1. Introduction
In 2015, an edited volume by J. Brockman was published entitled ‘
This Idea Must Die: Scientific Theories That Are Blocking Progress’ [
1]. This work comprises a large collection of short, wide-ranging essays written by scientists and philosophers covering ideas or theories that the authors maintain should be killed off for the reason captured in the sub-title of the book itself, i.e., that they are blocking progress in that particular discipline or, indeed, in science and philosophy more broadly. However, as the chemist and philosopher Eric Scerri remarked in 2019 in ‘
Five Ideas in Chemical Education that Must Die’ [
2], not one chemist was amongst the many (175!) authors who contributed to Brockman’s collection, an omission to which he took some exception, as is evident from the short quote reproduced below [
2].
“Remarkably, there is not a single chemist among the authors, thus reinforcing the misconception that chemistry lacks any philosophical substance or profound intellectual content. The steady growth in interest in the philosophy of chemistry since the early 1990s demands that this situation be rectified by considering what chemical ideas may also be in need of ‘killing off’.”
As promised in the title of his article, Scerri went on to consider five ideas in chemistry which, in his opinion, it was time to discard. These were specific ideas associated with the topics of: (i) whether a pH of 7 always indicates neutrality; (ii) Le Châtelier’s principle; (iii) the occupation of 4s orbitals in first row transition metals; (iv) the anomalous electron configuration of chromium; and (v) which elements should (and should not) constitute Group 3 of the periodic table. The reader is referred to [
2] for the details of Scerri’s arguments which will not be reprised here, but to his list of five, we would like to add a sixth: the concept of hypervalence as applied to certain aspects of p-block element chemistry. The case for suggesting that this idea or concept, which will be defined and exemplified shortly, should be abandoned will form the core of this essay, but we state from the outset that our focus is principally on whether or not hypervalence has any merit in an educational context, and we fully concur with the recent statement made by Schwerdtfeger and Frenking and co-workers that, ‘
Chemical bonding models are not right or wrong, they are more or less useful’ [
3], and with a somewhat earlier remark by Dewar that, ‘
The only criterion of a model is usefulness, not its “truth”’ [
4]. It is not, therefore, about whether the concept of hypervalence is right or wrong, although we will note some intrinsic inconsistencies, it is more about whether or not it is useful. It will be argued here that it is not useful because we are not convinced that the term offers any tangible benefit in describing the compounds to which it is or could be applied. In developing our argument, it will be necessary to consider the value of the octet rule, about which we shall be rather more positive, albeit with certain provisos, and it will also be useful to examine some of the orbital-based bonding models, computational methods, and graphical representations that offer an alternative explication of the topics under scrutiny. We make no pretence at being comprehensive, or even especially rigorous, and will do no more than adumbrate some of the studies which relate to this subject. We have sought, however, to recount enough of the salient history to make our case and to highlight the contributions made by some of the key protagonists in this story.
2. Some Definitions and Examples
The noun
hypervalence and the adjective
hypervalent are most often encountered in descriptions of p-block element compounds which may be considered to have more than eight valence electrons around the central atom, which serves as the basis for the most general definition of hypervalence: hypervalent compounds are those which apparently violate the octet rule. Before introducing some examples, however, we must first be clear about the meaning of the root term,
valence. The origin of the concept of chemical valence itself is generally credited to Frankland and Kekulé and dates from the 1850s (
Appendix A Note 1) [
5,
6,
7], but we have adopted the simple and largely unambiguous definition provided by Sidgwick in the 1920s, which defines the chemical valence (or simply, valence) of an element as the number of electrons it uses in bonding (
Appendix A Note 2) [
8]. Thus, for example, the phosphorus atom in PF
3 is classified as trivalent because the phosphorus uses three of its five valence electrons in bonding, the remaining two being present as a non-bonding or lone pair, as shown in
A. In contrast, in PF
5 (
B), the phosphorus is described as pentavalent because it uses all five of its valence electrons (
Appendix A Note 3) [
9]. More generally, one can assign the valence of an element by simply subtracting the number of electrons in formal Lewis-type lone pairs (if present) from the total number of available valence electrons, in accordance with Parkin’s comment that the determination of an atom’s valence in any particular compound follows straightforwardly from a simple Lewis structure [
10]. It should be stressed, however, that valence is a classical concept, as noted by Smith [
11], which will be important to bear in mind when considering the molecular orbital energy-level diagrams discussed later. With regard to the prefix hyper-, this is defined by the Oxford English Dictionary as
over,
beyond,
above or
excessively [
12], and our objection to the term hypervalence arises, in part, from this definition. For similar reasons, we will also object to the use of the prefixes hypo- and sub-, which are sometimes encountered in this context (
Appendix A Note 4).
As a minor but important digression, it is worth highlighting at this point that the terms
valence and
oxidation state (or
oxidation number) are sometimes conflated and used interchangeably, but this is an error because their definitions are quite different, as the following examples will illustrate [
10,
11,
13,
14]. In the molecule PF
3, the phosphorus is trivalent with a formal oxidation state of P(III) such that in this instance, the valence and oxidation state have the same numerical value. However, in P
2F
4 (
C), the phosphorus remains trivalent but has a formal oxidation state of P(II) (
Appendix A Note 5) [
15,
16]. Any description of the phosphorus in P
2F
4 as subvalent (or even low-valent) is therefore equivocal. P
2F
4 can be described as a compound of P(II), but the phosphorus is not divalent, a genuine example of which would be isolated PF
2 radicals. We suggest, therefore, that using prefixes such as hypo- or sub- (or perhaps even low-) in association with the term valence is, at best, ambiguous and at worst, misleading, and can be incorrect if employed as synonyms for a low oxidation state, a circumstance not unknown in the chemical literature [
10,
11]. In this essay, we will argue that use of the prefix hyper- is also ambiguous, as well as being unnecessary, and conclude that the only prefixes of any educational virtue in the context of valence are those of Greek provenance which denote a number such as in trivalent and pentavalent; only the purists object to a Greek prefix in association with a Latin stem [
5].
![Chemistry 04 00082 i001]()
As promised above, the curtain will now be raised to reveal some examples of compounds typically classified as hypervalent, and a far from exhaustive list is presented in
Table 1. It is evident that most comprise fluorides, oxides, and oxyhalides of the elements of Groups 14–18, but other examples include coordination complexes as well as certain poly-alkyl and poly-aryl species. For the sake of clarity, the number in parentheses for each compound is the formal number of valence electrons associated with the central element centre but having listed some examples and offered a concise definition, the origins of the term hypervalence must now be examined.
3. Historical Origins and Early Bonding Models
A brief but excellent summary of the origin and history of the concept of hypervalence has been provided by Jensen (
Appendix A Note 6) [
17], which begins with reference to the introduction of the term itself by Musher in 1969, who proposed a description of the bonding for those compounds of Groups 15, 16, 17, and 18 in which the valence of the element concerned exceeded 3, 2, 1, and 0, respectively (
Appendix A Note 7) [
18]. Thus, according to the proposal set out by Musher, trivalent PF
3, for example, would be considered as a compound containing ‘normal’ covalent P–F bonds (which he designated as CV bonds), whereas the description of pentavalent PF
5 incorporates so-called ‘hypervalent’ (HV) bonds associated with the linear F–P–F unit in structure
B. PF
5 is, therefore, according to Musher, considered to be a compound featuring hypervalence (
Appendix A Notes 8,9), a definition of hypervalence also adopted by Weinhold and Landis, whose contributions we will return to later [
19]. Innocuous as Musher’s proposal may have seemed, it would lead to decades of debate which had, in fact, already started almost half a century earlier.
Thus, a long-standing point of contention had been whether or not compounds such as PF
5 (PCl
5 was the example used in the early literature) or SF
6 violate the octet rule. This well-known rule had originated with Lewis and, separately, with Langmuir in the 1910s, Lewis having first described his ‘rule of eight’, whereas it was Langmuir who later introduced the term ‘octet’ [
20,
21]. As Jensen relates, however [
17], the discussion between the two regarding violations of the rule had begun in earnest in the 1920s, with Lewis [
22] favouring a description where all bonds were two-centre, two-electron (2c,2e) bonds (based on his even more foundational ‘rule of two’ that defines the electron pair bond), whereas Langmuir [
23] argued for the primacy of the octet rule even in those compounds that appeared to violate it.
If we consider PF5 as an example, an insistence on all P–F bonds being 2c,2e bonds requires an expansion of the octet to accommodate 10 valence electrons around the phosphorus (5 from the phosphorus and 1 each from the five fluorine atoms), where each of the P–F bonds shown in B would be of the 2c,2e type. However, an alternative, partially ionic description of the bonding, shown in D, allows for retention of the octet around phosphorus. In this depiction, any one of the fluorine atoms could carry the negative charge and the structure of PF5 would involve a ‘resonance’ between the five possibilities which also implicitly recognises the polarity of the P–F bonds based on the greater electronegativity of fluorine compared with phosphorus. A more detailed consideration of these two bonding models for PF5, along with others set out below, provide the foundations upon which our argument concerning hypervalence will be constructed.
