

Acid Site Density as a Kinetic Descriptor of Catalytic Reactions over Zeolites

Laboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University, 20500 Turku/Åbo, Finland
Chemistry 2022, 4(4), 1609-1623; https://doi.org/10.3390/chemistry4040105
Submission received: 5 November 2022
/
Revised: 21 November 2022
/
Accepted: 22 November 2022
/
Published: 23 November 2022
(This article belongs to the Special Issue Heterogeneous Catalysis — A Themed Issue in Honor of Prof. Dr. Avelino Corma)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:A mathematical framework for the quantitative description of site density dependence of catalytic data (activity and selectivity) was developed considering that changes in the electrostatic contribution to the Gibbs energy of an elementary reaction on the acid sites in zeolites depend on the proximity of these sites. For the two-step sequence with the most abundant surface intermediate, an expression for turnover frequency explicitly containing the acid site density was derived. The treatment was extended to linear sequences of elementary reaction and analysis of the acid site density on selectivity in parallel and consecutive reactions, allowing to quantitatively relate the ratio between products for such reactions. Experimental data on Prins condensation of isopulegol with acetone and transformations of syngas over mesoporous H-ZSM-5 supported cobalt nanoparticles to a mixture of iso- and normal hydrocarbons were used as a show case.
1. Introduction
Solid Brønsted acid catalysts, such as zeolites, are of immense importance for oil refining and transformations of chemicals [1,2,3]. Significant efforts have been made on the development of novel materials and mechanistic interpretation of catalytic data. Several parameters, including the structure, morphology and the Si/Al ratio, presence of various ions, etc., can be used to fine-tune the number, nature, strength and distribution of acid sites [4,5,6,7].
Kinetic analysis of catalytic reactions over zeolites often is limited by only considering the acid sites as isolated and noninteracting with each other, allowing to apply the classical approaches of heterogeneous catalytic kinetics [8,9,10]. On the other hand, there is a little doubt that active sites in zeolitic materials possess different acid strength and thus activity, which is manifested by analysis of acidity with ammonia or pyridine [11,12,13] or calorimetry [14].
One of the descriptors, which can be used to reflect the complexity of catalytic reactions over zeolites, is the acid site density [15,16], as the dependence of turnover frequency (TOF) as a function of this parameter indicates a structure sensitivity similar to the dependence of TOF vs. the metal dispersion in the case of catalysis over supported metals. Recently, it was demonstrated that TOF calculated per proton can be independent of the acid site density, decrease, or even pass through the maximum [15,17,18,19]. Moreover, in [19], it was shown that for the Prins cyclization of (-) isopulegol with acetone, also selectivity, expressed as the ratio of products, depends on the acid site concentration.
2. Site Density Dependence for Adsorption
An explanation proposed for the maxima in TOF vs. the acid site density was related [17] to changes in the ionic strength depending itself on the volumetric densities of hydronium ions, and thus on the concentration of Brønsted acid sites. The general applicability of the Debye–Hückel or the semi-empirical Truesdell–Jones equations to the nanoconfined space of zeolites might be questionable [20], and thus an alternative approach to account for the spatial constraints in such a nanoconfined space was proposed [21].
More precisely, along the nonelectrostatic contribution to the Gibbs energy of the solid surface with acid sites, also the electrostatic one was considered:
where is the charge of acid sites/hydronium ions, is the average distance between these ions/acid sites, is the permittivity in vacuum, dielectric constant, e is the charge of the electron, NA is Avogadro’s number, and is lumped constant .
Changes in the electrostatic contribution to the Gibbs energy upon adsorption on the acid sites with a partial donation of protons to the adsorbate were expressed [21] as
where is the increment of the electrostatic contribution to the Gibbs energy upon adsorption on the acid sites. From Equation (2), changes in the electrostatic contribution depend on proximity of sites or the average distance between the acid sites . From the relationship between the Gibbs energy and the equilibrium constant the equilibrium constant for adsorption can be easily expressed:
Further linking the rate constant k with the equilibrium constants K through the linear free energy relationship [22] the rate constant of adsorption takes the form
where α is the Polanyi parameter (0 < α < 1). The average distance between acid sites can be calculated through the density of acid sites (mol/g) defined via the overall surface area divided by the effective area around the surface site. This effective area in the simplest case is taken as a circle with the diameter equal to the average distance between acid sites giving, thus,
and subsequently
Finally, the rate of adsorption can be expressed via the Brønsted acid site density:
where
3. Two-Step Sequence
Quite often, heterogeneous catalytic kinetics is expressed by the two-step mechanism with two kinetically significant steps [23,24], and one most abundant surface intermediate I:
where A1 and A2 are reactants, B1, and B2 are products, * is the surface vacant site, and I is an adsorbed intermediate.
