experimental investigation of gas-liquid distribution in periodic open cellular structures as potential catalyst supports. Catal. Today 2016, 273, 161–171. [Google Scholar] [CrossRef]
- Busse, C.; Freund, H.; Schwieger, W. Intensification of heat transfer in catalytic reactors by additively manufactured periodic open cellular structures (POCS). Chem. Eng. Proc. 2018, 124, 199–214. [Google Scholar] [CrossRef]
- Lämmermann, M.; Horak, G.; Schwieger, W.; Freund, H. Periodic open cellular structures (POCS) for intensification of multiphase reactors: Liquid holdup and two-phase pressure drop. Chem. Eng. Process. 2018, 126, 178–189. [Google Scholar] [CrossRef]
- Klumpp, M.; Inayat, A.; Schwerdtfeger, J.; Körner, C.; Singer, R.F.; Freund, H.; Schwieger, W. Periodic open cellular structures with ideal cubic cell geometry: Effect of porosity and cell orientation on pressure drop behavior. Chem. Eng. J. 2014, 242, 364–378. [Google Scholar] [CrossRef]
- Parra-Cabrera, C.; Achille, C.; Kuhn, S.; Ameloot, R. 3D printing in chemical engineering and catalytic technology: Structured catalysts, mixers and reactors. Chem. Soc. Rev. 2018, 47, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Coppens, M.-O. A nature-inspired approach to reactor and catalysis engineering. Curr. Opin. Chem. Eng. 2012, 1, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Mazumder, S. Toward simulation of full-scale monolithic catalytic converters with complex heterogeneous chemistry. Comput. Chem. Eng. 2010, 34, 135–145. [Google Scholar] [CrossRef]
- Mazumder, S. Modeling full-scale monolithic catalytic converters: Challenges and possible solutions. J. Heat Transfer 2006, 129, 526–535. [Google Scholar] [CrossRef]
- Groppi, G.; Belloli, A.; Tronconi, E.; Forzatti, P. A comparison of lumped and distributed models of monolith catalytic combustors. Chem. Eng. Sci. 1995, 50, 2705–2715. [Google Scholar] [CrossRef]
- Hayes, R.E.; Kolaczkowski, S.T. A study of Nusselt and Sherwood numbers in a monolith reactor. Catal. Today 1999, 47, 295–303. [Google Scholar] [CrossRef]
- Fox, R.O. CFD models for analysis and design of chemical reactors. In Advances in Chemical Engineering; Marin, G.B., Ed.; Academic Press: Salt Lake City, UT, USA, 2006; Volume 31, pp. 231–305. [Google Scholar]
- Nien, T.; Mmbaga, J.P.; Hayes, R.E.; Votsmeier, M. Hierarchical multi-scale model reduction in the simulation of catalytic converters. Chem. Eng. Sci. 2013, 93, 362–375. [Google Scholar] [CrossRef]
- Heidebrecht, P.; Pfafferodt, M.; Sundmacher, K. Multiscale modelling strategy for structured catalytic reactors. Chem. Eng. Sci. 2011, 66, 4389–4402. [Google Scholar] [CrossRef]
- Bertrand, F.; Devals, C.; Vidal, D.; Préval, C.S.d.; Hayes, R.E. Towards the simulation of the catalytic monolith converter using discrete channel-scale models. Catal. Today 2012, 188, 80–86. [Google Scholar] [CrossRef]
- Štěpánek, J.; Kočí, P.; Marek, M.; Kubíček, M. Catalyst simulations based on coupling of 3D CFD tool with effective 1D channel models. Catal. Today 2012, 188, 87–93. [Google Scholar] [CrossRef]
- Kamkeng, A.D.N.; Wang, M.; Hu, J.; Du, W.; Qian, F. Transformation technologies for CO2 utilisation: Current status, challenges and future prospects. Chem. Eng. J. 2021, 409, 128138. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- le Saché, E.; Pastor-Pérez, L.; Haycock, B.J.; Villora-Picó, J.J.; Sepúlveda-Escribano, A.; Reina, T.R. Switchable catalysts for chemical CO2 recycling: A step forward in the methanation and reverse water–gas shift reactions. ACS Sustain. Chem. Eng. 2020, 8, 4614–4622. [Google Scholar] [CrossRef]
- Whitlow, J.E.; Parrish, C.F. Operation, modeling and analysis of the reverse water gas shift process. AIP Conf. Proc. 2003, 654, 1116–1123. [Google Scholar]
- van der Wiel, D.; Zilka, J.; Wang, Y.; Tonkovich, A.; Wegeng, R. Carbon Dioxide Conversions in Microreactors. In Proceedings of the 4th International Conference on Microreaction Technology, Atlanta, GA, USA, 23–25 June 2014; AIChE Spring National Meeting: Atlanta, GA, USA, 2000; p. 187. [Google Scholar]
- Gaudillere, C.; Navarrete, L.; Serra, J.M. CO2 hydrogenation on Ru/Ce based catalysts dispersed on highly ordered micro-channelled 3YSZ monoliths fabricated by freeze-casting. Int. J. Hydrogen Energy 2017, 42, 895–905. [Google Scholar] [CrossRef]