![Chemistry 04 00082 i002]()
Examining first structures of type
D, these were proposed by Pauling in the 1940s and further discussed by Coulson in the 1960s in terms of the Valence Bond (VB) theory of electronic structure [
24,
25]. It is fair to say that representations of this nature involving resonance between ionic or partly ionic forms have become rather less favoured in recent decades, although they have some merit, as noted above, in drawing attention to the polar nature of the bonds concerned. We will return again to this ionic representation, but for the fully covalent description implicit in structure
B, the nature of the orbitals involved becomes all-important and the accompanying electronic structure has been described in terms of both VB theory and Molecular Orbital (MO) theory, the essentials of which should now be considered.
The first point to note is that Musher’s original bonding model involved only the valence s and p orbitals in a manner that did not result in compounds such as PF
5 violating the octet rule. In fact, Musher’s model bore some resemblance to one proposed by Sugden in the 1920s and 1930s, in which the axial P–F bonds (i.e., those in the linear F–P–F unit) were represented as two-centre, one-electron (2c,1e) bonds, thereby retaining an octet overall (three equatorial 2c,2e bonds and two axial 2c,1e bonds) [
26]. This was later developed in the 1940s and 1950s by, amongst others, Rundle and Pimentel into the three-centre, four-electron (3c,4e) bonding model [
27,
28]. Thus, in PF
5, the three equatorial P–F bonds may be described as 2c,2e bonds formed using sp
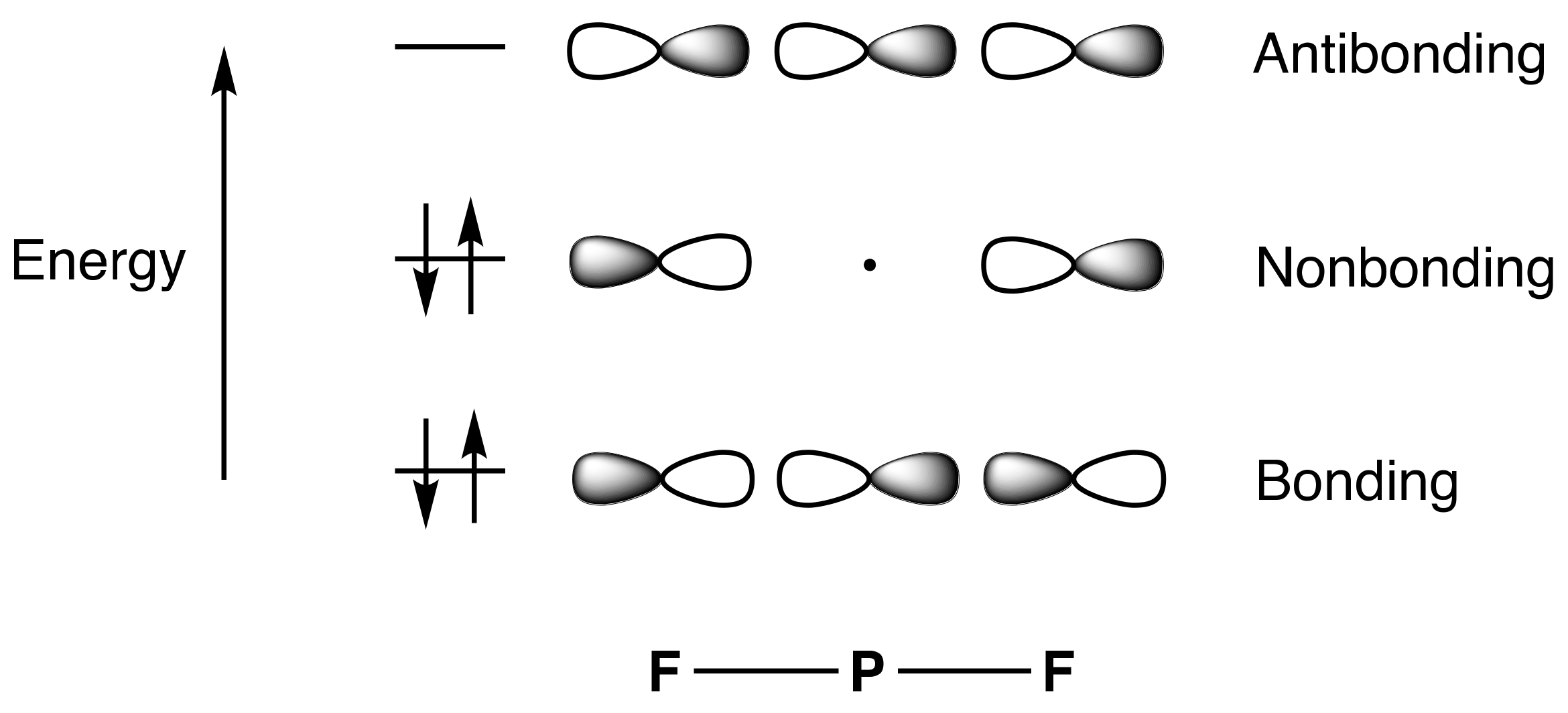
2 hybrids on the phosphorus, whereas the linear, axial F–P–F unit would comprise a 3c,4e interaction involving the remaining phosphorus p orbital. In this arrangement, the bonding orbital is delocalised over all three atoms but, crucially, the non-bonding orbital is localised only on the two fluorine atoms, with no contribution from the central phosphorus. A total of eight electrons are therefore associated with the phosphorus centre: three 2c,2e equatorial P–F bond pairs and two from the bonding 3c,4e pair for the F–P–F unit. A rendition of the 3c,4e MOs for the axial F–P–F unit is presented in
Figure 1, which shows the filled low-energy, three-centre bonding orbital and the filled non-bonding orbital, the latter localised only on the two fluorine atoms (
Appendix A Note 10). To a first approximation, therefore, both this multicentre bonding model (or indeed Musher’s original 2c,1e model) and the ionic alternative (
D) already obviate any concerns that the octet rule is violated, at least for PF
5, and thereby undermine the foundations on which the concept of hypervalence is based. However, there are other models to consider before reaching any premature conclusions.
Remaining with PF
5, an alternative bonding model involves the use of vacant d orbitals, and this was discussed as early as the 1940s by Pauling, although he was more in favour of the ionic bonding description shown in
D [
24]. Thus, although the primary valence orbitals for phosphorus are the 3s and the three 3p orbitals, the vacant 3d orbitals are potentially available for bonding. On this basis, using a single d orbital, a set of five dsp
3 hybrids can be constructed which allows for each P–F bond to be a 2c,2e bond and PF
5 would therefore have 10 valence electrons associated with the phosphorus centre: 5 from the phosphorus and 1 from each of the five fluorine atoms (as noted above and implied in
B) if each solid line is taken to represent a 2c,2e bond (
Appendix A Note 11) [
29]. Similarly, the molecule SF
6 would have six 2c,2e S–F bonds arising from six equivalent sulphur-centred d
2sp
3 hybrids. We can assert that this hybridisation model reflects a prejudice for retaining the 2c,2e bonds favoured by Lewis and others and which underlies the modern definition of hypervalence applied to compounds with more than eight valence electrons. However, this proposal hinges critically on the energetic availability (and, to some extent, the radial extension) of 3d orbitals in these and cognate examples.
From at least the 1980s with Kutzelnigg [
30], and certainly by the early 1990s from authors such as Magnusson [
31] and Reed and Schleyer [
32], 3d orbitals in this context have been demonstrated by means of quantitative electronic structure calculations to be energetically unavailable (i.e., too high in energy (
Appendix A Note 12)); on this basis, these and many other authors, notably Gilheany [
33], have argued that any bonding model incorporating d orbital hybrids such as dsp
3 and d
2sp
3 should be abandoned. Curnow [
34,
35] and Galbraith [
36] (
Appendix A Note 13) each offer useful summaries, but a more recent, comprehensive overview of this topic has been included in a review by Schwerdtfeger, Frenking, and co-workers, from which we take the following quotation [
3].
“The extension of the valence space to d-orbitals, which was earlier suggested to explain the stability of so-called hypervalent molecules, was discarded on the basis of quantum chemical calculations. Atomic orbitals with higher angular momentum such as d and f functions only serve as polarization functions for the sp space, but they are not genuine valence orbitals in main-group compounds.”
Having outlined some of the relevant history above, we are now in a better position to state the kernel of our argument. Thus, any bonding model for compounds such as PF5 and SF6 based on 2c,2e bonds involving d orbitals and d orbital hybridisation must be considered untenable. This assertion, together with the merits of the simple 3c,4e description or an alternative ionic representation, both of which we have outlined for PF5, challenge the foundations upon which the concept of hypervalence is built because there is no longer any reasonable basis for maintaining that the octet rule is violated, at least at the level of sophistication examined so far. However, the localised 3c,4e bonding picture is a rather simplistic model and not one well suited to a description of higher symmetry species such as SF6. We should therefore consider some rather more substantive MO-based analyses and will commence with SF6 and PF5 before looking at further examples of EFn species. As will become clear, matters start to become a little more complicated with regard to the octet rule, and also with how the valence of the central atom is determined but not, we suggest, in a manner which undermines our critique of hypervalence.