1. * + A1 ↔ *I + B1
2. *I + A2↔ * + B2
A1 + A2 ↔ B1 + B2
2. *I + A2↔ * + B2
A1 + A2 ↔ B1 + B2
The reaction rate for this mechanism being well-known is presented below for turnover frequency per acid site [25]:
where CA1, etc., are concentrations of reagents, and ki is the rate constants.
The rate constant for the first step in mechanism (9) directly follows from Equation (7), namely,
where α1 is the Polanyi parameter of the first step. Similarly, for the backward reaction of the second step, one obtains
The rate constant for the backward reaction of the first step is obtained from Equation (11) and the acid site density dependence of the adsorption equilibrium constant:
leading to
Analogously, it holds for the forward reaction of the second step:
The TOF per acid site can be expressed subsequently:
When both steps are irreversible, Equation (16) can be transformed into
It can be demonstrated that Equation (18) exhibits a maximum of TOF as a function of the acid site density. To illustrate this, a minimum of the reciprocal value of TOF can be determined by taking d(1/TOF)/d equal to zero:
Giving then
Equation (21), after some manipulations, is
and
can rewritten in a form allowing to obtain an explicit expression for the acid site density at which the maximum in TOF is observed:
When the Polanyi parameters of the steps are equal to each other, Equation (25) can be simplified:
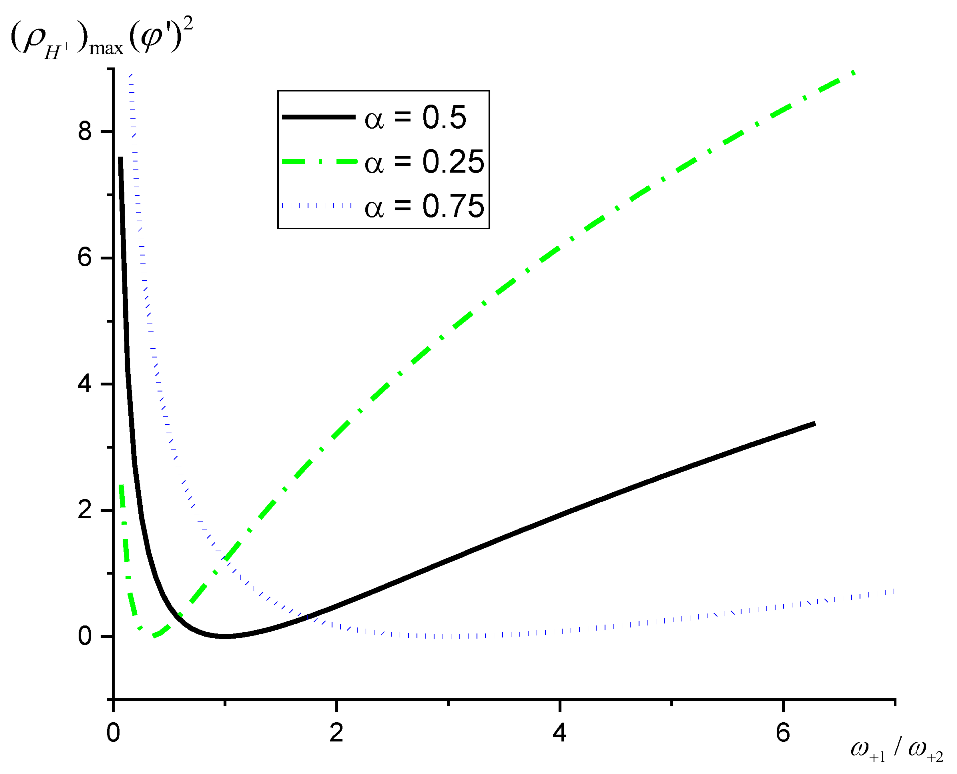
Figure 1 illustrates that the value of the acid site density at which maxima in the TOF are observed depends on the values of the Polanyi parameter and the frequencies of steps. The latter implies that not only the rate constants vary depending on the catalyst, but also the concentrations of reagents can have an impact on the experimentally observed values of the acid site density corresponding to the maxima in TOF. For high values of the Polanyi parameter (e.g., 0.75), such dependence will be less pronounced.
Comparison of the experimental data for reactions occurring over catalysts with different acid site density is typically performed at the same values of the reaction parameters (i.e., temperature and concentration of reagents). The current theoretical analysis highlights that the acid site density, at which TOF exhibits a maximum, is a function not only on the catalyst, but the process parameters as well. This apparently urges a detailed experimental exploration of this hypothesis.