- Fujita, S.-i.; Usui, M.; Takezawa, N. Mechanism of the reverse water gas shift reaction over Cu/ZnO catalyst. J. Catal. 1992, 134, 220–225. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Evans, J.; Feria, L.; Vidal, A.B.; Liu, P.; Nakamura, K.; Illas, F. CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: Production of CO, methanol, and methane. J. Catal. 2013, 307, 162–169. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 reduction on transition metal (Fe, Co, Ni, and Cu) surfaces: In comparison with homogeneous catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Baibars, F.; Le Sache, E.; Arellano-García, H.; Gu, S.; Reina, T.R. CO2 valorisation via Reverse Water-Gas Shift reaction using advanced Cs doped Fe-Cu/Al2O3 catalysts. J. CO2 Util. 2017, 21, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Study of reverse water gas shift reaction by TPD, TPR and CO2 hydrogenation over potassium-promoted Cu/SiO2 catalyst. Appl. Catal. A Gen. 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Study of iron-promoted Cu/SiO2 catalyst on high temperature reverse water gas shift reaction. Appl. Catal. A Gen. 2004, 257, 97–106. [Google Scholar] [CrossRef]
- Chen, C.S.; Lin, J.H.; You, J.H.; Chen, C.R. Properties of Cu(thd)2 as a precursor to prepare Cu/SiO2 catalyst using the atomic layer epitaxy technique. J. Am. Chem. Soc. 2006, 128, 15950–15951. [Google Scholar] [CrossRef]
- Zhou, G.; Dai, B.; Xie, H.; Zhang, G.; Xiong, K.; Zheng, X. CeCu composite catalyst for CO synthesis by reverse water–gas shift reaction: Effect of Ce/Cu mol ratio. J. CO2 Util. 2017, 21, 292–301. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Chen, Y.; Yang, S. Reverse water–gas shift reaction over co-precipitated Co–CeO2 catalysts: Effect of Co content on selectivity and carbon formation. Int. J. Hydrogen Energy 2017, 42, 3682–3689. [Google Scholar] [CrossRef]
- Tang, R.; Zhu, Z.; Li, C.; Xiao, M.; Wu, Z.; Zhang, D.; Zhang, C.; Xiao, Y.; Chu, M.; Genest, A.; et al. Ru-catalyzed reverse water gas shift reaction with near-unity selectivity and superior stability. ACS Mater. Lett. 2021, 3, 1652–1659. [Google Scholar] [CrossRef]
- Tang, Y.; Asokan, C.; Xu, M.; Graham, G.W.; Pan, X.; Christopher, P.; Li, J.; Sautet, P. Rh single atoms on TiO2 dynamically respond to reaction conditions by adapting their site. Nat. Commun. 2019, 10, 4488. [Google Scholar] [CrossRef] [Green Version]
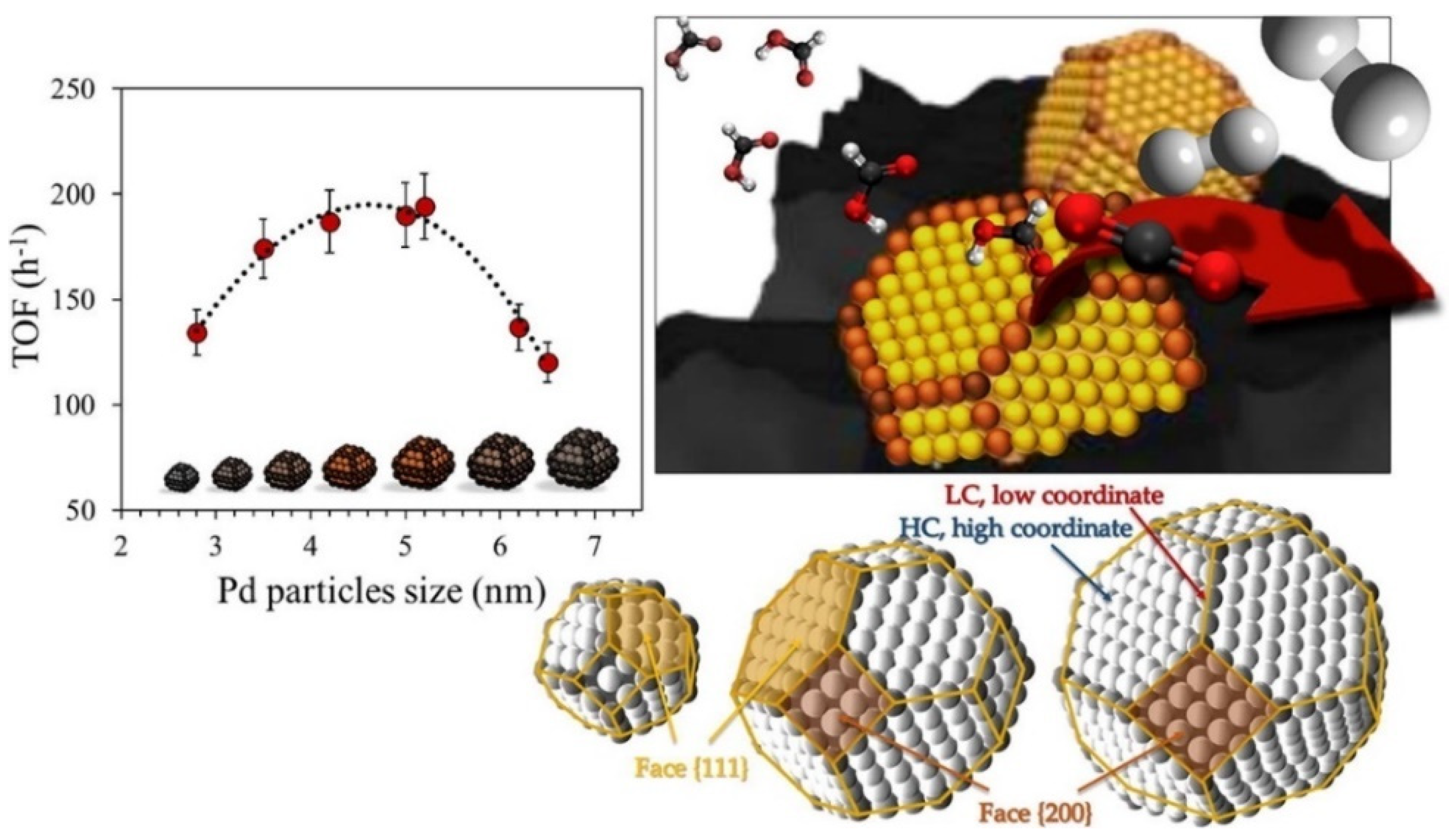
- Lebarbier, V.; Dagle, R.; Datye, A.; Wang, Y. The effect of PdZn particle size on reverse-water–gas-shift reaction. Appl. Catal. A Gen. 2010, 379, 3–6. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, M.; Ma, P.; Zheng, Y.; Chen, J.; Li, H.; Zhang, X.; Zheng, K.; Kuang, Q.; Xie, Z.-X. Atomically dispersed Pt/CeO2 catalyst with superior CO selectivity in reverse water gas shift reaction. Appl. Catal. B Environ. 2021, 291, 120101. [Google Scholar] [CrossRef]
- Panagiotopoulou, P. Hydrogenation of CO2 over supported noble metal catalysts. Appl. Catal. A Gen. 2017, 542, 63–70. [Google Scholar] [CrossRef]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Lee, H.H.; Hong, S.C. The effect of the morphological characteristics of TiO2 supports on the reverse water–gas shift reaction over Pt/TiO2 catalysts. Appl. Catal. B Environ. 2012, 119-120, 100–108. [Google Scholar] [CrossRef]
- Vourros, A.; Garagounis, I.; Kyriakou, V.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Marnellos, G.E.; Konsolakis, M. Carbon dioxide hydrogenation over supported Au nanoparticles: Effect of the support. J. CO2 Util. 2017, 19, 247–256. [Google Scholar] [CrossRef]
- Wang, L.C.; Tahvildar Khazaneh, M.; Widmann, D.; Behm, R.J. TAP reactor studies of the oxidizing capability of CO2 on a Au/CeO2 catalyst—A first step toward identifying a redox mechanism in the Reverse Water–Gas Shift reaction. J. Catal. 2013, 302, 20–30. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Song, W.; Liu, B.; Zheng, H.; Deng, J.; Zhong, W.; Liu, J.; Gong, X.-Q.; Zhao, Z. Elucidation of the high CO2 reduction selectivity of isolated Rh supported on TiO2: A DFT study. Catal. Sci. Technol. 2016, 6, 6128–6136. [Google Scholar] [CrossRef]
- Avanesian, T.; Gusmão, G.S.; Christopher, P. Mechanism of CO2 reduction by H2 on Ru(0001) and general selectivity descriptors for late-transition metal catalysts. J. Catal. 2016, 343, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Sabatier, P.; Senderens, J. Hydrogénation directe des oxydes du carbone en présence de divers métaux divisés. Comptes Rendus 1902, 134, 689–691. [Google Scholar]
- Xiaoding, X.; Moulijn, J.A. Mitigation of CO2 by chemical conversion: Plausible chemical reactions and promising products. Energy Fuels 1996, 10, 305–325. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Jacquemin, M.; Beuls, A.; Ruiz, P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157, 462–466. [Google Scholar] [CrossRef]