4. Molecular Orbital Analyses
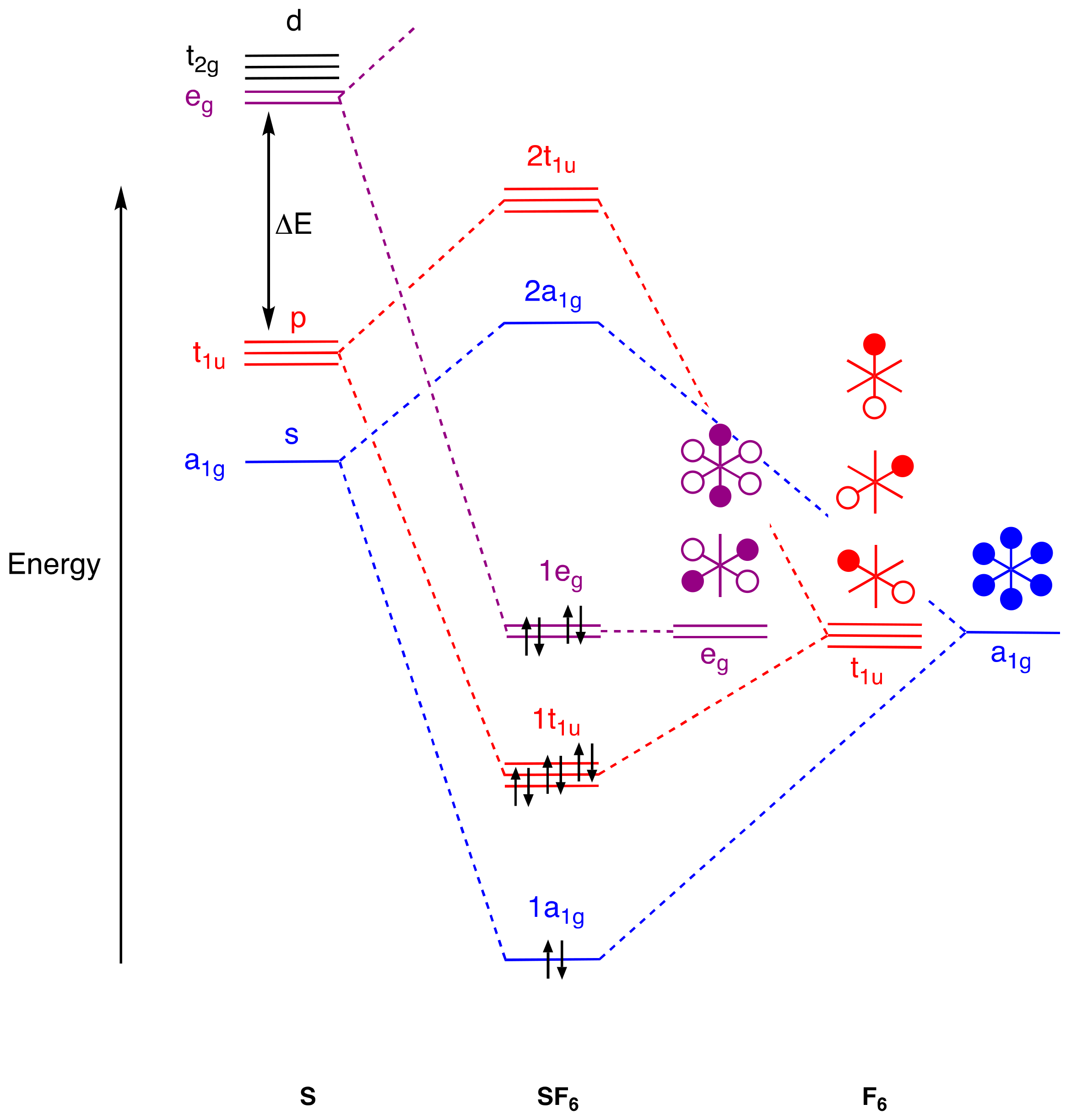
Looking first at SF
6, a simple, σ-only qualitative MO energy-level diagram is shown in
Figure 2 [
37,
38,
39,
40] (
Appendix A Note 14). If we focus initially on those interactions shown in blue and red, it is clear, using group theory arguments based on the O
h point group, that SF
6 has four occupied bonding orbitals (1a
1g and 1t
1u). An occupied pair of non-bonding orbitals localised on the fluorine atoms (1e
g) is also present (shown in purple). This amounts to 12 electrons overall but, importantly for the argument advanced here, only 8 electrons (i.e., an octet) are associated with the sulphur centre itself. If we now consider the sulphur 3d
z2 and 3d
x2–y2 orbitals (also shown in purple), these do have the correct symmetry to overlap with the fluorine-based e
g set, but the crucial factor is the energy gap indicated in
Figure 2 as ΔE. Thus, if ΔE is large as a result of the sulphur 3d orbitals being high in energy, in accordance with the calculations described in the previous section [
31,
32], the corresponding involvement of the d orbitals in any bonding is negligible, the more so, the larger is ΔE. This is illustrated in
Figure 2 by the 1e
g MOs being drawn at the same energy as the fluorine-based e
g set.
It is worth mentioning at this point that there are a few 14-electron species with O
h symmetry, such as the anions [SbCl
6]
3− and [TeCl
6]
2−, that have an additional pair of valence electrons compared with SF
6 which occupy the antibonding 2a
1g orbital shown in
Figure 2, the energetic accessibility of which becomes increasingly favoured for species incorporating heavier p-block elements (
Appendix A Note 15). Inasmuch as this orbital has a component located on the central atom, it can certainly be argued that there are more than eight electrons associated with the central element in apparent violation of the octet rule, but we will return to the matter of the octet rule later.
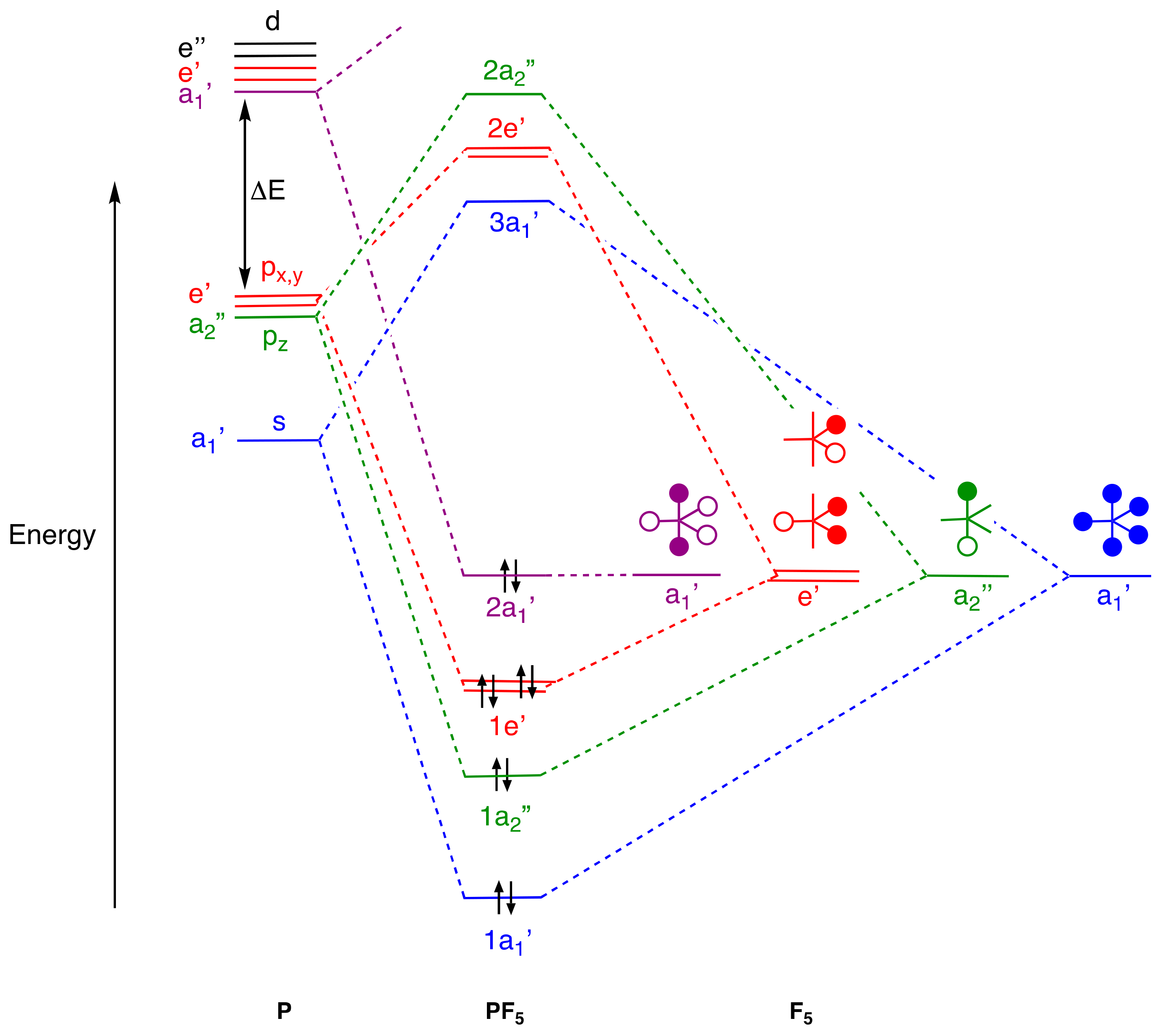
Turning now to PF
5, a qualitative, σ-only MO energy-level diagram is shown in
Figure 3 [
37,
38,
39,
40] which reveals four bonding orbitals labelled 1a
1’, 1a
2’’, and 1e’ (assigned based on the D
3h point group) shown in blue, green, and red which involve the phosphorus s, p
z, and p
x,y orbitals, respectively. However, there are two fluorine-based orbitals that have a
1’ symmetry. One of these interacts with the phosphorus 3s orbital to yield the strongly bonding MO labelled 1a
1’ shown in blue, whereas the other a
1’ orbital (shown in purple) is essentially non-bonding with respect to the phosphorus 3s orbital and becomes the MO labelled 2a
1’. This latter a
1’ orbital does have the correct symmetry to overlap with the phosphorus 3d
z2 atomic orbital (also of a
1’ symmetry in D
3h) but assuming, for reasons already given, that the energy separation ΔE shown in
Figure 3 is large, this interaction will be negligible. The relative contributions that all of the orbitals make to the bonding picture in trigonal bipyramidal molecules related to PF
5 will vary according to the elements involved, but the essential picture of four pairs of electrons that constitute the primary bonding manifold is likely to persist, which remains consistent with our thesis, thus far at least, on the usefulness of the octet rule. Much the same comment regarding PF
5 has been made by Schwerdtfeger, Frenking, and co-workers [
3].
