

Operando NAP-XPS Studies of a Ceria-Supported Pd Catalyst for CO Oxidation
 ,
,  , , , , , and
, , , , , and 
Abstract
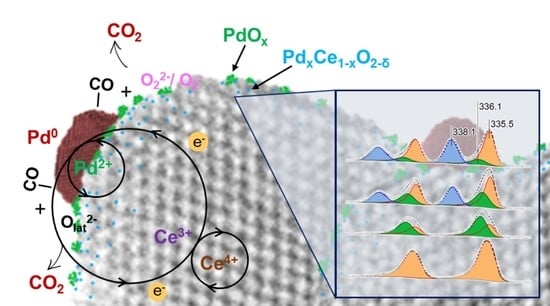
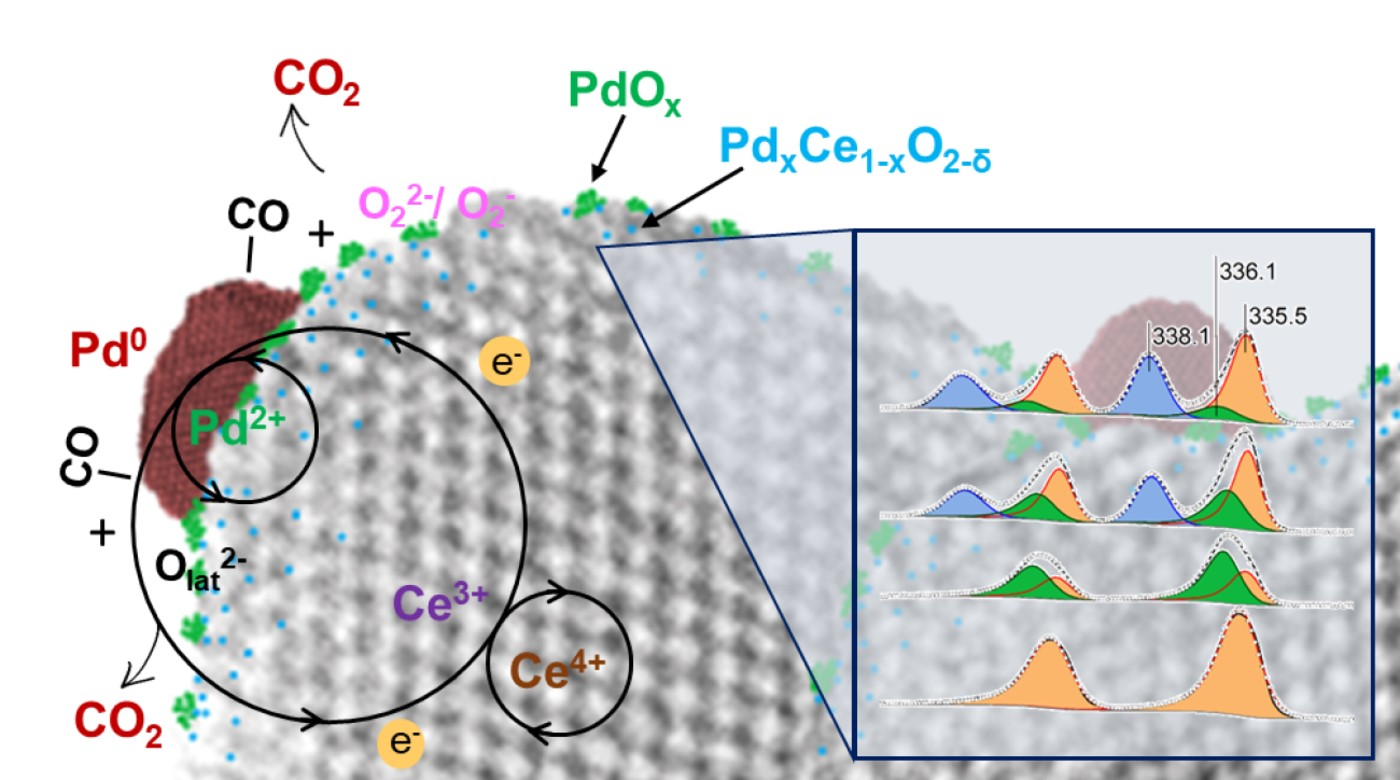
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
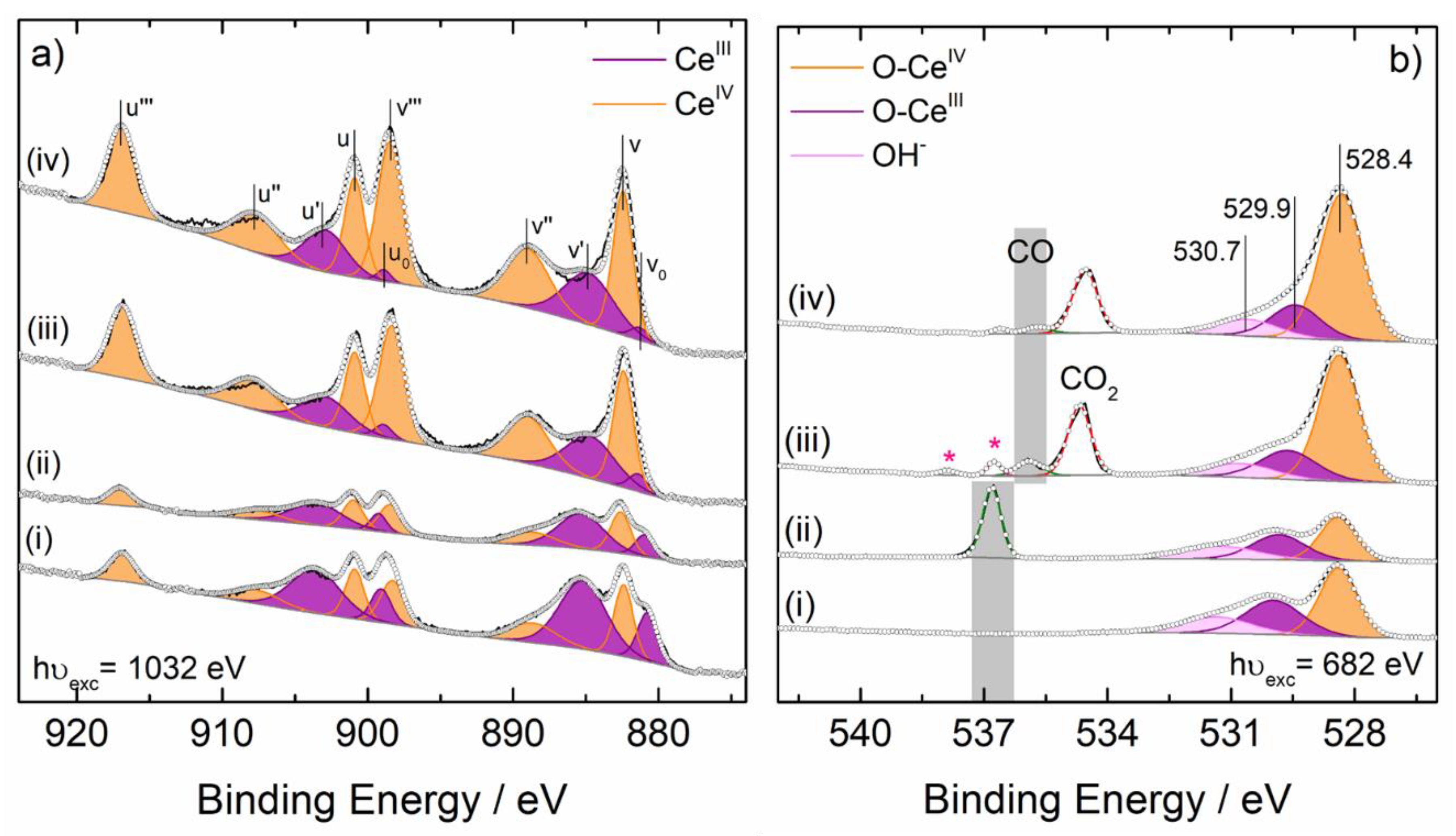{kind=link}
{kind=link}
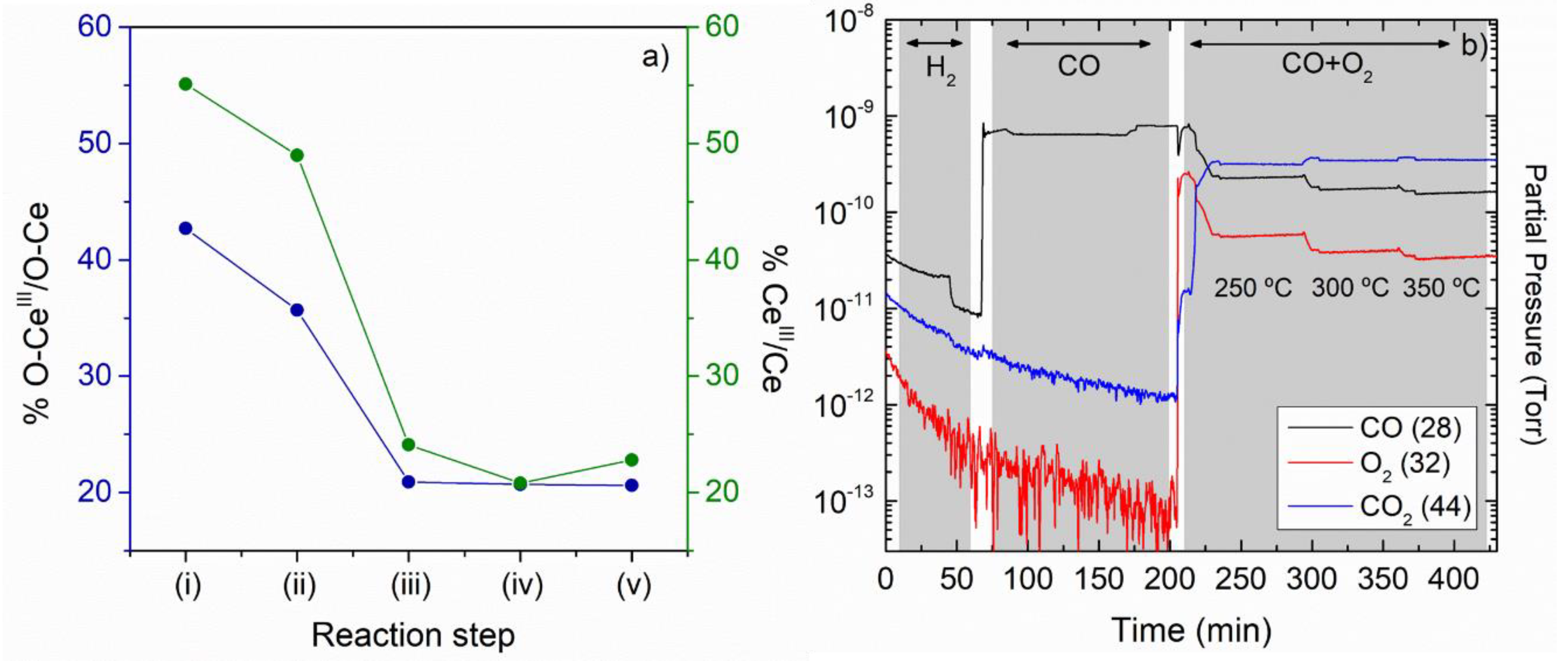
{kind=link}
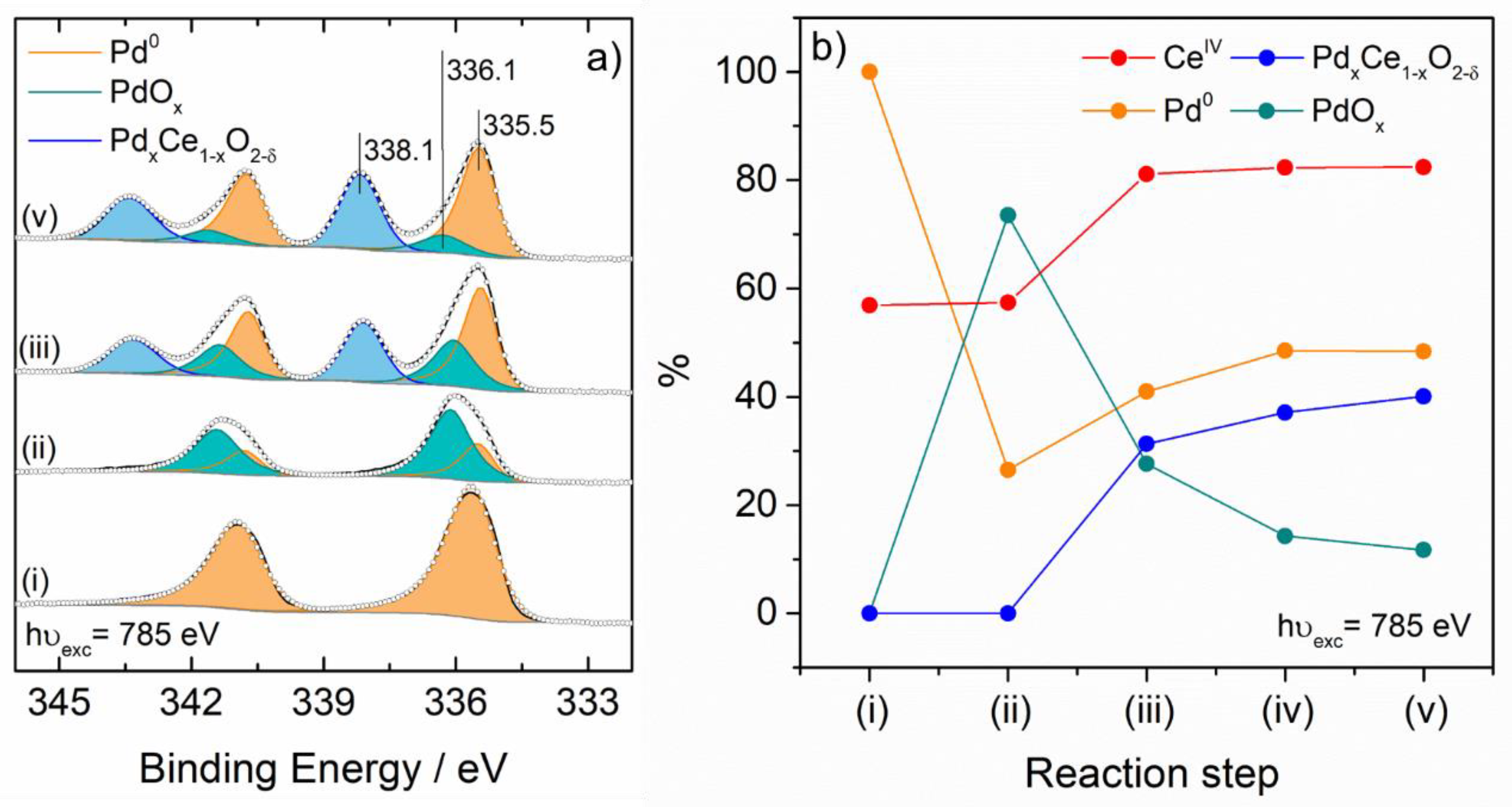{kind=link}
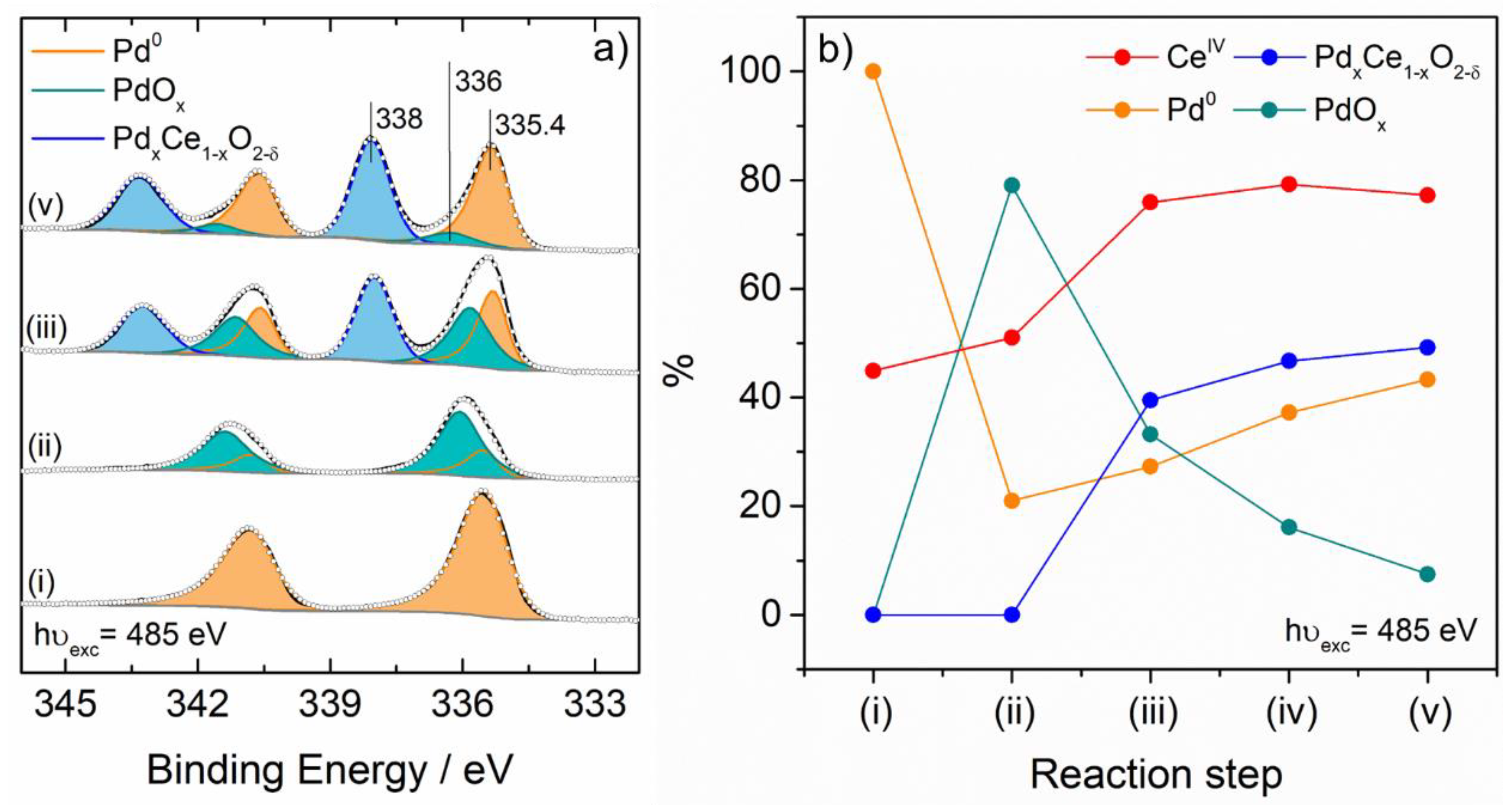{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalyst Characterization
3. Results and Discussion
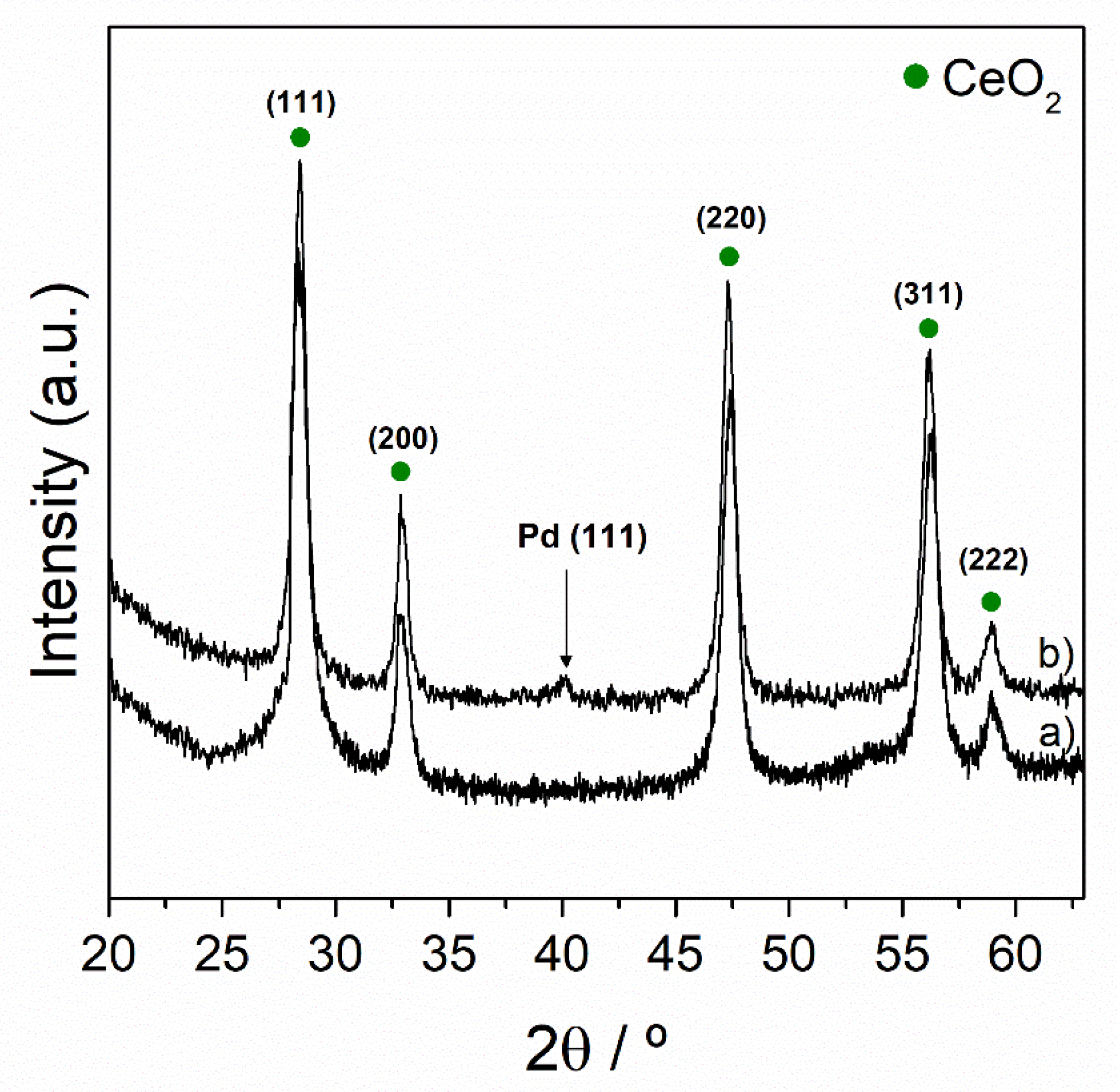
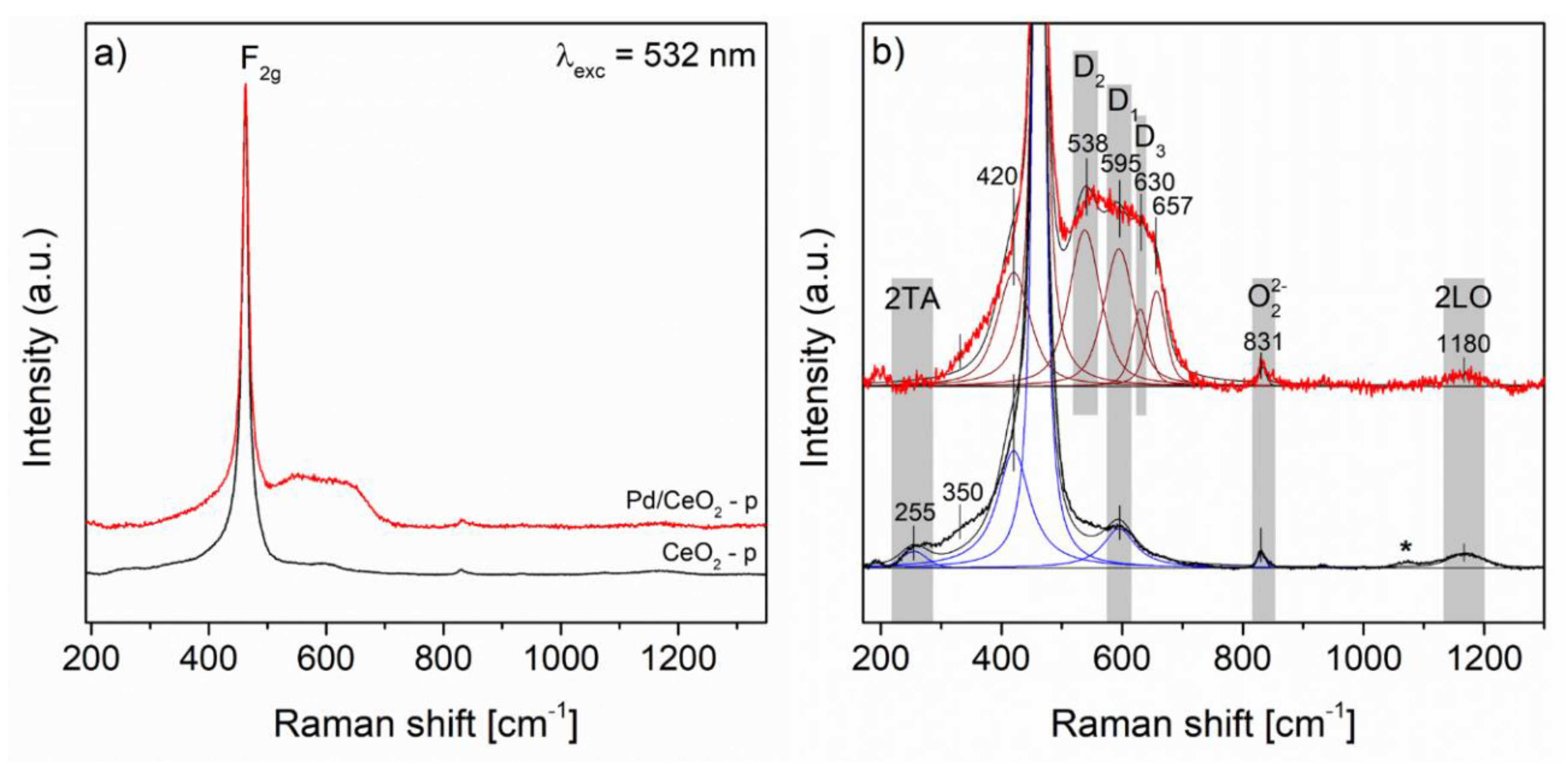
3.1. Characterization
3.2. Operando NAP-XPS
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aneggi, E.; Boaro, M.; de Leitenburg, C.; Dolcetti, G.; Trovarelli, A. Insights into the redox properties of ceria-based oxides and their implications in catalysis. J. Alloys Compd. 2006, 412, 1096–1102. [Google Scholar] [CrossRef]
- Vivier, L.; Duprez, D. Ceria-Based Solid Catalysts for Organic Chemistry. ChemSusChem 2010, 3, 654–678. [Google Scholar] [CrossRef] [PubMed]
- Bueno-López, A.; Krishna, K.; Makkee, M.; Moulijn, J.A. Active oxygen from CeO2 and its role in catalysed soot oxidation. Catal. Lett. 2005, 99, 203–205. [Google Scholar] [CrossRef]
- Freund, H.-J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Trovarelli, A.; de Leitenburg, C.; Boaro, M.; Dolcetti, G. The utilization of ceria in industrial catalysis. Catal. Today 1999, 50, 353–367. [Google Scholar] [CrossRef]
- Kim, G. Ceria-promoted three-way catalysts for auto exhaust emission control. Ind. Eng. Chem. Prod. Res. Dev. 1982, 21, 267–274. [Google Scholar] [CrossRef]
- Kaspar, J.; Fornasiero, P.; Balducci, G.; Di Monte, R.; Hickey, N.; Sergo, V. Effect of ZrO2 content on textural and structural properties of CeO2–ZrO2 solid solutions made by citrate complexation route. Inorg. Chim. Acta 2003, 349, 217–226. [Google Scholar] [CrossRef]
- Sun, C.; Li, H.; Chen, L. Nanostructured ceria-based materials: Synthesis, properties, and applications. Energy Environ. Sci. 2012, 5, 8475–8505. [Google Scholar] [CrossRef]
- Bueno-Loópez, A.; Krishna, K.; Makkee, M.; Moulijn, J.A. Enhanced soot oxidation by lattice oxygen via La3+-doped CeO2. J. Catal. 2005, 230, 237–248. [Google Scholar] [CrossRef]