- Dubois, L.H.; Somorjai, G.A. Comments on “why CO2 does not dissociate on Rh at low temperature” by W.H. Weinberg. Surf. Sci. 1983, 128, L231–L235. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Türks, D.; Mena, H.; Armbruster, U.; Martin, A. Methanation of CO2 on Ni/Al2O3 in a Structured Fixed-Bed Reactor—A Scale-Up Study. Catalysts 2017, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Jalama, K. Carbon dioxide hydrogenation over nickel-, ruthenium-, and copper-based catalysts: Review of kinetics and mechanism. Catal. Rev. 2017, 59, 95–164. [Google Scholar] [CrossRef]
- Agnelli, M.; Swaan, H.M.; Márquez-Alvarez, C.; Martín, G.A.; Mirodatos, C. CO hydrogenation on a nickel catalyst: II. A mechanistic study by transient kinetics and infrared spectroscopy. J. Catal. 1998, 175, 117–128. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Wissing, L.; Skjøth-Rasmussen, M.S. High temperature methanation: Catalyst considerations. Catal. Today 2013, 215, 233–238. [Google Scholar] [CrossRef]
- Ghaib, K.; Nitz, K.; Ben-Fares, F.-Z. Chemical methanation of CO2: A Review. ChemBioEng Rev. 2016, 3, 266–275. [Google Scholar] [CrossRef]
- Xu, Y.; Wan, H.; Du, X.; Yao, B.; Wei, S.; Chen, Y.; Zhuang, W.; Yang, H.; Sun, L.; Tao, X.; et al. Highly active Ni/CeO2/SiO2 catalyst for low-temperature CO2 methanation: Synergistic effect of small Ni particles and optimal amount of CeO2. Fuel Process. Technol. 2022, 236, 107418. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Roger, A.-C. Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2 mixed oxide catalysts prepared by sol–gel method. Appl. Catal. A Gen. 2009, 369, 90–96. [Google Scholar] [CrossRef]
- Safdar, M.; González-Castaño, M.; Penkova, A.; Centeno, M.A.; Odriozola, J.A.; Arellano-García, H. CO2 methanation on Ni/YMn1−xAlxO3 perovskite catalysts. Appl. Mater. Today 2022, 29, 101577. [Google Scholar] [CrossRef]
- Vogt, C.; Groeneveld, E.; Kamsma, G.; Nachtegaal, M.; Lu, L.; Kiely, C.J.; Berben, P.H.; Meirer, F.; Weckhuysen, B.M. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 2018, 1, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Gil, A.; Díaz, A.; Gandía, L.M.; Montes, M. Influence of the preparation method and the nature of the support on the stability of nickel catalysts. Appl. Catal. A Gen. 1994, 109, 167–179. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Chemistry of nickel-alumina catalysts. J. Catal. 1976, 45, 41–53. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Li, J.; Gu, F.; Xu, G.; Zhong, Z.; Su, F. Nickel catalysts supported on calcium titanate for enhanced CO methanation. Catal. Sci. Technol. 2013, 3, 490–499. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 Methanation: The effect of catalysts and reaction conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Aldana, P.A.U.; Ocampo, F.; Kobl, K.; Louis, B.; Thibault-Starzyk, F.; Daturi, M.; Bazin, P.; Thomas, S.; Roger, A.C. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201–207. [Google Scholar]
- Navarro-Jaén, S.; Szego, A.; Bobadilla, L.F.; Laguna, Ó.H.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Operando spectroscopic evidence of the induced effect of residual species in the reaction intermediates during CO2 hydrogenation over ruthenium nanoparticles. ChemCatChem 2019, 11, 2063–2068. [Google Scholar] [CrossRef] [Green Version]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S. Production of synthetic natural gas (SNG) from coal and dry biomass—A technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
- Navarro, J.C.; Centeno, M.A.; Laguna, O.H.; Odriozola, J.A. Policies and motivations for the CO2 Valorization through the Sabatier reaction using structured catalysts. A review of the most recent advances. Catalysts 2018, 8, 578. [Google Scholar]
- Lawson, S.; Li, X.Z.; Thakkar, H.V.; Rownaghi, A.A.; Rezaei, F. Recent advances in 3D printing of structured materials for adsorption and catalysis applications. Chem. Rev. 2021, 121, 6246–6291. [Google Scholar] [CrossRef] [PubMed]
- Danaci, S.; Protasova, L.; Snijkers, F.; Bouwen, W.L.; Bengaouer, A.; Marty, P. Innovative 3D-manufacture of structured copper supports post-coated with catalytic material for CO2 methanation. Chem. Eng. Process. 2018, 127, 168–177. [Google Scholar] [CrossRef]
- Danaci, S.; Protasova, L.; Try, R.; Bengaouer, A.; Marty, P. Experimental and numerical investigation of heat transport and hydrodynamic properties of 3D-structured catalytic supports. Appl. Therm. Eng. 2017, 126, 167–178. [Google Scholar] [CrossRef]