4. Christiansen Sequence
An extension of the reaction mechanism discussed above is a Christiansen sequence containing a linear step of isomerization in the adsorbed state [26]:
1.* +A1 ↔ *I1 + B1
2.* I1 ↔ *I2
3. * I2 + A2↔ * + B2
A1 + A2 ↔ B1 + B2
2.* I1 ↔ *I2
3. * I2 + A2↔ * + B2
A1 + A2 ↔ B1 + B2
Such a type of generic mechanism can be relevant in the context of skeletal isomerization, cracking or alkylation reactions. In a simplified treatment of this reaction mechanism, it can be assumed that or the increment of the electrostatic contribution to the Gibbs energy upon adsorption on the acid sites is the same for both intermediates I1 and I2.
The equilibrium constant of the first step is then
For the third step of Equation (27), it can be written as
As the overall constant does not depend on the acid site density, it is apparently clear that for the isomerization step 2 in (27), the equilibrium constant does not depend on the acid site density either.
With the frequencies of steps in the particular case of the three-step reaction on acid sites,
When in the reaction mechanism all steps are irreversible Equation (30) can be further simplified to
Often, the value of the Polanyi parameter is equal to 0.5 [25], implying that and
The acid site density can be determined in a similar fashion as for the two-step sequence (Equation (20)), giving
Thus,
leading to an expression of the acid site density at the maximum TOF when the Polanyi parameters are equal to 0.5.
Similar to the treatment above, the acid site density, at which TOF exhibits a maximum, depends on the frequencies of steps and thus process parameters.
The treatment above considered that the increment of the electrostatic contribution to the Gibbs energy of adsorbed species is the same, which should not be necessarily the case upon adsorption on the acid sites for both intermediates I1 and I2.
In such an instance, instead of Equation (8), the relevant expressions for intermediates I1 and I2 will be
resulting in the expressions for turnover frequencies of steps 1 and 3 of the reaction mechanism (27):
Considering the modified equilibrium constant of these steps,
An expression for the equilibrium constant of the second step is
Implying that
For the three-step catalytic sequence of all irreversible steps, it holds that
which for is simplified to
Apparently, Equation (43) also exhibits a maximum in TOF as a function of the acid site density, the determination of which requires solving the following rather complex equation:
5. Parallel Reactions: Coupling between Cycles
The conceptual ideas in the analysis above can be applied to elucidation of the influence of acid site density on selectivity in parallel and consecutive reactions.
First, the case of kinetic coupling between catalytic cycles will be considered with a joint reaction intermediate:
1.* + A1 ↔ * I1 + B1 1 1
2. * I1 + A2 ↔ * + B2 1 0
3. * I1 + A3↔ * + B3 0 1
N(1) A1 + A2 ↔ B1 + B2; N(2) A1 + A3 ↔ B1 + B3
2. * I1 + A2 ↔ * + B2 1 0
3. * I1 + A3↔ * + B3 0 1
N(1) A1 + A2 ↔ B1 + B2; N(2) A1 + A3 ↔ B1 + B3
The reaction scheme in Equation (47) reflects two reaction routes, N(1) and N(2), taking place simultaneously. On the right-hand side of the equations for the steps, the respective stoichiometric (Horiuti) numbers are given, which should be multiplied by the equations of steps to yield the chemical equations along the different routes. For example, after multiplying the equations of the first and the second steps in Equation (45) by unity and the third step by zero and summing up all concentrations on the left and right sides, concentrations of the surface species are cancelled, giving the equation for the first route, i.e., A1 + A2 ↔ B1 + B2.