- Soler, L.; Casanovas, A.; Escudero, C.; Pérez-Dieste, V.; Aneggi, E.; Trovarelli, A.; Llorca, J. Ambient Pressure Photoemission Spectroscopy Reveals the Mechanism of Carbon Soot Oxidation in Ceria-Based Catalysts. ChemCatChem 2016, 8, 2748–2751. [Google Scholar] [CrossRef] [Green Version]
- Slavinskaya, E.M.; Boronin, A.I.; Danilova, I.G.; Amosov, Y.I.; Ivanova, A.S.; Kuznetsov, P.A.; Polukhina, I.A.; Gulyaev, R.V.; Stadnichenko, A.I.; Koshcheev, S.V.; et al. Synthesis and physicochemical characterization of palladium-cerium oxide catalysts for the low-temperature oxidation of carbon monoxide. Kinet. Catal. 2009, 50, 819–823. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.; Graham, G. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Hilaire, S.; Wang, X.; Luo, T.; Gorte, R.; Wagner, J. A comparative study of water-gas-shift reaction over ceria supported metallic catalysts. Appl. Catal. A Gen. 2001, 215, 271–278. [Google Scholar] [CrossRef]
- Ramírez-López, R.; Elizalde-Martinez, I.; Balderas-Tapia, L. Complete catalytic oxidation of methane over Pd/CeO2–Al2O3: The influence of different ceria loading. Catal. Today 2010, 150, 358–362. [Google Scholar] [CrossRef]
- Rossell, M.D.; Caparrós, F.J.; Angurell, I.; Muller, G.; Llorca, J.; Seco, M.; Rossell, O. Magnetite-supported palladium single-atoms do not catalyse the hydrogenation of alkenes but small clusters do. Catal. Sci. Technol. 2016, 6, 4081–4085. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Hirabayashi, T.; Dohmae, K.; Takagi, N.; Minami, T.; Shinjoh, H.; Matsumoto, S.I. Sintering inhibition mechanism of platinum supported on ceria-based oxide and Pt-oxide–support interaction. J. Catal. 2006, 242, 103–109. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, X.; Meng, D.; Guo, Y.; Guo, Y.; Lu, G. Effect of Ceria Crystal Plane on the Physicochemical and Catalytic Properties of Pd/Ceria for CO and Propane Oxidation. ACS Catal. 2016, 6, 2265–2279. [Google Scholar] [CrossRef]
- Boronin, A.; Slavinskaya, E.; Danilova, I.; Gulyaev, R.; Amosov, Y.; Kuznetsov, P.; Polukhina, I.; Koscheev, S.; Zaikovskii, V.; Noskov, A. Investigation of palladium interaction with cerium oxide and its state in catalysts for low-temperature CO oxidation. Catal. Today 2009, 144, 201–211. [Google Scholar] [CrossRef]