- Danaci, S.; Protasova, L.; Lefevere, J.; Bedel, L.; Guilet, R.; Marty, P. Efficient CO2 methanation over Ni/Al2O3 coated structured catalysts. Catal. Today 2016, 273, 234–243. [Google Scholar] [CrossRef]
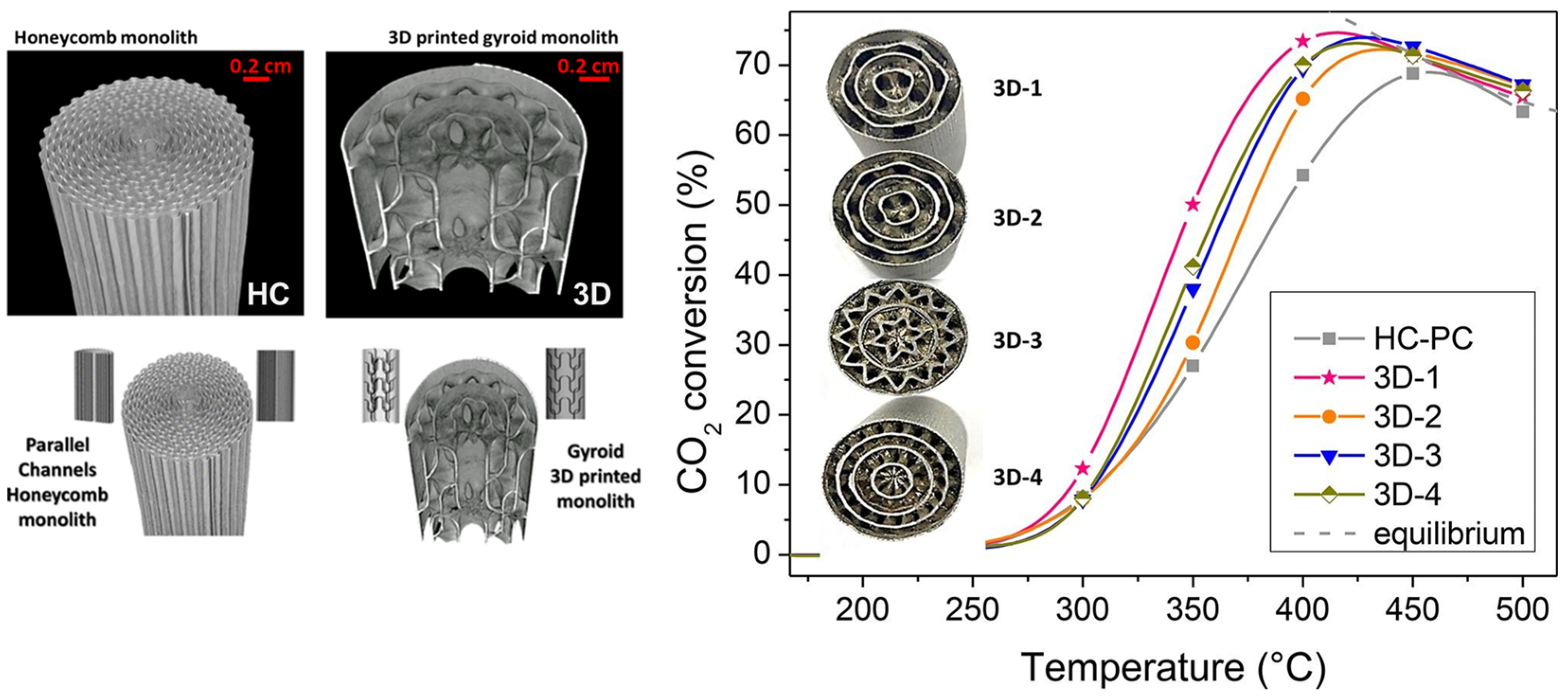
- González-Castaño, M.; Baena-Moreno, F.; Carlos Navarro de Miguel, J.; Miah, K.U.M.; Arroyo-Torralvo, F.; Ossenbrink, R.; Odriozola, J.A.; Benzinger, W.; Hensel, A.; Wenka, A.; et al. 3D-printed structured catalysts for CO2 methanation reaction: Advancing of gyroid-based geometries. Energy Conv. Manag. 2022, 258, 115464. [Google Scholar] [CrossRef]
- Baena-Moreno, F.M.; González-Castaño, M.; Navarro de Miguel, J.C.; Miah, K.U.M.; Ossenbrink, R.; Odriozola, J.A.; Arellano-García, H. Stepping toward efficient microreactors for CO2 methanation: 3D-printed gyroid geometry. ACS Sustain. Chem. Eng. 2021, 9, 8198–8206. [Google Scholar] [CrossRef]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Eppinger, J.; Huang, K.-W. Formic acid as a hydrogen energy carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source—recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Pérez-Fortes, M.; Schöneberger, J.C.; Boulamanti, A.K.; Harrison, G.; Tzimas, E. Formic acid synthesis using CO2 as raw material: Techno-economic and environmental evaluation and market potential. Int. J. Hydrogen Energy 2016, 41, 16444–16462. [Google Scholar] [CrossRef]
- Inoue, Y.; Izumida, H.; Sasaki, Y.; Hashimoto, H. Catalytic fixation of carbon dioxide to formic acid by transition-metal complexes under mild conditions. Chem. Lett. 1976, 5, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Behr, A.; Nowakowski, K. Book Series: Advances in Inorganic Chemistry; CO2 Chemistry; Elsevier: Amsterdam, The Netherlands, 2014; Volume 66. [Google Scholar]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Gunasekar, G.H.; Park, K.; Jung, K.-D.; Yoon, S. Recent developments in the catalytic hydrogenation of CO2 to formic acid/formate using heterogeneous catalysts. Inorg. Chem. Front. 2016, 3, 882–895. [Google Scholar] [CrossRef]
- Song, H.; Zhang, N.; Zhong, C.; Liu, Z.; Xiao, M.; Gai, H. Hydrogenation of CO2 into formic acid using a palladium catalyst on chitin. New J. Chem. 2017, 41, 9170–9177. [Google Scholar] [CrossRef]
- Upadhyay, P.R.; Srivastava, V. Selective hydrogenation of CO2 gas to formic acid over nanostructured Ru-TiO2 catalysts. RSC Adv. 2016, 6, 42297–42306. [Google Scholar] [CrossRef]
- Nguyen, L.T.M.; Park, H.; Banu, M.; Kim, J.Y.; Youn, D.H.; Magesh, G.; Kim, W.Y.; Lee, J.S. Catalytic CO2 hydrogenation to formic acid over carbon nanotube-graphene supported PdNi alloy catalysts. RSC Adv. 2015, 5, 105560–105566. [Google Scholar] [CrossRef]
- Maru, M.S.; Ram, S.; Shukla, R.S.; Khan, N.-u.H. Ruthenium-hydrotalcite (Ru-HT) as an effective heterogeneous catalyst for the selective hydrogenation of CO2 to formic acid. Mol. Catal. 2018, 446, 23–30. [Google Scholar] [CrossRef]
- Maru, M.S.; Ram, S.; Adwani, J.H.; Shukla, R.S. Selective and Direct Hydrogenation of CO2 for the Synthesis of Formic Acid over a Rhodium Hydrotalcite (Rh-HT) Catalyst. ChemistrySelect 2017, 2, 3823–3830. [Google Scholar] [CrossRef]
- Mori, K.; Taga, T.; Yamashita, H. Isolated single-atomic Ru catalyst bound on a layered double hydroxide for hydrogenation of CO2 to formic acid. ACS Catal. 2017, 7, 3147–3151. [Google Scholar] [CrossRef]
- Umegaki, T.; Satomi, Y.; Kojima, Y. Catalytic properties of palladium nanoparticles for hydrogenation of carbon dioxide into formic acid. J. Japan Inst. Energy 2017, 96, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551–12554. [Google Scholar] [CrossRef]
- Hao, C.; Wang, S.; Li, M.; Kang, L.; Ma, X. Hydrogenation of CO2 to formic acid on supported ruthenium catalysts. Catal. Today 2011, 160, 184–190. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Zhao, Y.; Ma, X. Hydrogenation of scCO2 to formic acid catalyzed by heterogeneous ruthenium(III)/Al2O3 catalysts. Chem. Lett. 2016, 45, 555–557. [Google Scholar] [CrossRef]
- He, C.-S.; Gong, L.; Zhang, J.; He, P.-P.; Mu, Y. Highly selective hydrogenation of CO2 into formic acid on a nano-Ni catalyst at ambient temperature: Process, mechanisms and catalyst stability. J. CO2 Util. 2017, 19, 157–164. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, T.; Wang, X.; Hao, R.; Wang, H. CO2 hydrogenation to formate over nano-scale zero-valent nickel catalyst under atmospheric pressure. Chem. Eng. J. 2018, 347, 860–869. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Vrijburg, W.L.; Hensen, E.J.M.; Pidko, E.A. On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates. J. Catal. 2016, 343, 97–105. [Google Scholar] [CrossRef]