It is interesting to note that the simple 3c,4e description of the axial F–P–F unit is reflected in the combination of the filled 1a
2’’ and 2a
1’ orbitals in the MO analysis shown in
Figure 3, but the localised 3c,4e model is not obviously discernible in the MO-based analyses of more delocalised systems such as SF
6, or in the other EF
n species described below. We should not necessarily expect it to be, however; the 3c,4e model is a much simpler model than the more expansive MO-derived schemes.
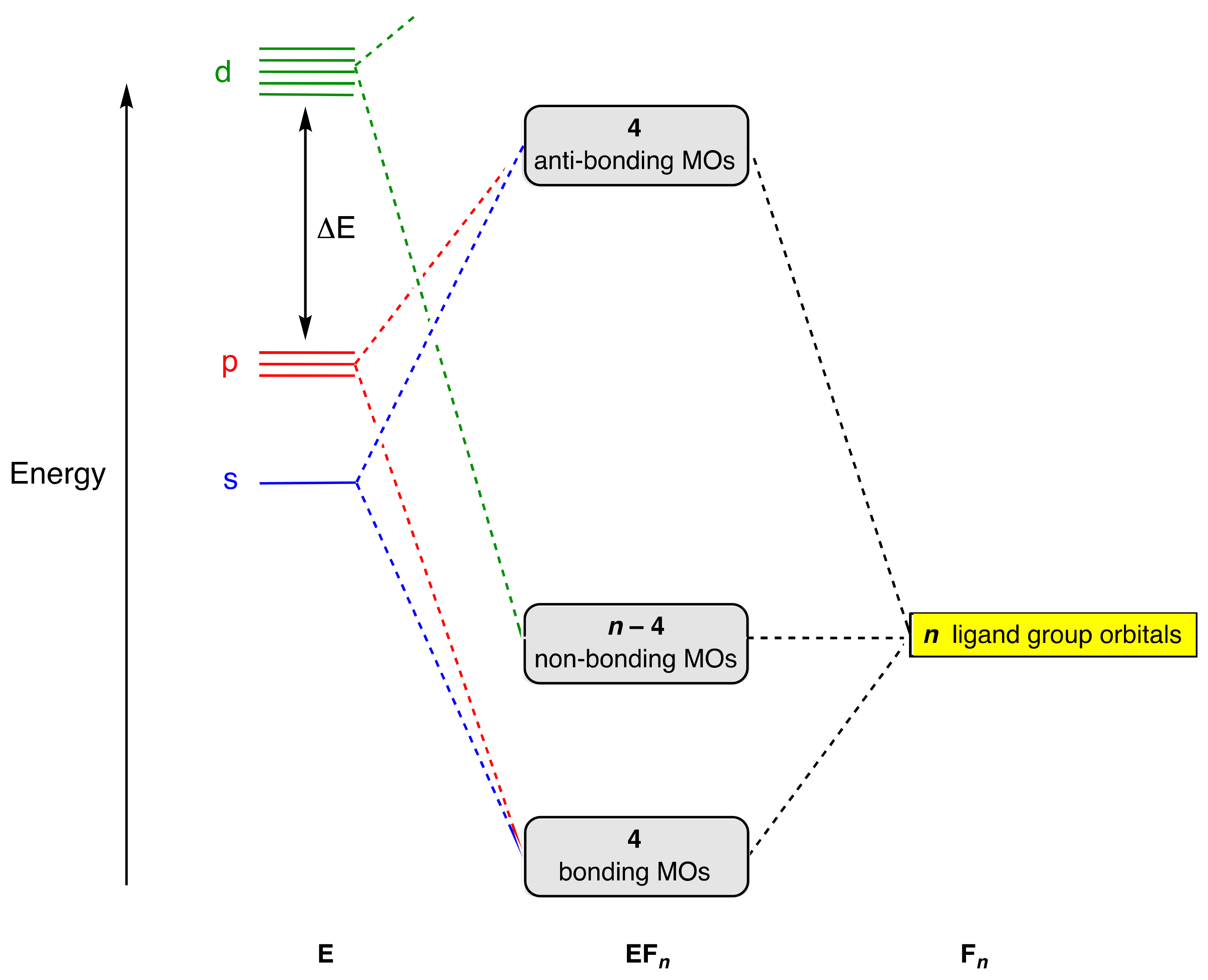
We can, in fact, take this MO-based approach one step further. Thus, extrapolating from the MO diagrams for SF
6 and PF
5 presented in
Figure 2 and
Figure 3, an approximate general MO scheme can be constructed, as shown in
Figure 4, which covers the compounds EF
n, where
n = 4–8, in which E achieves its maximum or group valence [
9]. This MO scheme reveals that, in each case, a closed-shell structure (where all bonding and non-bonding orbitals are filled) is obtained for an electron count of 2
n. Importantly for the argument advanced here, however, in each example, the central atom E has an octet of electrons in four strongly bonding orbitals, whereas the largely non-bonding electrons are predominantly or exclusively localised on the fluorine atoms, as discussed in detail for SF
6 and PF
5 above. The extent of d orbital participation will depend on ΔE, but because this is assumed to be minor, any resulting stabilisation of the non-bonding MOs is negligible.