- Mayernick, A.D.; Janik, M.J. Ab initio thermodynamic evaluation of Pd atom interaction with CeO2 surfaces. J. Chem. Phys. 2009, 131, 084701. [Google Scholar] [CrossRef]
- Yang, Z.; Luo, G.; Lu, Z.; Hermansson, K. Oxygen vacancy formation energy in Pd-doped ceria: A DFT+U study. J. Chem. Phys. 2007, 127, 074704. [Google Scholar] [CrossRef] [PubMed]
- Colussi, S.; Gayen, A.; Camellone, M.F.; Boaro, M.; Llorca, J.; Fabris, S.; Trovarelli, A. Nanofaceted Pd-O Sites in Pd-Ce Surface Superstructures: Enhanced Activity in Catalytic Combustion of Methane. Angew. Chem. Int. Ed. 2009, 48, 8481–8484. [Google Scholar] [CrossRef] [PubMed]
- Priolkar, K.R.; Bera, P.; Sarode, P.R.; Hegde, M.S.; Emura, S.; Kumashiro, R.; Lalla, N.P. Formation of Ce1-xPdxO2-δ Solid Solution in Combustion-Synthesized Pd/CeO2 Catalyst: XRD, XPS, and EXAFS Investigation. Chem. Mater. 2002, 14, 2120–2128. [Google Scholar] [CrossRef]
- Gulyaev, R.; Stadnichenko, A.; Slavinskaya, E.; Ivanova, A.; Koscheev, S.; Boronin, A. In situ preparation and investigation of Pd/CeO2 catalysts for the low-temperature oxidation of CO. Appl. Catal. A Gen. 2012, 439–440, 41–50. [Google Scholar] [CrossRef]
- Pérez-Dieste, V.; Aballe, L.; Ferrer, S.; Nicolas, J.; Escudero, C.; Milán, A.; Pellegrin, E. Near Ambient Pressure XPS at ALBA. J. Phys. Conf. Ser. 2013, 425, 072023. [Google Scholar] [CrossRef] [Green Version]
- Garcia, X.; Soler, L.; Divins, N.J.; Vendrell, X.; Serrano, I.; Lucentini, I.; Prat, J.; Solano, E.; Tallarida, M.; Escudero, C.; et al. Ceria-Based Catalysts Studied by Near Ambient Pressure X-ray Photoelectron Spectroscopy: A Review. Catalysts 2020, 10, 286. [Google Scholar] [CrossRef] [Green Version]
- Colussi, S.; Trovarelli, A.; Vesselli, E.; Baraldi, A.; Comelli, G.; Groppi, G.; Llorca, J. Structure and morphology of Pd/Al2O3 and Pd/CeO2/Al2O3 combustion catalysts in Pd–PdO transformation hysteresis. Appl. Catal. A Gen. 2010, 390, 1–10. [Google Scholar] [CrossRef]
- Mai, H.-X.; Sun, L.-D.; Zhang, Y.-W.; Si, R.; Feng, W.; Zhang, H.-P.; Liu, A.H.-C.; Yan, C.-H. Shape-Selective Synthesis and Oxygen Storage Behavior of Ceria Nanopolyhedra, Nanorods, and Nanocubes. J. Phys. Chem. B 2005, 109, 24380–24385. [Google Scholar] [CrossRef]