A chemical example of such a mechanism can be the formation of carbocations in the first step with the subsequent splitting of different carbon–carbon bonds. For the sake of simplicity, just the irreversible steps in Equation (45) are considered. It can be easily demonstrated using the steady-state approximation,
that
leading subsequently to
The overall rate is
Selectivity to a product B2 can be easily obtained from Equations (48) and (49), giving
Analogously, it holds for the forward reaction of the second and third steps similar to Equation (15) that
Dependence of selectivity on the acid site density for the simplified case of three irreversible steps is thus
while the ratio between the products is defined as
The latter equation can be linearized as
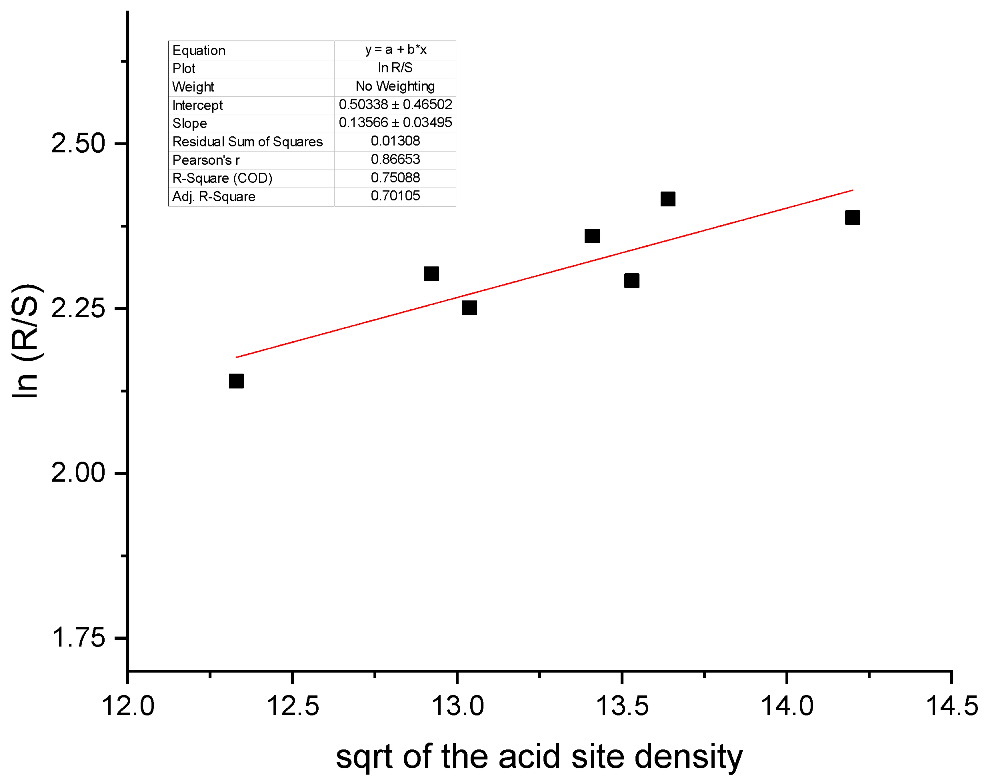
allowing to relate the rates and subsequently the ratios between products with the square root of the acid site density. Figure 2 illustrates the successful implementation of Equation (55) to treat the experimental ratio of 4R and 4S chromenols obtained from the Prins cyclization of (-)isopulegol with acetone [19] as a function of the square root of the total acid site density.
6. Parallel Reactions: Separate Cycles
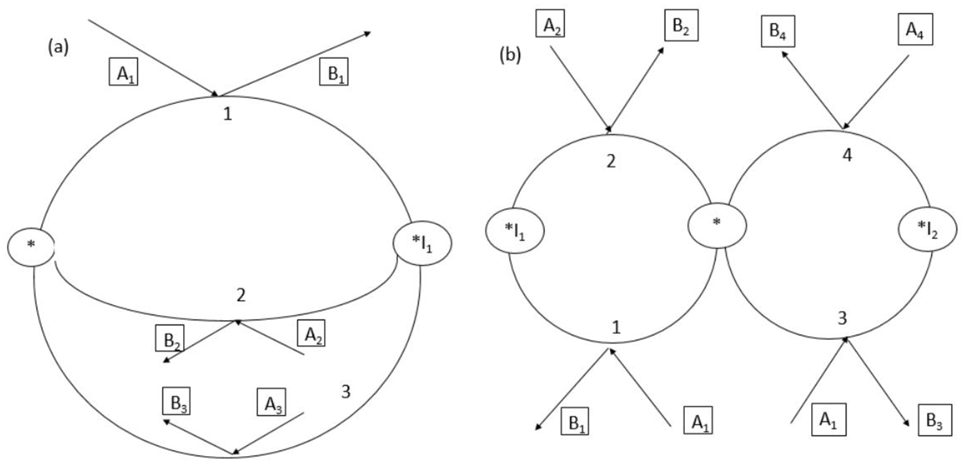
The mechanism given by Equation (45) represents an example of a catalytic sequence with a joint edge connecting the nodes of the graph (Figure 3a) using the formalism of the graph theory [27]. One route shown in Figure 3 comprises steps 1 and 2, while in the second route besides step 1 common for both routes, step 3 is involved.