Having examined the particular EF
n species above, there are, nevertheless, many so-called hypervalent compounds EF
n where the classical valence of the element E is less than the maximum possible or group valence and which have one, two, or three non-bonded or lone pairs associated with the central element; examples include SF
4, IF
3, IF
5, XeF
2, XeF
4, and XeF
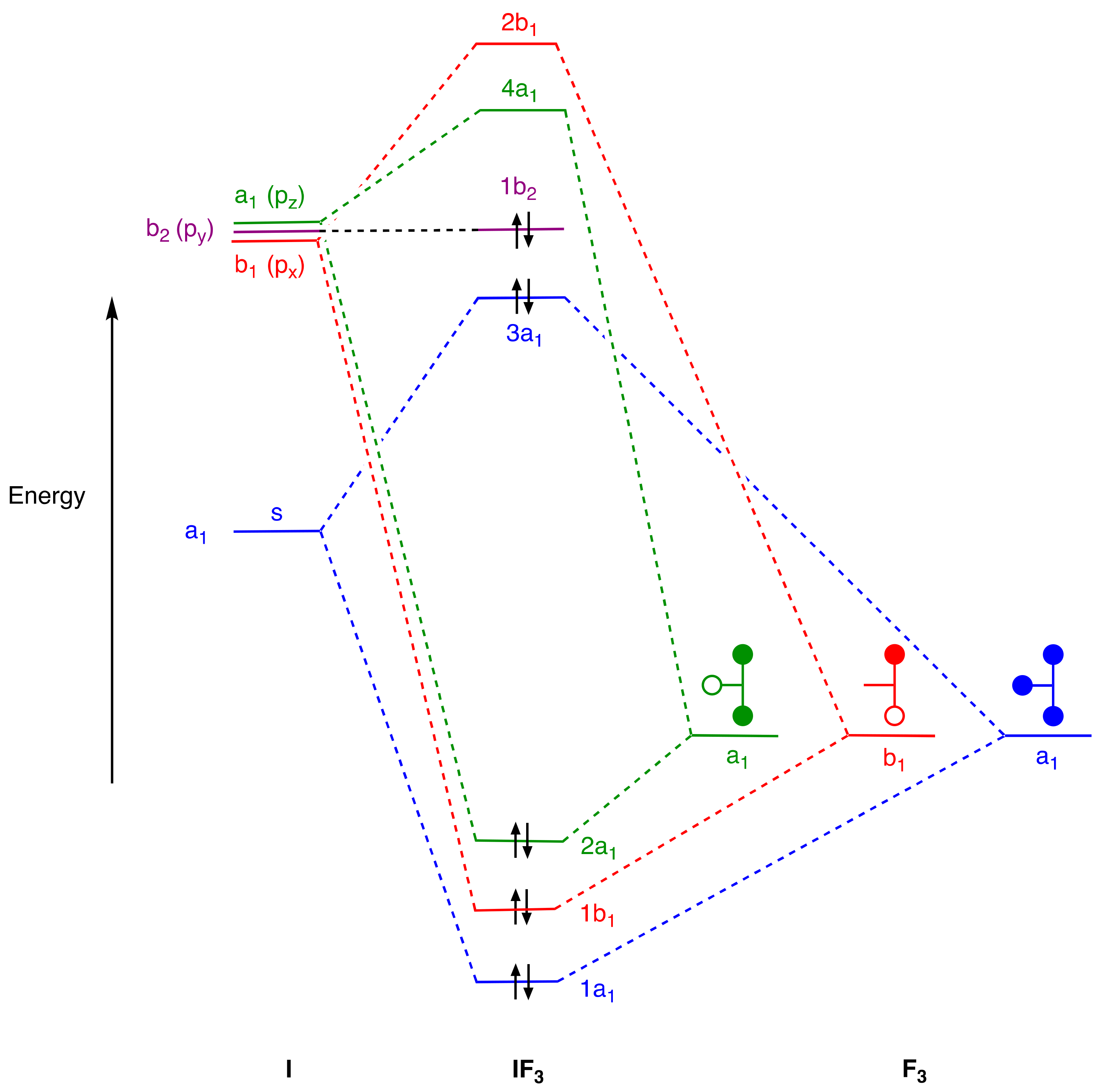
6, as well as numerous cationic and anionic derivatives. A brief examination of two such compounds is warranted in the context of this essay since this leads to some important general conclusions pertinent to our thesis, and accordingly, simple qualitative, σ-only, MO energy-level diagrams for IF
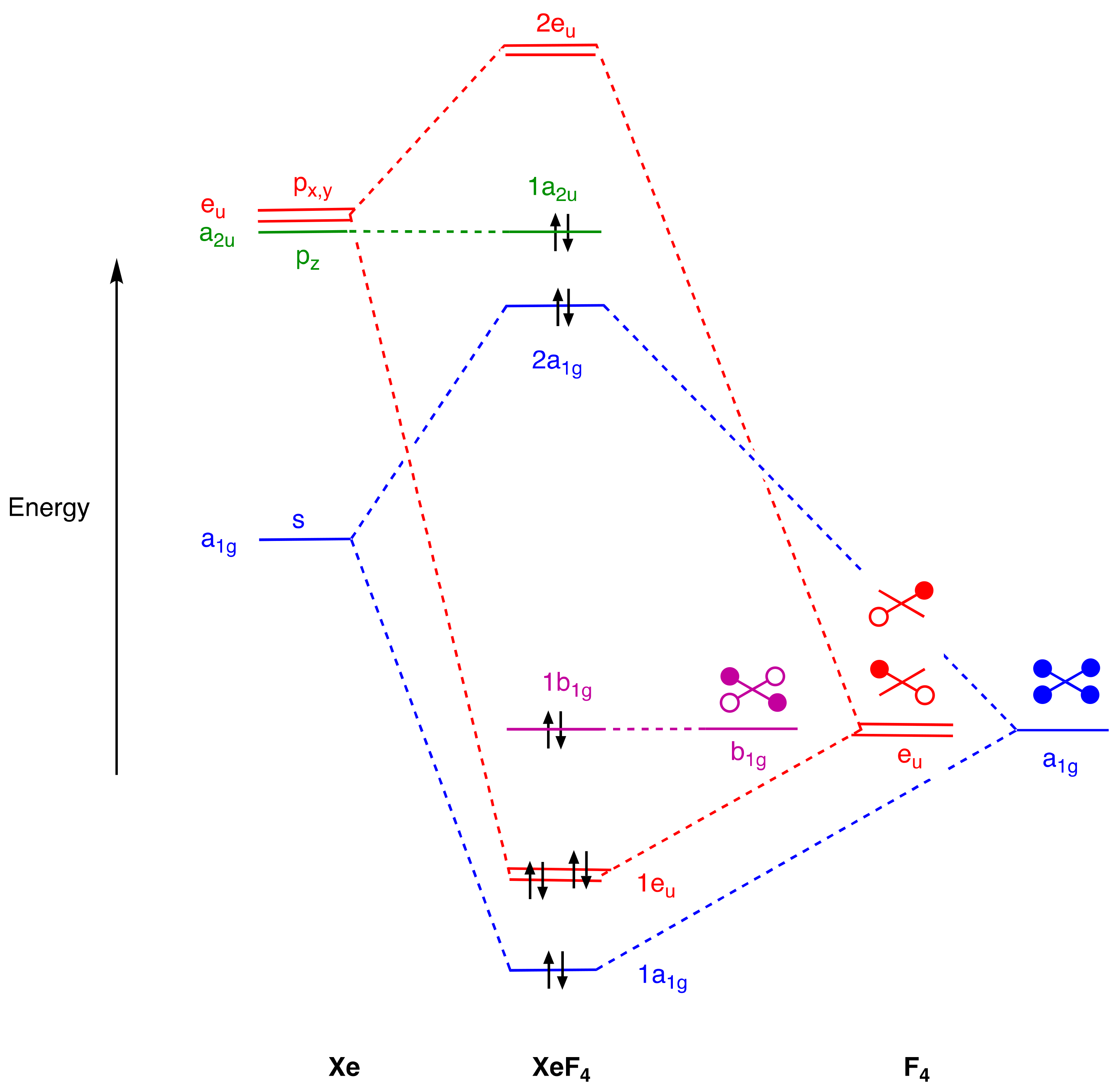
3 and XeF
4, ignoring d orbital involvement and any s–p mixing, are presented in
Figure 5 and
Figure 6, respectively [
37,
38,
39,
40]. A selection of similarly constructed MO energy-level diagrams for other lower-valent EF
n species has been collected in the
Supplementary Materials.
For IF3, in C2v symmetry, we observe three bonding orbitals (1a1, 1b1, and 2a1) and two occupied orbitals at higher energy (3a1 and 1b2). For XeF4, the analysis in D4h symmetry reveals a broadly similar picture with three bonding orbitals (1a1g and 1eu), and two higher-energy orbitals (2a1g and 1a2u), but now with a fluorine-based orbital (1b1g) which finds no symmetry match amongst the xenon s and p orbitals. Regarding the general conclusions anticipated above, there are two points that emerge which are important to highlight.
First, for both IF
3 and XeF
4, it is clear from
Figure 5 and
Figure 6 that, in each case, there are 10 electrons (5 pairs) in orbitals that have a component associated with the central element. An account of the bonding in terms of a simple octet rule therefore becomes less tenable for these and related lower-valent EF
n species once we embark on this type of MO analysis (for more on other examples, see the
Supplementary Materials), but as we will argue later when considering π-bonding interactions, this offers little in the way of any reprieve for the concept of hypervalence. It is also apparent from
Figure 5 and
Figure 6 that, unlike in the example of PF
5, one cannot straightforwardly extract any component that constitutes an isolated 3c,4e interaction (simplistically, one such interaction would be present in IF
3 and an orthogonal pair in XeF
4) (
Appendix A Note 16).
The second point relates to the determination of a classical valence and whether or not this is reflected in the MO diagrams for the EF
n species we have considered. There is nothing particularly contentious in relation to the sulphur and phosphorus valences in SF
6 and PF
5, but what of IF
3 and XeF
4, which would be classified as having trivalent iodine and tetravalent xenon according to the definition of valence outlined earlier? For IF
3, if we accept that the 1b
2 orbital is non-bonding but also consider the 3a
1 orbital to be predominantly non-bonding, because it is so close in energy to the iodine s (a
1) orbital, one arrives, approximately, at a situation with three bonding and two non-bonding orbitals consistent with a classical Lewis structure and a formal trivalent classification. For the 3a
1 orbital, this non-bonding designation becomes rather more obvious once s–p mixing is allowed, where it takes on more of the form of a lone pair [
37]. One can offer a similar argument in support of a tetravalent xenon in XeF
4 (with 1a
2u and 2a
1g assigned as lone pairs on Xe), and although we should reiterate that valence is a classical concept, its determination can be seen to emerge from a more sophisticated MO analysis, at least at this level of approximation. Further discussion of these and other MO diagrams in terms of classical valence is given in the
Supplementary Materials.