- Soler, L.; Casanovas, A.; Urrich, A.; Angurell, I.; Llorca, J. CO oxidation and COPrOx over preformed Au nanoparticles supported over nanoshaped CeO2. Appl. Catal. B Environ. 2016, 197, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Mullins, D.; Overbury, S.; Huntley, D. Electron spectroscopy of single crystal and polycrystalline cerium oxide surfaces. Surf. Sci. 1998, 409, 307–319. [Google Scholar] [CrossRef]
- Päiväsaari, J.; Putkonen, M.; Niinistö, L. Cerium dioxide buffer layers at low temperature by atomic layer deposition. J. Mater. Chem. 2002, 12, 1828–1832. [Google Scholar] [CrossRef]
- Wu, Z.; Li, M.; Howe, J.; Meyer, I.H.M.; Overbury, S.H. Probing Defect Sites on CeO2 Nanocrystals with Well-Defined Surface Planes by Raman Spectroscopy and O2 Adsorption. Langmuir 2010, 26, 16595–16606. [Google Scholar] [CrossRef] [PubMed]
- Filtschew, A.; Hofmann, K.; Hess, C. Ceria and Its Defect Structure: New Insights from a Combined Spectroscopic Approach. J. Phys. Chem. C 2016, 120, 6694–6703. [Google Scholar] [CrossRef]
- Agarwal, S.; Zhu, X.; Hensen, E.J.M.; Lefferts, L.; Mojet, B.L. Defect Chemistry of Ceria Nanorods. J. Phys. Chem. C 2014, 118, 4131–4142. [Google Scholar] [CrossRef]
- Taniguchi, T.; Watanabe, T.; Sugiyama, N.; Subramani, A.K.; Wagata, H.; Matsushita, N.; Yoshimura, M. Identifying Defects in Ceria-Based Nanocrystals by UV Resonance Raman Spectroscopy. J. Phys. Chem. C 2009, 113, 19789–19793. [Google Scholar] [CrossRef]
- Li, L.; Chen, F.; Lu, J.-Q.; Luo, M.-F. Study of Defect Sites in Ce1–xMxO2−δ (x = 0.2) Solid Solutions Using Raman Spectroscopy. J. Phys. Chem. A 2011, 115, 7972–7977. [Google Scholar] [CrossRef]
- Lin, H.-L.; Wu, C.-Y.; Chiang, R.-K. Facile synthesis of CeO2 nanoplates and nanorods by [100] oriented growth. J. Colloid Interface Sci. 2010, 341, 12–17. [Google Scholar] [CrossRef]
- Sartoretti, E.; Novara, C.; Giorgis, F.; Piumetti, M.; Bensaid, S.; Russo, N.; Fino, D. In situ Raman analyses of the soot oxidation reaction over nanostructured ceria-based catalysts. Sci. Rep. 2019, 9, 3875. [Google Scholar] [CrossRef] [Green Version]