On the contrary, in the mechanism with two parallel routes featured in Figure 3b, there is only one common node corresponding to the vacant acid site and separate cycles with stepss 1, 2 and 3, 4. Subsequently, in the most general case, the reaction mechanism can be written in the following way:
1.* + A1 ↔ * I1 + B1 1 0
2. *I1 + A2 ↔ * + B2 1 0
3.* +A1 ↔ * I2 + B3 0 1
4. *I2 + A3↔ * + B4 0 1
N(1): A1 + A2 ↔ B1 + B2; N(2): A1 + A3 ↔ B3 + B4
2. *I1 + A2 ↔ * + B2 1 0
3.* +A1 ↔ * I2 + B3 0 1
4. *I2 + A3↔ * + B4 0 1
N(1): A1 + A2 ↔ B1 + B2; N(2): A1 + A3 ↔ B3 + B4
There are obviously many variations of the mechanism in Equation (56), such as the cases when A2 is the same as A3 reflecting for example different adsorption modes through different functional groups, or when B1 is the same as B3. The corresponding equations can be easily derived from a more general consideration.
For the sake of simplicity, steps 2 and 4 are considered irreversible, giving
where is the coverage of vacant site and , are coverages of respective intermediates. The rates along different routes can be calculated considering the site balance resulting this in
with
The ratio between the rates along different routes is thus
The expressions for the rate constants as a function of the site density follow from the general considerations discussed above, giving
The ratio of rates takes a relatively complicated form:
An expression similar to Equation (54) can be obtained when all steps are irreversible with, however, different dependencies on the concentration of substrates:
Equation (63) can be also linearized in the same way as Equation (55).
7. Consecutive Reactions
The last example is related to a network of consecutive reactions as visualized in Equation (64):
1.* + A ≡ * A 1 1 0
2. * A → * B 1 1 0
3. * B ≡ B + * 1 0 −1
4. * B → * C 0 1 1
5. * C ≡ *+ C 0 1 1
N(1): A ↔ B; N(2): A ↔ C; N(3): B ↔ C
2. * A → * B 1 1 0
3. * B ≡ B + * 1 0 −1
4. * B → * C 0 1 1
5. * C ≡ *+ C 0 1 1
N(1): A ↔ B; N(2): A ↔ C; N(3): B ↔ C
To make the illustration more apparent, only two products are considered and moreover steps 1, 3 and 5 are considered to be at quasi-equilibria and steps 2 and 4 are assumed to be irreversible. The expressions for the rate and equilibrium constants naturally follow from the considerations above:
It should be noted that the routes N(1) to N(3) are not independent ones, as route N(3) can be obtained by the subtraction of route N(1) from the route N(2) as discussed previously in the literature for similar reaction networks [28]. Subsequently, the reaction network can be described with just two routes having the reaction rates
which should be solved together with the generation equations for the components
where is the catalyst bulk density. Subsequently, selectivity toward the product B can be obtained
Or,
which can be written in the following way:
With the expressions for constants,
Equation (70) gives a possibility to describe dependence of selectivity to the reactant B concentration as a function of conversion derived previously in the literature for a similar case [29]:
where is the conversion. Equation (73) can be presented in a more explicit form:
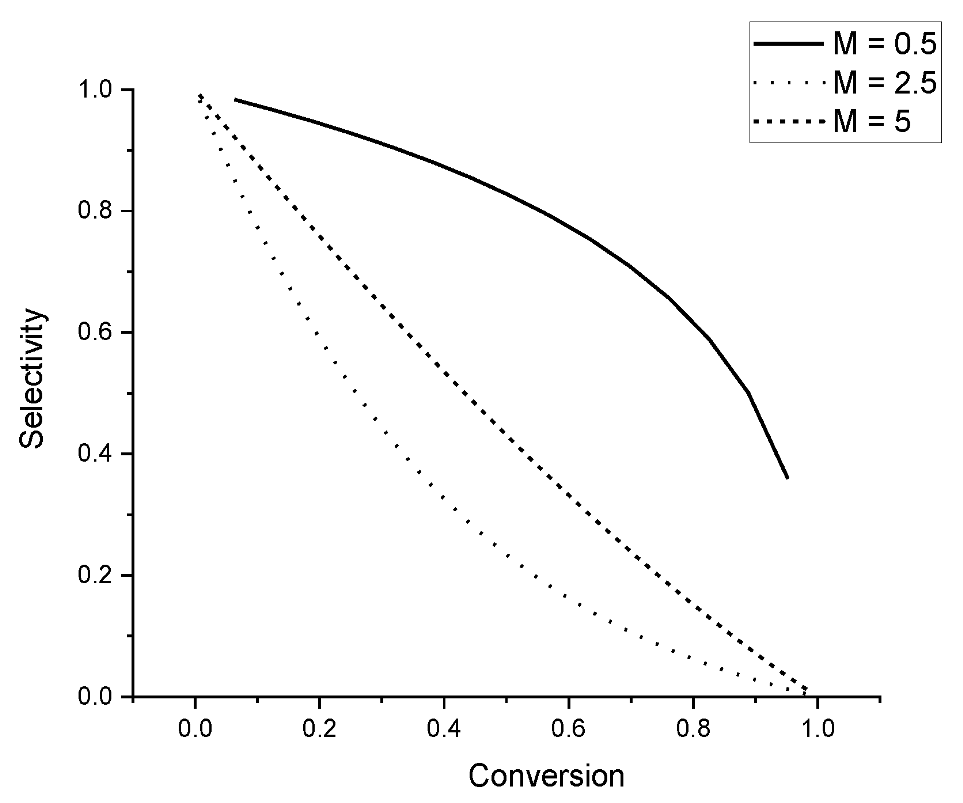
As visible from Figure 4, selectivity declines with an increase in the value of M, which reflects in essence how fast the second route is compared to the first one. At the same level of conversion, a higher acid site density will apparently result in low selectivity.