- Su, J.; Yang, L.; Lu, M.; Lin, H. Highly efficient hydrogen storage system based on ammonium bicarbonate/formate redox equilibrium over palladium nanocatalysts. ChemSusChem 2015, 8, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Lu, M.; Lin, H. High yield production of formate by hydrogenating CO2 derived ammonium carbamate/carbonate at room temperature. Green Chem. 2015, 17, 2769–2773. [Google Scholar] [CrossRef]
- Bi, Q.-Y.; Lin, J.-D.; Liu, Y.-M.; Du, X.-L.; Wang, J.-Q.; He, H.-Y.; Cao, Y. An aqueous rechargeable formate-based hydrogen battery driven by heterogeneous Pd catalysis. Angew. Chem. Int. Ed. 2014, 53, 13583–13587. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Megías-Sayago, C.; Ivanova, S.; Centeno, M.Á.; Odriozola, J.A. Functionalized biochars as supports for Pd/C catalysts for efficient hydrogen production from formic acid. Appl. Catal. B Environ. 2021, 282, 119615. [Google Scholar] [CrossRef]
- Santos, J.L.; León, C.; Monnier, G.; Ivanova, S.; Centeno, M.Á.; Odriozola, J.A. Bimetallic PdAu catalysts for formic acid dehydrogenation. Int. J. Hydrogen Energy 2020, 45, 23056–23068. [Google Scholar] [CrossRef]
- Santos, J.L.; Megías-Sayago, C.; Ivanova, S.; Centeno, M.Á.; Odriozola, J.A. Structure-sensitivity of formic acid dehydrogenation reaction over additive-free Pd NPs supported on activated carbon. Chem. Eng. J. 2021, 420, 127641. [Google Scholar] [CrossRef]
- Qin, Y.-l.; Wang, J.; Meng, F.-z.; Wang, L.-m.; Zhang, X.-B. Efficient PdNi and PdNi@Pd-catalyzed hydrogen generation via formic acid decomposition at room temperature. Chem. Commun. 2013, 49, 10028–10030. [Google Scholar] [CrossRef]
- Iglesia, E.; Boudart, M. Decomposition of formic acid on copper, nickel, and copper-nickel alloys: III. Catalytic decomposition on nickel and copper-nickel alloys. J. Catal. 1983, 81, 224–238. [Google Scholar] [CrossRef]
- Navlani-García, M.; Mori, K.; Salinas-Torres, D.; Kuwahara, Y.; Yamashita, H. New approaches toward the hydrogen production from formic acid dehydrogenation over Pd-based heterogeneous catalysts. Front. Mater. 2019, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Cazaña, F.; Galetti, A.; Meyer, C.; Sebastián, V.; Centeno, M.A.; Romeo, E.; Monzón, A. Synthesis of Pd-Al/biomorphic carbon catalysts using cellulose as carbon precursor. Catal. Today 2018, 301, 226–238. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Gärtner, F.; Junge, H.; Beller, M. Catalytic generation of hydrogen from formic acid and its derivatives: Useful hydrogen storage materials. Top. Catal. 2010, 53, 902–914. [Google Scholar] [CrossRef]
- Beloqui Redondo, A.; Morel, F.L.; Ranocchiari, M.; van Bokhoven, J.A. Functionalized ruthenium–phosphine Metal–Organic Framework for continuous vapor-phase dehydrogenation of formic acid. ACS Catal. 2015, 5, 7099–7103. [Google Scholar] [CrossRef]
- Lee, J.H.; Ryu, J.; Kim, J.Y.; Nam, S.-W.; Han, J.H.; Lim, T.-H.; Gautam, S.; Chae, K.H.; Yoon, C.W. Carbon dioxide mediated, reversible chemical hydrogen storage using a Pd nanocatalyst supported on mesoporous graphitic carbon nitride. J. Mater. Chem. A 2014, 2, 9490–9495. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Wang, H.-L.; Ping, Y.; Jiang, Q. Au@Pd core–shell nanoclusters growing on nitrogen-doped mildly reduced graphene oxide with enhanced catalytic performance for hydrogen generation from formic acid. J. Mater. Chem. A 2013, 1, 12721–12725. [Google Scholar] [CrossRef]
- Caiti, M.; Padovan, D.; Hammond, C. Continuous production of hydrogen from formic acid decomposition over heterogeneous nanoparticle catalysts: From batch to continuous flow. ACS Catal. 2019, 9, 9188–9198. [Google Scholar] [CrossRef]
- Hafeez, S.; Sanchez, F.; Al-Salem, S.M.; Villa, A.; Manos, G.; Dimitratos, N.; Constantinou, A. Decomposition of additive-free formic acid using a Pd/C catalyst in flow: Experimental and CFD modelling studies. Catalysts 2021, 11, 341. [Google Scholar] [CrossRef]
- Katuri, P.; Maralla, Y.S.S.; Tumma, B.N. Production of performic acid through a capillary microreactor by heterogeneous catalyst. Int. J. Chem. Reactor Eng. 2020, 20. In press. [Google Scholar] [CrossRef]