- Paunović, N.; Dohčević-Mitrović, Z.; Scurtu, R.; Aškrabić, S.; Prekajski, M.; Matović, B.; Popović, Z.V. Suppression of inherent ferromagnetism in Pr-doped CeO2 nanocrystals. Nanoscale 2012, 4, 5469–5476. [Google Scholar] [CrossRef]
- Piumetti, M.; Andana, T.; Bensaid, S.; Russo, N.; Fino, D.; Pirone, R. Study on the CO Oxidation over Ceria-Based Nanocatalysts. Nanoscale Res. Lett. 2016, 11, 165. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Carabineiro, S.A.; Iliopoulou, E.; Andriopoulou, C.; Kallithrakas-Kontos, N.; Boghosian, S.; Konsolakis, M. Ceria nanoparticles shape effects on the structural defects and surface chemistry: Implications in CO oxidation by Cu/CeO2 catalysts. Appl. Catal. B Environ. 2018, 230, 18–28. [Google Scholar] [CrossRef]
- Gulyaev, R.V.; Kardash, T.Y.; Malykhin, S.E.; Stonkus, O.A.; Ivanova, A.S.; Boronin, A.I. The local structure of PdxCe1−xO2−x−δ solid solutions. Phys. Chem. Chem. Phys. 2014, 16, 13523–13539. [Google Scholar] [CrossRef] [PubMed]
- Popović, Z.; Dohčević-Mitrović, Z.; Šćepanović, M.; Grujić-Brojčin, M.; Aškrabić, S. Raman scattering on nanomaterials and nanostructures. Ann. Phys. 2011, 523, 62–74. [Google Scholar] [CrossRef]
- Yao, Y.F.Y. Ceria in automotive exhaust catalysts I. Oxygen storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- Giordano, F.; Trovarelli, A.; de Leitenburg, C.; Giona, M. A Model for the Temperature-Programmed Reduction of Low and High Surface Area Ceria. J. Catal. 2000, 193, 273–282. [Google Scholar] [CrossRef]
- Kurnatowska, M.; Kepinski, L.; Mista, W. Structure evolution of nanocrystalline Ce1−xPdxO2−y mixed oxide in oxidizing and reducing atmosphere: Reduction-induced activity in low-temperature CO oxidation. Appl. Catal. B Environ. 2012, 117–118, 135–147. [Google Scholar] [CrossRef]
- Romeo, M.; Bak, K.; El Fallah, J.; Le Normand, F.; Hilaire, L. XPS Study of the reduction of cerium dioxide. Surf. Interface Anal. 1993, 20, 508–512. [Google Scholar] [CrossRef]
- Jain, R.; Dubey, A.; Ghosalya, M.K.; Gopinath, C.S. Gas–solid interaction of H2–Ce0.95Zr0.05O2: New insights into surface participation in heterogeneous catalysis. Catal. Sci. Technol. 2015, 6, 1746–1756. [Google Scholar] [CrossRef]