An analytic expression for the concentration of the intermediate product B in a consecutive reaction network of the type presented in Equation (64) was derived in the literature [29]:
where is the initial concentration of the reactant A. If the initial concentrations of B and C are equal to zero, the concentration of the component C easily follows from the reaction stoichiometry:
giving thus an expression for the ratio between the final and the intermediate products:
Equation (77) presents a dependence of the ratio between the final and the intermediate products as a function on conversion δ and can be used in a more explicit form illustrating a dependence on the acid site density:
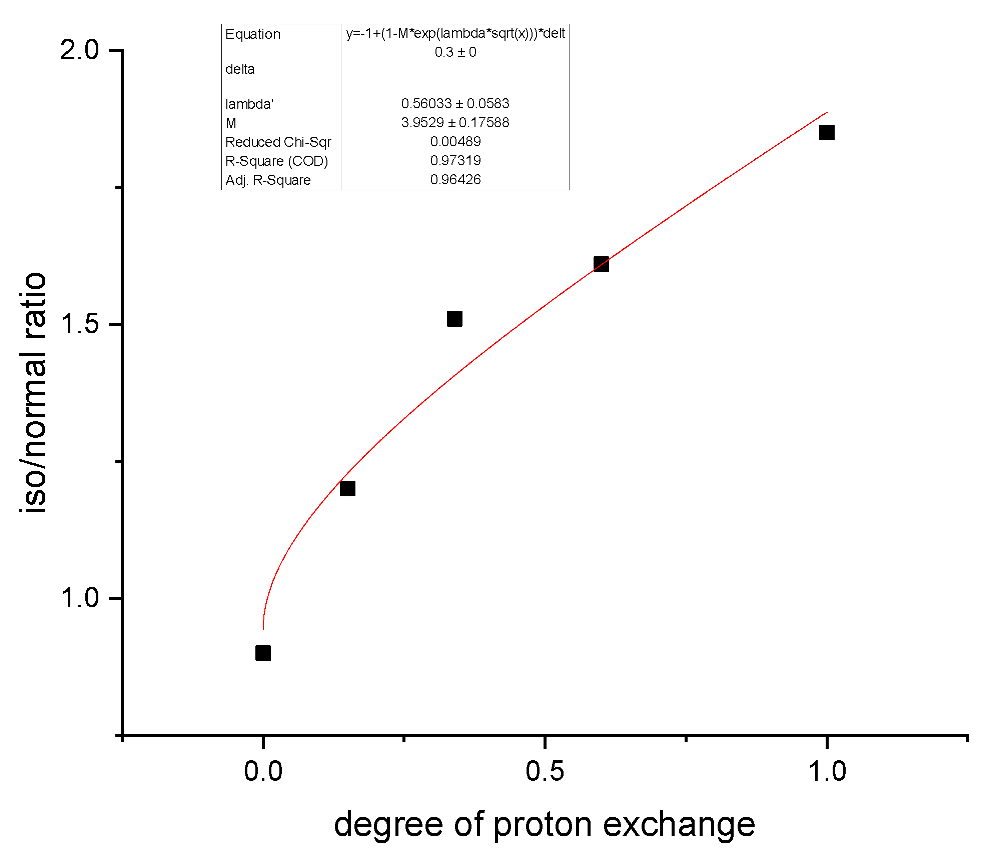
As an example of Equation (78) utilization to treat the experimental data, the ratio of iso to normal hydrocarbons obtained in transformations of syngas over mesoporous H-ZSM-5 supported cobalt nanoparticles [30] is considered. In that study, the materials were prepared with a different degree of proton exchange, which serves as a proxy for the acid site density. Obviously, the reaction network is much more complicated, but for the illustration properties, the iso compounds can be considered the final, while the normal hydrocarbons the intermediate, products. The experimental data in [30] were reported at the same conversion of ca. 30%, thus allowing to probe directly the applicability of Equation (78). The calculations along with the experimental data are presented in Figure 5, clearly confirming that an equation of type (78) can explain the distribution of products in a consecutive reaction in a quantitative way with high precision.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The paper is dedicated to 70th anniversary of Avelino Corma.