In concluding this MO-based analysis, we might now be tempted to pronounce that our argument against the concept of hypervalence is justified. Based on the energetic unavailability of d orbitals and the simplicity of the 3c,4e (or ionic) bonding model, we would argue that our case is certainly secure at this level, but there is a little more work still required since, as we have shown in this section, an octet-based description of many (although not all) EF
n compounds does not survive a more detailed MO analysis. Before considering this matter in more detail, however, we must first review the work of Gillespie, as well as Robinson, who have both been critical of the term hypervalence, although for rather different reasons from those advanced here [
41,
42,
43,
44,
45,
46] (
Appendix A Note 17).
5. Historical Criticisms
Much of Gillespie’s criticism of hypervalence initially stemmed from what he considered to be a general misunderstanding of the original ‘rule of eight’ proposed, as stated earlier, by Lewis [
20,
22], and later described by Langmuir [
21,
23] as the octet rule. Thus, Lewis originally considered that an electron pair in a bond could be counted equally for both atoms in terms of its overall electron count such that in the molecule XY shown in
E, both X and Y can be said to have eight electrons and obey the octet rule, regardless of any polarity in the X–Y bond resulting from a difference in electronegativity between X and Y. In an ionic structure such as
F, however, whereas atom Y would retain eight electrons, atom X would now have only six electrons. On this basis, for a polar covalent bond where Y is more electronegative than X, it could be argued that X would have fewer than eight but more than six electrons associated with it, which Gillespie describes in terms of a modified octet rule that an atom obeys by having eight or fewer electrons, although he was also critical of this modified rule because the value of eight is not particularly privileged in this scheme.
![Chemistry 04 00082 i003]()
Thus, Gillespie’s criticism of the octet (or modified octet) rule was that it was only ever an empirical observation by Lewis, albeit one of broad significance, but one where Lewis himself recognised exceptions such as PCl5 and SF6 (which have more than eight electrons), and also BF3 (which has fewer than eight electrons). Too much emphasis had therefore been afforded to the rule being more fundamental than was actually warranted. This was also the basis of Gillespie’s aversion to structures such as D which, he claimed, were arbitrarily drawn merely to support the octet rule itself, i.e., there was no defensible reason for preferring D over B, or indeed over an even more ionic description such as G. Gillespie was similarly critical of drawing BF3 in the manner shown in H, arguing that this representation was also biased by an unwarranted adherence to the octet rule.
![Chemistry 04 00082 i004]()
These arguments, together with the many compounds such as PF
5 and SF
6 which ostensibly have 10 and 12 electrons around the central element, respectively (
Table 1), were the crux of Gillespie’s hypothesis that the usefulness of the octet rule had become diminished, particularly as more and more examples of compounds which violate it had been characterised. It is interesting, however, that Gillespie and Robinson did propose a duodecet rule to complement the octet rule. Briefly, their proposal was that the stability of compounds such as CF
4 and SF
6 could be attributed to the presence of 8 and 12 valence electrons around the carbon and sulphur centres, respectively, further supported, in their view, by the observation that the 6- and 10-electron compounds BF
3 and PF
5 readily accept a fluoride ion to form the much more stable (or less reactive) 8- and 12-electron species [BF
4]
− and [PF
6]
− [
41,
46]. The fact that 8-electron SiF
4 reacts with fluoride to form the 12-electron [SiF
6]
2− anion to some extent undermines the simplicity of this idea, the basis of which is more easily rationalised in terms of accessible coordination numbers (four for the 2p elements and six for the 3p elements), but we will return to the matter of coordination numbers later.
In seeking to better understand the bonding in a molecule such as PF
5, in addition to rejecting the value of any ionic structures such as
D, Gillespie accepted that d orbital hybridisation had been shown to be untenable for the reasons already described. However, he was also critical of the 3c,4e bonding model on the basis that it was predicated on there being a fundamental difference between a 2c,2e and a 3c,4e bond, thereby affording a different nature to the equatorial and axial P–F bonds in PF
5. Thus, as noted earlier, in the Rundle/Pimentel model, the equatorial P–F bonds are presumed to be 2c,2e bonds, which implies a bond order of 1, whereas the axial P–F bonds, using the 3c,4e model, would each have a bond order of only 0.5. However, as Gillespie stressed, the measured P–F bond lengths in PF
5 are actually quite similar (P–F
(eq) 1.543 Å and P–F
(ax) 1.577 Å) which is not consistent with them having markedly different bond orders (although axial and equatorial bond lengths differ rather more in PCl
5 and SF
4) [
41] (
Appendix A Notes 18,19).
The approach to understanding the bonding in species such as PF
5 and SF
6 that Gillespie preferred was largely based on insights derived from a computational method known as the electron localisation function (ELF), which can be expressed in terms of localised orbitals and derived bond orders [
42,
43]. The details of the ELF function are described in [
42,
43] and will not be elaborated upon here, but crucially, the results calculated for the equatorial P–F bonds in PF
5 are quite similar to those determined for the axial P–F bonds. This similarity was the principal basis for Gillespie proposing that there is little difference between these two bond types and, thus, that the 2c,1e bonds proposed by Musher or the 3c,4e bonds proposed by Rundle, Pimentel, and others are neither useful nor necessary.
A further output from ELF calculations is a total valence shell population for the central atom,
Nv(A), where A is the central element, which for PF
5 is calculated to be 5.33, whereas for the hypothetical PMe
5,
Nv(A) is determined to be 9.42, the large difference being due to the differing polarities of the P–F and P–C bonds [
43]. Accordingly, together with a number of similar examples, Gillespie argued that both hypervalence and the octet rule have little explanatory value either in terms of the nature of the bonding or with regard to the total valence electron count at the p-block element centre, thereby undermining the purported difference between compounds which are presumed to obey the octet rule and those hypervalent compounds which do not. Gillespie does, however, note the contrast between the ELF method and both the natural population analysis (NPA) described by Reed and Schleyer [
32] and the atomic overlap matrix (AOM) method discussed by Cioslowski and Mixon [
47], both of which find total bond orders of less than four (i.e., fewer than eight electrons) for the hypervalent molecules they consider. We will not expatiate on these differences here because they are beyond the scope of this essay but will briefly reference the work of Cooper and Gerratt and their report of a spin-coupled valence bond analysis of, amongst other examples, PF
5, some of the conclusions of which are captured in the following quotation [
48].
“In spite of its continuing overwhelming appeal we suggest that the familiar octet rule should be demoted. We retain only an eight-electron rule (cf. the 18-electron rule of transition metal chemistry), which indicates that a formal electron count of eight around a central atom is favorable. We assert here the new democracy principle, which, stated very simply, suggests that “it is the democratic right of every valence electron to take part in chemical bonding if it wants!””
The authors themselves recognise that their democracy principle may be a little too anthropomorphic, but although they are critical of the octet rule in much the same way as Gillespie was, their eight-electron rule does not depart too radically from the position we have taken here.
A more recent contribution to the bonding in hypervalent compounds, which also covers some of the history recounted here, has been offered by Durrant [
49] who has proposed a quantitative scale of hypervalency based on atomic charges and a derived electron count at the central atom calculated using Bader’s Quantum Theory of Atoms in Molecules [
50]. This is somewhat analogous to the ELF approach adopted by Gillespie, albeit using a different computational method. To quote Durrant,
“In order to calculate the overall electron count … we may now define a parameter called the valence electron equivalent, γ, as ‘the formal shared electron count at a given atom, obtained by any combination of valid ionic and covalent resonance forms that reproduces the observed charge distribution’.”
and
“It follows that for any given atom X, if γ(X) = 8, the atom obeys the original Lewis octet rule. If γ(X) < 8, the atom obeys the ‘modified octet rule’, but if γ(X) > 8, neither form of the octet rule is obeyed and the atom is hypervalent.”
We will not comment further on Durrant’s study except to note that γ(S) calculated for sulphate, SO42−, is 4.34, whereas γ(Cl) for perchlorate, ClO4−, is computed to be 9.11 and on this basis, sulphate is not considered to be hypervalent, whereas the isoelectronic perchlorate is. We submit that although this quantitative analysis has much to reveal about atomic charges within molecules (which are often quite different from valence-bond-type formal charges), to classify one member of an isoelectronic pair as hypervalent and the other not may cause some confusion in an educational context.