- Hafeez, S.; Al-Salem, S.M.; Bansode, A.; Villa, A.; Dimitratos, N.; Manos, G.; Constantinou, A. Computational investigation of microreactor configurations for hydrogen production fromformic acid decomposition using a Pd/C catalyst. Ind. Eng. Chem. Res. 2022, 61, 1655–1665. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl ether (DME) as an alternative fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Arcoumanis, C.; Bae, C.; Crookes, R.J.; Kinoshita, E. The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel 2008, 87, 1014–1030. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the development of novel cobalt Fischer−Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Shikada, T.; Ohno, Y.; Ogawa, T.; Ono, M.; Mizuguchi, M.; Tomura, K.; Fujimoto, K. Direct synthesis of dimethyl ether form synthesis gas. In Studies in Surface Science and Catalysis; Parmaliana, A., Sanfilippo, D., Frusteri, F., Vaccari, A., Arena, F., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Volume 119, pp. 515–520. [Google Scholar]
- Ohno, Y.; Yoshida, M.; Shikada, T.; Inokoshi, O.; Ogawa, T.; Inoue, N. New Direct Synthesis Technology for DME (Dimethyl Ether) and Its Application Technology; JFE Technical Report, nº 8; JFE Holdings Inc.: Tokyo, Japan, October 2006. [Google Scholar]
- Shikada, T.; Ohno, Y.; Ogawa, T.; Mizuguchi, M.; Ono, M.; Fujimoto, K. Catalyst for Producing Dimethyl Ether, Method for Producing Catalyst and Method for Producing Dimethyl Ether. U.S. Patent 7,033,972 B2, 25 April 2006. [Google Scholar]
- Rostrup-Nielsen, T.; Madsen, J. Process for the Preparation of Pure Dimethyl Ether. U.S. Patent 7,910,630 B2, 22 March 2011. [Google Scholar]
- Joensen, F.; Madsen, J.; Erik, P. Process for the Preparation of Dimethyl Ether. WO 2014032973 A1, 6 March 2014. [Google Scholar]
- Lewnard, J.J.; Hsiung, T.H.; White, J.F.; Bhatt, B.L. Liquid Phase Process for Dimethyl Ether Synthesis. U.S. Patent 5,218,003, 8 June 1993. [Google Scholar]
- Peng, X. Use of Aluminum Phosphate as the Dehydration Catalyst in Single Step Dimethyl Ether Process. U.S. Patent 5,753,716, 19 May 1998. [Google Scholar]
- Pagani, G. Process for the Production of Dimethyl Ether. U.S. Patent 4,098,809, 4 July 1978. [Google Scholar]
- Bell, W.K.; Chang, C.D. Dimethyl Ether Synthesis Catalysts. U.S. Patent 4,423,155, 27 December 1983. [Google Scholar]
- Khandan, N.; Kazemeini, M.; Aghaziarati, M. Direct production of dimethyl ether from synthesis gas utilizing bifunctional catalysts. Appl. Petrochem. Res. 2012, 1, 21–27. [Google Scholar] [CrossRef]
- Bridger, G.W.; Spencer, M.S. Catalyst Handbook, 2nd ed.; Twigg, M.V., Ed.; Wofle Press: London, UK, 1989. [Google Scholar]
- Stiefel, M.; Ahmad, R.; Arnold, U.; Döring, M. Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions. Fuel Process. Technol. 2011, 92, 1466–1474. [Google Scholar] [CrossRef]
- Cheiky, M. Low Pressure Dimethyl Ether Synthesis Catalyst. U.S. Patent 2013/0211147 A1, 15 August 2013. [Google Scholar]
- García-Trenco, A.; Martínez, A. Direct synthesis of DME from syngas on hybrid CuZnAl/ZSM-5 catalysts: New insights into the role of zeolite acidity. Appl. Catal. A Gen. 2012, 411–412, 170–179. [Google Scholar] [CrossRef] [Green Version]
- García-Trenco, A.; Martínez, A. The influence of zeolite surface-aluminum species on the deactivation of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis. Catal. Today 2014, 227, 144–153. [Google Scholar] [CrossRef]
- García-Trenco, A.; Martínez, A. A rational strategy for preparing Cu–ZnO/H-ZSM-5 hybrid catalysts with enhanced stability during the one-step conversion of syngas to dimethyl ether (DME). Appl. Catal. A Gen. 2015, 493, 40–49. [Google Scholar] [CrossRef]
- García-Trenco, A.; Valencia, S.; Martínez, A. The impact of zeolite pore structure on the catalytic behavior of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis. Appl. Catal. A Gen. 2013, 468, 102–111. [Google Scholar] [CrossRef] [Green Version]
- García-Trenco, A.; Vidal-Moya, A.; Martínez, A. Study of the interaction between components in hybrid CuZnAl/HZSM-5 catalysts and its impact in the syngas-to-DME reaction. Catal. Today 2012, 179, 43–51. [Google Scholar] [CrossRef]
- Xiao, K.; Wang, Q.; Qi, X.; Zhong, L. For better industrial Cu/ZnO/Al2O3 methanol synthesis catalyst: A Compositional Study. Catal. Lett. 2017, 147, 1581–1591. [Google Scholar] [CrossRef]
- Sun, J.; Yang, G.; Ma, Q.; Ooki, I.; Taguchi, A.; Abe, T.; Xie, Q.; Yoneyama, Y.; Tsubaki, N. Fabrication of active Cu–Zn nanoalloys on H-ZSM5 zeolite for enhanced dimethyl ether synthesis via syngas. J. Mater. Chem. A 2014, 2, 8637–8643. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Sanz, O.; Banús, E.D.; Goya, A.; Larumbe, H.; Delgado, J.J.; Monzón, A.; Montes, M. Stacked wire-mesh monoliths for VOCs combustion: Effect of the mesh-opening in the catalytic performance. Catal. Today 2017, 296, 76–83. [Google Scholar] [CrossRef]
- Merino, D.; Sanz, O.; Montes, M. Effect of catalyst layer macroporosity in high-thermal-conductivity monolithic Fischer-Tropsch catalysts. Fuel 2017, 210, 49–57. [Google Scholar] [CrossRef]
- Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors: New Technology for Modern Chemistry; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2000. [Google Scholar]
- Hessel, V.; Renken, A.; Schouten, J.C.; Yoshida, J. (Eds.) Micro Process Engineering. A Comprehensive Handbook; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2009. [Google Scholar]
- Hu, J.; Wang, Y.; Cao, C.; Elliott, D.C.; Stevens, D.J.; White, J.F. Conversion of biomass syngas to DME using a microchannel reactor. Ind. Eng. Chem. Res. 2005, 44, 1722–1727. [Google Scholar] [CrossRef]
- Hayer, F.; Bakhtiary-Davijany, H.; Myrstad, R.; Holmen, A.; Pfeifer, P.; Venvik, H.J. Synthesis of dimethyl ether from syngas in a microchannel reactor—Simulation and experimental study. Chem. Eng. J. 2011, 167, 610–615. [Google Scholar] [CrossRef]
- Dagle, R.A.; Wang, Y.; Baker, E.G.; Hu, J. Dimethyl Ether Production from Methanol and/or Syngas. U.S. Patent 8,957,259 B2, 17 February 2015. [Google Scholar]
- Venvik, H.J.; Yang, J. Catalysis in microstructured reactors: Short review on small-scale syngas production and further conversion into methanol, DME and Fischer-Tropsch products. Catal. Today 2017, 285, 135–146. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A. Direct synthesis of DME over nanostructured CuO–ZnO–Al2O3/HZSM-5 catalyst washcoated on high pressure microreactor: Effect of catalyst loading and process condition on reactor performance. Chem. Eng. J. 2015, 262, 1175–1186. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Dai, Y.; Zhang, J.; Yang, Y. The thermodynamics analysis and experimental validation for complicated systems in CO2 hydrogenation process. J. Energy Chem. 2016, 25, 1027–1037. [Google Scholar] [CrossRef]
- Noriyuki, I.; Kouji, H.; Atsushi, S.; Tadashi, H.; Yuichi, M. Unique temperature dependence of acetic acid formation in CO2 hydrogenation on Ag-promoted Rh/SiO2 catalyst. Chem. Lett. 1994, 23, 263–264. [Google Scholar]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. State of the art and perspectives in catalytic processes for CO2 conversion into chemicals and fuels: The distinctive contribution of chemical catalysis and biotechnology. J. Catal. 2016, 343, 2–45. [Google Scholar] [CrossRef]
- Bristow, T.G. Integrated Process for Making Acetic Acid. WO2014096254, 26 June 2014. [Google Scholar]
- Somiari, I.; Manousiouthakis, V.I. Coproduction of acetic acid and hydrogen/power from natural gas with zero carbon dioxide emissions. AIChE J. 2018, 64, 860–876. [Google Scholar] [CrossRef] [Green Version]
- Dagle, R.A.; Hu, J.; Jones, S.B.; Wilcox, W.; Frye, J.G.; White, J.F.; Jiang, J.; Wang, Y. Carbon dioxide conversion to valuable chemical products over composite catalytic systems. J. Energy Chem. 2013, 22, 368–374. [Google Scholar]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Law, D.J.; Iglesia, E. Site requirements and elementary steps in dimethyl ether carbonylation catalyzed by acidic zeolites. J. Catal. 2007, 245, 110–123. [Google Scholar] [CrossRef]
- Xu, B.-Q.; Sachtler, W.M.H. Rh/NaY: A selective catalyst for direct synthesis of acetic acid from syngas. J. Catal. 1998, 180, 194–206. [Google Scholar] [CrossRef]
- Han, L.; Mao, D.; Yu, J.; Guo, Q.; Lu, G. C2-oxygenates synthesis through CO hydrogenation on SiO2-ZrO2 supported Rh-based catalyst: The effect of support. Appl. Catal. A Gen. 2013, 454, 81–87. [Google Scholar] [CrossRef]
- Yu, J.; Mao, D.; Lu, G.; Guo, Q.; Han, L. Enhanced C2 oxygenate synthesis by CO hydrogenation over Rh-based catalyst supported on a novel SiO2. Catal. Commun. 2012, 24, 25–29. [Google Scholar]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Iglesia, E. Selective carbonylation of dimethyl ether to methyl acetate catalyzed by acidic zeolites. Angew. Chem. Int. Ed. 2006, 45, 1617–1620. [Google Scholar] [CrossRef] [PubMed]
- Volkova, G.G.; Plyasova, L.M.; Shkuratova, L.N.; Budneva, A.A.; Paukshtis, E.A.; Timofeeva, M.N.; Likholobov, V.A. Solid superacids for halide-free carbonylation of dimethyl ether to methyl acetate. Stud. Surf. Sci. Catal. 2004, 147, 403–408. [Google Scholar]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Blasco, T.; Boronat, M.; Concepción, P.; Corma, A.; Law, D.; Vidal-Moya, J.A. Carbonylation of methanol on metal–acid zeolites: Evidence for a mechanism involving a multisite active center. Angew. Chem. Int. Ed. 2007, 46, 3938–3941. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Allian, A.D.; Sunley, G.J.; Law, D.J.; Iglesia, E. Specificity of sites within eight-membered ring zeolite channels for carbonylation of methyls to acetyls. J. Am. Chem. Soc. 2007, 129, 4919–4924. [Google Scholar] [CrossRef]