Conflicts of Interest
The author declares no conflict of interest.
References
- Cejka, J.; Corma, A.; Zones, S. Zeolites and Catalysis: Synthesis, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Murzin, D.Y. Chemical Reaction Technology; de Gruyter: Berlin, Germany, 2022. [Google Scholar]
- Jess, A.; Wasserschied, P. Chemical Technology; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Li, C.; Ferri, P.; Paris, C.; Moliner, M.; Boronat, M.; Corma, A. Design and synthesis of the active site environment in zeolite catalysts for selectively manipulating mechanistic pathways. JACS 2021, 143, 10718–10726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, J.; Corma, A. Applications of zeolites to C1 chemistry: Recent advances, challenges, and opportunities. Adv. Mat. 2020, 32, 2002927. [Google Scholar] [CrossRef] [PubMed]
- Dib, E.; Grand, J.; Gedeon, A.; Mintova, S.; Fernandez, C. Control the position of framework defects in zeolites by changing the symmetry of organic structure directing agents. Microporous Mesoporous Mater. 2021, 315, 110899. [Google Scholar] [CrossRef]
- Corma, A.; Li, C.; Moliner, M. Building zeolites from pre-crystallized units: Nanoscale architecture. Ang. Chem. Int. Ed. 2018, 57, 15330–15353. [Google Scholar]
- Laidler, K.J. Chemical Kinetics, 3rd ed.; Pearson: London, UK, 1987. [Google Scholar]
- Fogler, H.S. The Elements of Chemical Reaction Engineering, 5th ed.; Prentice Hall: Hoboken, NJ, USA, 2016; p. 992. [Google Scholar]
- Masel, R.J. Chemical Kinetis and Catalysis; Wiley: Weinheim, Germany, 2011. [Google Scholar]
- Kubicka, D.; Kumar, N.; Venäläinen, T.; Karhu, H.; Kubickova, I.; Österholm, H.; Murzin, D.Y. The metal-support interactions in zeolite-supported noble metals: The influence of metal crystallites on the support acidity. J. Phys. Chem. B 2006, 110, 4937–4946. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.; Corma, A. What is measured when measuring acidity in zeolites with probe molecules? ACS Catal. 2019, 9, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Derouane, E.G.; Védrine, J.C.; Pinto, R.R.; Borges, P.M.; Costa, L.; Lemos, M.A.N.D.A.; Lemos, L.; Ribeiro, F.R. The acidity of zeolites: Concepts, measurements and relation to catalysis: A review on experimental and theoretical methods for the study of zeolite acidity. Cat. Rev. 2013, 55, 454–515. [Google Scholar] [CrossRef]
- Damjanovic, L.; Auroux, A. Handbook of Thermal Analysis and Calorimetry; Recent Advances, Techniques and Applications; Brown, M.E., Gallagher, P.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 5. [Google Scholar]
- Pfriem, N.; Hintermeier, P.H.; Kim, S.; Liu, Q.; Shi, H.; Milakovic, L.; Liu, Y.; Haller, G.L.; Barath, E.; Liu, Y.; et al. Role of the ionic environment in enhancing the activity of reacting molecules in zeolite pores. Science 2021, 372, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Li, J.; Wei, Y.; He, Y.; Xu, L.; Liu, Z. Influence of acid site density on the three-staged MTH induction reaction over HZSM-5 zeolite. RSC Adv. 2016, 6, 52284–52291. [Google Scholar] [CrossRef]
- Milakovic, L.; Hintermeier, P.H.; Liu, Y.; Barath, E.; Lercher, J.A. Influence of intracrystalline ionic strength in mfi zeolites on aqueous phase dehydration of methylcyclohexanols. Ang. Chem. Int. Ed. 2021, 60, 24806–24810. [Google Scholar] [CrossRef] [PubMed]
- Barakov, R.; Shcherban, N.; Mäki-Arvela, P.; Yaremov, P.; Bezverkhyy, I.; Wärnå, J.; Murzin, D.Y. Hierarchical beta zeolites as catalysts in α-pinene oxide isomerization. ACS Sust. Chem. Eng. 2022, 10, 6642–6656. [Google Scholar] [CrossRef]
- Laluc, M.; Barakov, R.; Mäki-Arvela, P.; Shcherban, N.; Murzin, D.Y. Catalytic activity of hierarchical beta zeolites in the Prins cyclization of (−)-isopulegol with acetone. Appl. Cat. A. Gen. 2021, 618, 118131. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Z.; Wei, G.; Gu, Y.; Shi, H. A critical assessment of the roles of water molecules and solvated ions in acid-base-catalyzed reactions at solid-water interfaces. Chin. J. Catal. 2022, 43, 1964–1990. [Google Scholar] [CrossRef]