8. The Question of π-Bonding
Let us consider the molecule POF
3. Although simple electron-counting procedures (including VSEPR) would assign 10 electrons to the phosphorus centre in representation
I, only 8 electrons are associated with the phosphorus atom in
J and
K (
Appendix A Note 21). To have to describe
I as hypervalent, but not
J or
K, cannot be supported in any educational setting. One rarely comes across the depiction shown in
K, but the distinction between
I and
J has received much attention from, for example, Reed and Schleyer [
32], as well as from Gilheany [
33]. Both representations have their merits, but in terms of the bonding in the P–O unit, the best description is probably one based on structure
J but in which oxygen lone pairs in p orbitals are able to back-donate to vacant P–F σ* orbitals (shown in
Figure 7) as discussed extensively by Reed and Schleyer [
32] and by Gilheany [
33]. This type of π-bonding interaction is generally described as negative hyperconjugation and involves
n(or π)→σ* donation (
Appendix A Note 22). Any π-interactions of this type are not explicitly shown in
J but are implicit in the P=O bond drawn in
I, albeit in a way that does not properly reflect the symmetry of the molecule. Thus, in C
3v symmetry, there are two degenerate π-type lone pairs on oxygen and a degenerate pair of P–F σ* orbitals which can each act as acceptors of π symmetry. Both these interactions are shown in
Figure 7 and, in combination, can be illustrated diagrammatically as drawn in
L. Both
I and
L would necessarily be classed as hypervalent, and each reflects a degree of π-bonding between the oxygen and the phosphorus which would assign 10 and 12 electrons to the phosphorus centre, respectively, and which it is useful to consider in a little more depth.
![Chemistry 04 00082 i005]()
Thus, one should recognise that any π-back-donation from the oxygen in POF
3 leads to an increase in the number of electrons associated with the phosphorus centre as implied in representations
I and
L, and which would be evident from a more detailed MO treatment which explicitly takes account of π-bonding interactions. However, π-back-donation is not restricted to P–O species and is possible in PF
5 from π-type fluorine lone pairs to P–F σ* orbitals of the correct symmetry (not shown in
Figure 3), which similarly results in an increase over and above the eight electrons assigned in our earlier discussion. Is there not, therefore, a case for redeeming the term hypervalent based on an expansion beyond the octet due to this type of π-bonding as well as for the MO-derived reasons we considered for lower-valent EF
n species such as IF
3 and XeF
4? We argue that there should be no such redemption. If one wishes to account for electrons involved in π-back-bonding, then it is apparent that in the phosphorus molecules considered, electron counts at the phosphorus centre would exceed eight, in line with some of the calculated valence electron counts for related species outlined earlier [
42,
43,
49] but this would also be the case for molecules such as CF
4 which, as Reed and Schleyer made clear [
32], has significant π-donation from fluorine lone pairs to C–F σ* orbitals. This, we contend, is quite a powerful argument against the hypervalent label because any accounting of these π-interactions in CF
4 would lead to a number greater than eight around the carbon centre, but it makes little sense to consider that CF
4 has violated the octet rule and should therefore be classed as hypervalent, nor indeed has the carbon exceeded its usual valence of four. Almost every molecule (excluding hydrides and alkyls) would be hypervalent on this basis. Moreover, we should not take the σ-only MO-derived electron counts greater than 8 (as described earlier for IF
3 and XeF
4) as a reason to defend hypervalence since this would lead to a molecule such as SF
4 (see the
Supplementary Materials for an MO diagram of this species) with 10 electrons around the sulphur being classed as hypervalent, whereas SF
6, with only 8 sulphur-based electrons, would not be classified as such. We likewise reject the classification of hypervalent for the 14-electron complexes [SbCl
6]
3− and [TeCl
6]
2− referred to earlier.
11. Depictions of 3c,4e Bonds
Having stressed the value of the simple 3c,4e model to describe the bonding in molecules such as PF
5 (or others with linear X–E–X units) and the central part it plays in our thesis that hypervalence is a redundant concept, at least at an introductory level, let us now turn to how best this type of interaction might be represented without recourse to MO diagrams (
Appendix A Note 24). In
B, the ubiquitous solid line drawn between pairs of atoms which very often represents a 2c,2e bond (and has a long history (
Appendix A Note 25) clearly does not do so in each of the axial P–F bonds, for the reasons already discussed. Thus, in the linear axial F–P–F arrangement in PF
5, the two lines together represent a 3c,4e interaction, and the formal bond order of each P–F bond according to this model is 0.5 (
Appendix A Note 18). This would also be true in compounds with formally 10 valence electrons having linear X–E–X arrangements, such as in SF
4 and XeF
2. To make this point, these interactions have sometimes been drawn with dotted/dashed lines, as shown for PF
5 and SF
4 in
M and
N [
34,
35,
42]. Attractive though this might be as a way of differentiating between 2c,2e and 3c,4e interactions, we would argue that although dotted/dashed lines might have some value in these examples, their use would be unhelpful in more delocalised species such as SF
6, XeF
6, or IF
7 since all bonds would need to be drawn in this way. Moreover, dotted/dashed lines are often used to represent transition states in, for example, S
N2 reactions at a carbon centre, and it is therefore important, certainly in an educational setting, to avoid any suggestion that structures
M or
N represent a transition state. We note that an alternative symbolism has been advanced by Weinhold and Landis as shown, using PF
5 as an example, in
O which is sometimes referred to as an I bond [
19].
![Chemistry 04 00082 i006]()
A dotted/dashed line representation has also been used by Green and Parkin in their classification of 3c,4e interactions within the Covalent Bond Classification (CBC) [
57], but we highlight here their insightful description of 3c,4e interactions of the type found in PF
5 as examples of an X–L–X interaction based on the standard CBC framework definitions, where L contributes two electrons and X contributes one electron [
58,
59,
60] (
Appendix A Note 26). We note also that the CBC method would describe PF
5 as an MX
5 species according to the rules set out in [
58,
59,
60] and that in [
57], the case is made to highlight the presence of any hypervalent interaction by adding an H designation leading to an overall descriptor of the general form MLXZH, more specifically ML
lX
xZ
zH
h, where the lower-case subscripts
l,
x,
z, and
h indicate the numbers of L, X, Z, and H functions, respectively. The term
h is defined as the Hypervalent Index, which is the number of such interactions, although it is recognised that the situation becomes more complex for higher-order interactions, as found in molecules such as SF
6. In this scheme, PF
5 would therefore be designated as MX
5H to indicate that there is one hypervalent 3c,4e interaction present. Moreover, a modification to the definition of the CBC electron number (EN) is presented in [
57] such that EN =
m + 2
l +
x − 2
h (where
m is the number of valence electrons on M), with the hyperelectron number (HEN) being defined as HEN = EN + 2
h. Although we might argue for something other than the H designation
per se (because H denotes hypervalent), we recognise the merit of their scheme in highlighting the number of 3c,4e interactions and the value of the X–L–X descriptor, not least as a means of differentiating between other types of 3c,4e bonding (
Appendix A Note 26).
12. Conclusions and Recommendations
Having presented an outline history and overview of the topic of hypervalence, in addition to many of the bonding models which have historically been considered, and having made our position clear about the value of its continued use, it is time to summarise our conclusions and offer some recommendations. Thus, we contend that the concept of hypervalence, especially in an educational context, is not only unnecessary and potentially misleading, but also suffers from certain internal inconsistencies. For the many compounds which most would agree obey the octet rule (or even a modified octet rule) after counting the valence electrons around the central element, there is little argument to be had especially if one ignores any consequences of π-back-bonding due to negative hyperconjugation in molecules such as CF
4. For those compounds in which electron counting allocates 10 or 12 valence electrons, simple arguments based on a 3c,4e bonding model for linear X–E–X units or polar bonds in oxo compounds such as
+P–O
− are consistent with there being no more than 8 electrons significantly associated with the central element which obviates the need for any hypervalent label. Although consideration of more sophisticated MO analyses, including any explicit inclusion of negative hyperconjugation, will be at the expense of retaining an octet-based description for many, although not all, species, this fails to rescue a hypervalent classification in any meaningful or useful sense since numerous compounds that are not generally considered to be hypervalent would now have to be included within the curtilage of a much-expanded classification that would likely exclude few EX
n species, except when X = hydride or alkyl. Moreover, use of the prefix hyper- in species such as [SiF
5]
− or [SiF
6]
2− (isoelectronic with PF
5 and SF
6, respectively) lacks any logical basis since, according to the definition of valence, the silicon is tetravalent in each case, just as it is in SiF
4. Likewise, any description of hypothetical molecules such as [CH
6]
2+ or of isolable complexes such as [C(AuL)
6]
2+ (L = PPh
3) as containing hypervalent carbon should be rejected; like silicon, carbon is never more than tetravalent. Thus, we repeat our assertion that the only prefix which should be employed before the term valence/valent be a numerical one, so as to merely denote the actual valence. As some of the authors previously referenced have suggested (notably Schleyer), if the use of the prefix hyper- is to be retained, the term hypercoordinate has more merit than hypervalent for those species with a coordination number greater than four, but this too, we argue, is of only marginal descriptive advantage [
32].
We therefore summarise these arguments and others in a list of recommendations set out below, which are written from the point of view of how best to teach students at an introductory level about those molecules encountered in p-block element chemistry to which our arguments apply.