- Militello, M.C.; Simko, S.J. Elemental Palladium by XPS. Surf. Sci. Spectra 1994, 3, 387–394. [Google Scholar] [CrossRef]
- Militello, M.C.; Simko, S.J. Palladium Oxide (PdO) by XPS. Surf. Sci. Spectra 1997, 3, 395–401. [Google Scholar] [CrossRef]
- Blomberg, S.; Hoffmann, M.J.; Gustafson, J.; Martin, N.M.; Fernandes, V.R.; Borg, A.; Liu, Z.; Chang, R.; Matera, S.; Reuter, K.; et al. In SituX-Ray Photoelectron Spectroscopy of Model Catalysts: At the Edge of the Gap. Phys. Rev. Lett. 2013, 110, 117601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haerle, R.; Riedo, E.; Pasquarello, A.; Baldereschi, A. sp2/sp3hybridization ratio in amorphous carbon from C1score-level shifts: X-ray photoelectron spectroscopy and first-principles calculation. Phys. Rev. B 2001, 65, 045101. [Google Scholar] [CrossRef]
- Oh, S.-H.; Hoflund, G.B. Chemical State Study of Palladium Powder and Ceria-Supported Palladium during Low-Temperature CO Oxidation. J. Phys. Chem. A 2006, 110, 7609–7613. [Google Scholar] [CrossRef] [PubMed]
- Pillo, T.; Zimmermann, R.; Steiner, P.; Hüfner, S. The electronic structure of PdO found by photoemission (UPS and XPS) and inverse photoemission (BIS). J. Phys. Condens. Matter 1997, 9, 3987–3999. [Google Scholar] [CrossRef]
- Slavinskaya, E.; Gulyaev, R.; Zadesenets, A.; Stonkus, O.; Zaikovskii, V.; Shubin, Y.V.; Korenev, S.; Boronin, A. Low-temperature CO oxidation by Pd/CeO2 catalysts synthesized using the coprecipitation method. Appl. Catal. B Environ. 2014, 166–167, 91–103. [Google Scholar] [CrossRef]
- Muravev, V.; Spezzati, G.; Su, Y.-Q.; Parastaev, A.; Chiang, F.-K.; Longo, A.; Escudero, C.; Kosinov, N.; Hensen, E.J.M. Interface dynamics of Pd–CeO2 single-atom catalysts during CO oxidation. Nat. Catal. 2021, 4, 469–478. [Google Scholar] [CrossRef]
- Schiller, F.; Ilyn, M.; Pérez-Dieste, V.; Escudero, C.; Huck-Iriart, C.; del Arbol, N.R.; Hagman, B.; Merte, L.R.; Bertram, F.; Shipilin, M.; et al. Catalytic Oxidation of Carbon Monoxide on a Curved Pd Crystal: Spatial Variation of Active and Poisoning Phases in Stationary Conditions. J. Am. Chem. Soc. 2018, 140, 16245–16252. [Google Scholar] [CrossRef] [Green Version]