- Boronat, M.; Martínez-Sánchez, C.; Law, D.; Corma, A. Enzyme-like specificity in zeolites: A unique site position in mordenite for selective carbonylation of methanol and dimethyl ether with CO. J. Am. Chem. Soc. 2008, 130, 16316–16323. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.; Martínez, C.; Corma, A. Mechanistic differences between methanol and dimethyl ether carbonylation in side pockets and large channels of mordenite. Phys. Chem. Chem. Phys. 2011, 13, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Reule, A.A.C.; Shen, J.; Semagina, N. Copper affects the location of zinc in bimetallic ion-exchanged mordenite. ChemPhysChem 2018, 19, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Luzgin, M.V.; Kazantsev, M.S.; Volkova, G.G.; Stepanov, A.G. Solid-state NMR study of the kinetics and mechanism of dimethyl ether carbonylation on cesium salt of 12-tungstophosphoric acid modified with Ag, Pt, and Rh. J. Catal. 2013, 308, 250–257. [Google Scholar] [CrossRef]
- Xue, H.; Huang, X.; Ditzel, E.; Zhan, E.; Ma, M.; Shen, W. Coking on micrometer- and nanometer-sized mordenite during dimethyl ether carbonylation to methyl acetate. Chin. J. Catal. 2013, 34, 1496–1503. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, Y.; Du, L.; Liu, J.; Yao, J. Review of the applications of microreactors. Renew. Sustain. Energy Rev. 2015, 47, 519–539. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Global Anthropogenic CO2 Emission | Current CO2 Utilisation | ||
|---|---|---|---|
| Sector | Emissions (%) | Interest Products | CO2 Used (Mton/Year) |
| Energy (electricity, heat, and transport) | 73.2% | Urea | 114 |
| Inorganic carbonates | 70 | ||
| Agriculture, forestry, and land use | 18.4% | Methanol | 10 |
| Formaldehyde | 5 | ||
| Dimethyl ether (DME) | 1.5 | ||
| Direct industrial processes (construction, chemicals, and petrochemicals) | 5.2% | Methyl tert-butyl ether (MTBE) | 3.5 |
| Algae for biodiesel production | 2 | ||
| Formic acid | 0.9 | ||
| Waste (wastewater and landfills) | 3.2% | Polycarbonates | 1.5 |
| Polymers | 4 | ||
| Total in Mton/year | 32,189.7 (100%) | Total | 212.4 |
| Promising Products | Chemical Reaction Process | Uses and Applications |
|---|---|---|
| Syngas | Gas mixture used for synthesis of larger hydrocarbons via FTS | |
| Methane | Synthetic natural gas used for syngas production and ammonia synthesis | |
| Formic acid | Production of chemicals in textile and rubber industries. Utilisation as hydrogen carrier and energy vector | |
| Acetic acid | Food industry, cosmetics, manufacturing of plastics, additives, synthesis of acetic anhydride | |
| DME | Fuel alternative for diesel engines, energy vector, cosmetics, synthesis of olefins and aromatics | |
| Higher HCs | Production of synthetic liquid transportation fuels and additives | |
| Polycarbonates and cyclic carbonates |  | Plastics, electrolytes in batteries, electronic devices, automotive and aircraft components |
| Inorganic carbonates | Construction materials, drying agents, detergents, fire extinguishers, dusting powder, CO2 sequestering agents | |
| Urea | Production of fertilisers, plastics, and resins | |
| Methanol | Alternative transportation fuel, production of valuable chemicals (acetic acid, formaldehyde, and DME), H2 storage | |
| Polyurethanes |  | Production of elastomers, rubbers, adhesives, foams, coatings, and sealants |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobadilla, L.F.; Azancot, L.; Luque-Álvarez, L.A.; Torres-Sempere, G.; González-Castaño, M.; Pastor-Pérez, L.; Yu, J.; Ramírez-Reina, T.; Ivanova, S.; Centeno, M.A.; et al. Development of Power-to-X Catalytic Processes for CO2 Valorisation: From the Molecular Level to the Reactor Architecture. Chemistry 2022, 4, 1250-1280. https://doi.org/10.3390/chemistry4040083
Bobadilla LF, Azancot L, Luque-Álvarez LA, Torres-Sempere G, González-Castaño M, Pastor-Pérez L, Yu J, Ramírez-Reina T, Ivanova S, Centeno MA, et al. Development of Power-to-X Catalytic Processes for CO2 Valorisation: From the Molecular Level to the Reactor Architecture. Chemistry. 2022; 4(4):1250-1280. https://doi.org/10.3390/chemistry4040083
Chicago/Turabian StyleBobadilla, Luis F., Lola Azancot, Ligia A. Luque-Álvarez, Guillermo Torres-Sempere, Miriam González-Castaño, Laura Pastor-Pérez, Jie Yu, Tomás Ramírez-Reina, Svetlana Ivanova, Miguel A. Centeno, and et al. 2022. "Development of Power-to-X Catalytic Processes for CO2 Valorisation: From the Molecular Level to the Reactor Architecture" Chemistry 4, no. 4: 1250-1280. https://doi.org/10.3390/chemistry4040083