- Murzin, D.Y. Kinetics of heterogeneous single-site catalysis. ChemCatChem, 2022; In press. [Google Scholar]
- Arnaut, L.; Formosinho, S.; Burrows, H. Chemical Kinetics, From Molecular Structure to Chemical Reactivity; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Temkin, M.I. The optimal catalyst for the two-step reaction. Kinet. Katal. 1984, 25, 299–305. [Google Scholar]
- Boudart, M.; Tamaru, K. The step that determines the rate of a single catalytic cycle. Catal. Lett. 1991, 9, 15–22. [Google Scholar] [CrossRef]
- Temkin, M.I. The kinetics of some industrial heterogeneous catalytic reactions. Adv. Catal. 1979, 28, 173–291. [Google Scholar]
- Christiansen, J.A. The elucidation of reaction mechanisms by the method of intermediates in quasi-stationary concentrations. Adv. Catal. 1953, 5, 311–353. [Google Scholar]
- Yablonskii, G.; Bykov, V.; Gorban, A.; Elokhin, V. Kinetic models of catalytic reactions. In Comprehensive Chemical Kinetics; Compton, R.G., Ed.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 32, pp. 1–392. [Google Scholar]
- Murzin, D.Y.; Kul’kova, N.V.; Temkin, M.I. Kinetics of hydrogenation of benzene into cyclohexene and cyclohexane. Kinet. Catal. 1990, 31, 983. [Google Scholar]
- Murzin, D.Y.; Salmi, T. Catalytic Kinetics, Science and Engineering; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Cheng, K.; Zhang, L.; Kang, J.; Peng, X.; Zhang, Q.; Wang, Y. Selective transformation of syngas into gasoline-range hydrocarbons over mesoporous H-ZSM-5 supported cobalt nanoparticles. Chem. Eur. J. 2015, 21, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Dependence of as a function of the ratio of the frequencies of the first and second steps in the forward direction according to Equation (26).
Figure 1.
Dependence of as a function of the ratio of the frequencies of the first and second steps in the forward direction according to Equation (26).

Figure 2.
The ratio of reaction products in the Prins condensation of isopulegol with acetone as a function of the total concentration of acid sites. Points—experimental data from [19], calculations—Equation (55). The calculated values of
and are respectively 0.50338 ± 0.46502 and 0.13566 ± 0.03495.
Figure 2.
The ratio of reaction products in the Prins condensation of isopulegol with acetone as a function of the total concentration of acid sites. Points—experimental data from [19], calculations—Equation (55). The calculated values of
and are respectively 0.50338 ± 0.46502 and 0.13566 ± 0.03495.

Figure 3.
Mechanisms of parallel reactions: (a) with kinetic coupling and a joint edge, (b) with one joint node. A1,A2,A3,A4—reactants, B1,B2,B3,B4—products.
Figure 3.
Mechanisms of parallel reactions: (a) with kinetic coupling and a joint edge, (b) with one joint node. A1,A2,A3,A4—reactants, B1,B2,B3,B4—products.

Figure 4.
Dependence of selectivity vs. conversion according to Equation (73) for different values of M’.
Figure 4.
Dependence of selectivity vs. conversion according to Equation (73) for different values of M’.

Figure 5.
Dependence of the ratio of iso to normal hydrocarbons vs. a degree of proton exchange in transformations of syngas over mesoporous H-ZSM-5 supported cobalt nanoparticles. The experimental data are digitalized from [30].
Figure 5.
Dependence of the ratio of iso to normal hydrocarbons vs. a degree of proton exchange in transformations of syngas over mesoporous H-ZSM-5 supported cobalt nanoparticles. The experimental data are digitalized from [30].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Murzin, D.Y. Acid Site Density as a Kinetic Descriptor of Catalytic Reactions over Zeolites. Chemistry 2022, 4, 1609-1623. https://doi.org/10.3390/chemistry4040105
AMA Style
Murzin DY. Acid Site Density as a Kinetic Descriptor of Catalytic Reactions over Zeolites. Chemistry. 2022; 4(4):1609-1623. https://doi.org/10.3390/chemistry4040105
Chicago/Turabian StyleMurzin, Dmitry Yu. 2022. "Acid Site Density as a Kinetic Descriptor of Catalytic Reactions over Zeolites" Chemistry 4, no. 4: 1609-1623. https://doi.org/10.3390/chemistry4040105