- Teschner, D.; Borsodi, J.; Wootsch, A.; Révay, Z.; Havecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlogl, R. The Roles of Subsurface Carbon and Hydrogen in Palladium-Catalyzed Alkyne Hydrogenation. Science 2008, 320, 86–89. [Google Scholar] [CrossRef]
- Newton, M.A.; Di Michiel, M.; Kubacka, A.; Fernández-García, M. Combining Time-Resolved Hard X-ray Diffraction and Diffuse Reflectance Infrared Spectroscopy To Illuminate CO Dissociation and Transient Carbon Storage by Supported Pd Nanoparticles during CO/NO Cycling. J. Am. Chem. Soc. 2010, 132, 4540–4541. [Google Scholar] [CrossRef]
- Westerström, R.; Messing, M.E.; Blomberg, S.; Hellman, A.; Grönbeck, H.; Gustafson, J.; Martin, N.M.; Balmes, O.; van Rijn, R.; Andersen, J.N.; et al. Oxidation and reduction of Pd(100) and aerosol-deposited Pd nanoparticles. Phys. Rev. B 2011, 83, 115440. [Google Scholar] [CrossRef]
- Andersen, J.N.; Qvarford, M.; Nyholm, R.; Sorensen, S.L.; Wigren, C. Surface core-level shifts as a probe of the local overlayer structure: CO on Pd(100). Phys. Rev. Lett. 1991, 67, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Vélez, J.J.; Teschner, D.; Girgsdies, F.; Hävecker, M.; Streibel, V.; Willinger, M.G.; Cao, J.; Lamoth, M.; Frei, E.; Wang, R.; et al. The Role of Adsorbed and Subsurface Carbon Species for the Selective Alkyne Hydrogenation over a Pd-Black Catalyst: An Operando Study of Bulk and Surface. Top. Catal. 2018, 61, 2052–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, X.; Fang, D. A review on C1s XPS-spectra for some kinds of carbon materials. Full-Nanotub. Carbon Nanostruct. 2020, 28, 1048–1058. [Google Scholar] [CrossRef]
- Pozdnyakova, O.; Teschner, D.; Wootsch, A.; Kröhnert, J.; Steinhauer, B.; Sauer, H.; Toth, L.; Jentoft, F.; Knop-Gericke, A.; Paál, Z. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part II: Oxidation states and surface species on Pd/CeO2 under reaction conditions, suggested reaction mechanism. J. Catal. 2006, 237, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Otto, K.; Haack, L.; Devries, J. Identification of two types of oxidized palladium on γ-alumina by X-ray photoelectron spectroscopy. Appl. Catal. B Environ. 1992, 1, 1–12. [Google Scholar] [CrossRef]