- (i)
Introduce the definition of the term valence and stress that it is a classical concept, albeit a very useful one, which is most easily determined for a given element from a simple resonance or Lewis structure of the compound in which that element resides. It should be stressed that although prefixes which denote a number, such as in trivalent and pentavalent, are useful, those of a more subjective nature, such as hyper-, hypo- and sub-, afford no tangible benefit to any classification or description and should therefore not be used. The case against hyper- is further reinforced in some of the recommendations which follow.
- (ii)
Recognise the value of the octet rule as an important organising principle. As with all electron-counting rules, it is a simple heuristic but one of considerable scope and merit. Moreover, because the valence orbitals for p-block elements are the ns and three np orbitals, we should expect an 8-electron rule for much the same reason that we expect (and observe) an 18-electron rule for many d-block element compounds. There will always be exceptions, but it is a very good place to start. The fact that an octet-based description does not always survive an analysis involving more detailed MO methods is not a reason to ignore its merits at a simpler level.
- (iii)
Stress that vacant d orbitals play no significant role in p-block element chemistry because their energies, according to calculations, are too high, and bonding models based on d orbital occupancy or any use of d
nsp
3-type hybrids for compounds such as PF
5 and SF
6 should be abandoned [
61] (
Appendix A Note 27).
- (iv)
For compounds to which simple valence electron counting methods assign more than eight electrons and which contain linear X–E–X units such as PF5, SF4, XeF2, and XeF4, the 3c,4e bonding model for this unit should be introduced, emphasising that because the non-bonding orbital is not localised on the central atom, the actual electron counts in these species do not exceed eight.
- (v)
Recognise that for compounds where the coordination number exceeds four, some degree of multi-centre bonding will always be a feature, but any reference to the term hypercoordinate (as a possible alternative to hypervalent) is unhelpful because multi-centre bonding is also present in species with coordination numbers of four, or even less such as in SF4, XeF4, XeF2 IF3, or [I3]−, all of which contain linear X–E–X units.
- (vi)
Where more detailed insights into molecular electronic structure are required, this will be by means of simple, qualitative, MO energy-level diagrams derived from ligand group orbitals or, ultimately, by recourse to more quantitative electronic structure calculations, albeit sometimes at the expense of simple octet-based descriptions. This offers no reprieve for hypervalence, however, because too strict an adherence to classifying compounds as hypervalent if their electron count exceeds eight leads to a greatly increased catalogue of compounds, many of which would not normally be considered as such; see also point (ix) below.
- (vii)
With examples such as [SiF5]− and [SiF6]2−, it can be demonstrated that use of the term hypervalent is logically inconsistent because, according to the definition of the term valence, the silicon centre in these anions is tetravalent, as is the silicon in SiF4 itself. To describe these anions as hypervalent is therefore a misnomer.
- (viii)
For compounds with formal multiple bonds such as the P–O bond in POF3, the fact that the charge-separated +P–O− single bond representation allocates 8 electrons to the phosphorus centre whereas the P=O and −P≡O+ multiple bond depictions assign 10 and 12 electrons, respectively, highlights another inconsistency within the concept of hypervalence since in one portrayal of the bonding, the molecule would not be considered hypervalent, whereas in the other two, it would be described as such. The +P–O− representation is therefore preferred in this context (and with bonds to a terminal oxygen more generally) and brings a further advantage in that it reflects the polarity of the P–O bond. It should, nevertheless, be recognised that using the P=O form is useful in other contexts.
- (ix)
An additional advantage of the
+P–O
− representation (more generally,
+E–O
−) is that it provides a straightforward introduction to π-type negative hyperconjugation interactions which admits of the possibility of additional electron density at the central element, albeit not restricted to compounds that are traditionally classified as hypervalent, which further undermines the value of the term [
62]. (
Appendix A Note 28, 29)
Simple models are simple models, and they are enormously useful, especially in a teaching context. As with all simple models, there is a risk of them being applied outside of the envelope in which their use is appropriate, and it is often tempting to add additional auxiliary models to broaden their applicability or utility. This should be approached with some caution, however, because in so doing, one can very easily undermine the pedagogical value of the basic model to which the auxiliaries have been added. We argue that this is what has happened with hypervalence, and that by adopting the recommendations listed above, the term is seen to be both redundant and an unnecessary auxiliary to the concept of chemical valence and electron counting generally. As always, where simple models fail (as they always do when pressed too far), we should resort to qualitative or more quantitative electronic structure calculations in order to more closely interrogate the systems in which we are interested.
Do we really expect, on the basis of the arguments presented in this essay, that the term hypervalent will actually start to disappear from the literature? No, not really, nor are we about to embark on any crusade to have the term expunged from the chemical canon. It is too well established, especially in some research areas where we cannot credibly maintain that its use does any real harm. So-called hypervalent iodine compounds are widely used in organic synthesis, for example, and have been the subject of several books, numerous reviews, and countless papers which include ‘hypervalent iodine’ in the title [
63,
64,
65]. Although we would argue that the majority of these reagents would be better described as trivalent iodine compounds (some are pentavalent, although none are heptavalent), we have no expectation that any such change is realistic. Moreover, according to SciFinder, in the five years from 2017 to 2021, there were over 1250 papers with ‘hypervalent’ in the title or abstract such that any campaign to excise the usage of this term would likely be Sisyphean in nature. We do, however, make a plea that the concept of hypervalence in teaching and in teaching texts be abandoned (albeit that its presence in extant literature will have to be acknowledged), and maintain that in the vast majority of examples, enough insight into the categorisation and chemistry of p-block element compounds can be gained according to the recommendations set out above.
In concluding, we note two recent contributions which offer at least partial support for our position, although neither goes so far as to call for the abandonment of the term hypervalent. In the first of these, Crabtree draws attention to the familial link between hypervalency, secondary bonding, and hydrogen bonding, and cites both 3c,4e interactions and the MO scheme for SF
6 in support of the octet rule [
66]. In the second contribution, Harshman and Miliordos are more explicitly critical of the concept of hypervalence, based on computational studies which support octet-based descriptions arising from ionic representations similar to
D and depictions of E–O bonds such as
J or
K in strong preference to double-bonded alternatives such as
I [
67]. We shall no doubt see what others have to make of the arguments we have promoted in this essay in the hope that it is not too recondite a topic to warrant further debate.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
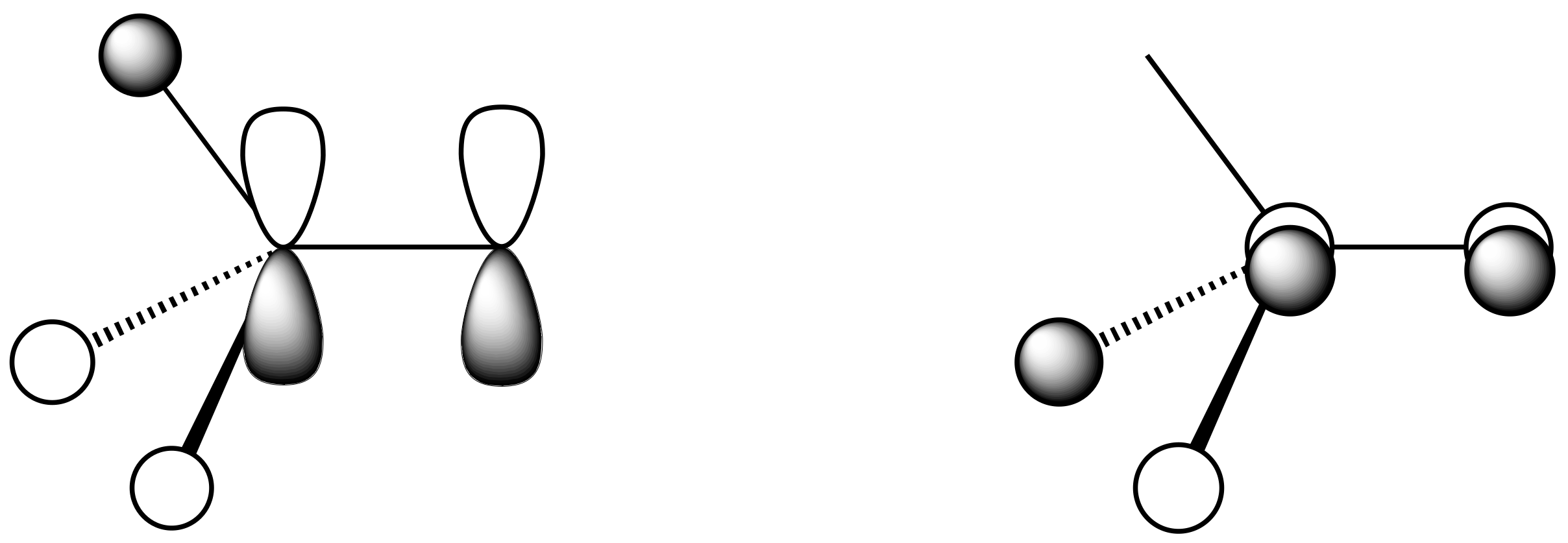{kind=link}