- Kibis, L.S.; Stadnichenko, A.I.; Koscheev, S.V.; Zaikovskii, V.I.; Boronin, A.I. Highly Oxidized Palladium Nanoparticles Comprising Pd4+ Species: Spectroscopic and Structural Aspects, Thermal Stability, and Reactivity. J. Phys. Chem. C 2012, 116, 19342–19348. [Google Scholar] [CrossRef]
- Bera, P.; Patil, K.; Jayaram, V.; Subbanna, G.; Hegde, M. Ionic Dispersion of Pt and Pd on CeO2 by Combustion Method: Effect of Metal–Ceria Interaction on Catalytic Activities for NO Reduction and CO and Hydrocarbon Oxidation. J. Catal. 2000, 196, 293–301. [Google Scholar] [CrossRef]
- Misch, L.M.; Kurzman, J.A.; Derk, A.R.; Kim, Y.-I.; Seshadri, R.; Metiu, H.; McFarland, E.W.; Stucky, G.D. C–H Bond Activation by Pd-substituted CeO2: Substituted Ions versus Reduced Species. Chem. Mater. 2011, 23, 5432–5439. [Google Scholar] [CrossRef]
- Zhou, G.-F.; Ma, J.; Bai, S.; Wang, L.; Guo, Y. CO catalytic oxidation over Pd/CeO2 with different chemical states of Pd. Rare Met. 2020, 39, 800–805. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Li, T.; Zhao, Z.; Gong, X.-Q.; Chen, Y.; Duan, A.; Jiang, G.; Wei, Y. Interfacial Effects of CeO2-Supported Pd Nanorod in Catalytic CO Oxidation: A Theoretical Study. J. Phys. Chem. C 2015, 119, 12923–12934. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Qu, W.; George, C.; Aouine, M.; Vernoux, P.; Tang, X. Well-defined palladium–ceria interfacial electronic effects trigger CO oxidation. Chem. Commun. 2018, 54, 10140–10143. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, X.; Soler, L.; Vendrell, X.; Serrano, I.; Herrera, F.; Prat, J.; Solano, E.; Tallarida, M.; Llorca, J.; Escudero, C. Operando NAP-XPS Studies of a Ceria-Supported Pd Catalyst for CO Oxidation. Chemistry 2023, 5, 1-18. https://doi.org/10.3390/chemistry5010001
Garcia X, Soler L, Vendrell X, Serrano I, Herrera F, Prat J, Solano E, Tallarida M, Llorca J, Escudero C. Operando NAP-XPS Studies of a Ceria-Supported Pd Catalyst for CO Oxidation. Chemistry. 2023; 5(1):1-18. https://doi.org/10.3390/chemistry5010001
Chicago/Turabian StyleGarcia, Xènia, Lluís Soler, Xavier Vendrell, Isabel Serrano, Facundo Herrera, Jordi Prat, Eduardo Solano, Massimo Tallarida, Jordi Llorca, and Carlos Escudero. 2023. "Operando NAP-XPS Studies of a Ceria-Supported Pd Catalyst for CO Oxidation" Chemistry 5, no. 1: 1-18. https://doi.org/10.3390/chemistry5010001