
Structure and Bonding in Planar Hypercoordinate Carbon Compounds


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
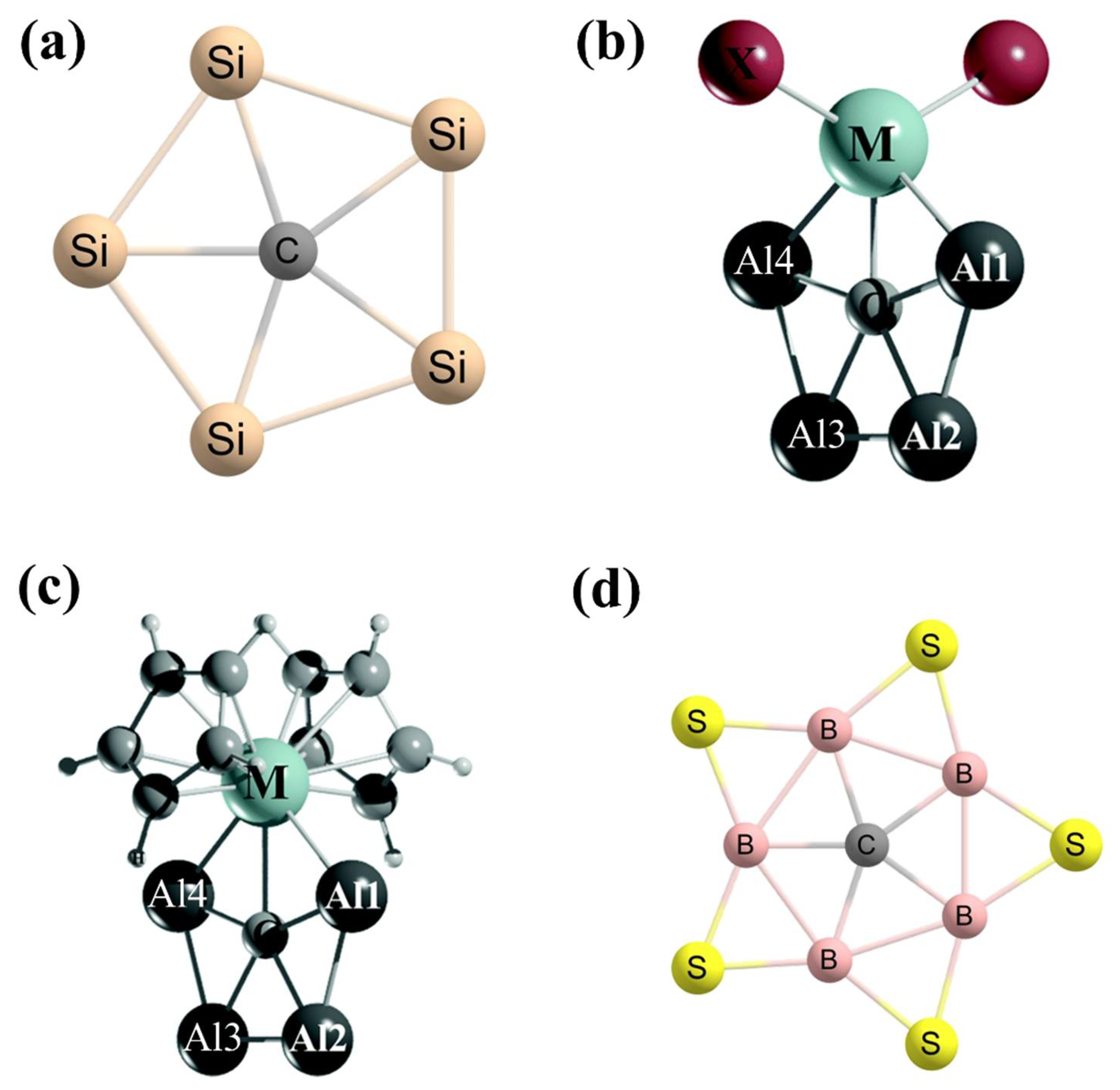
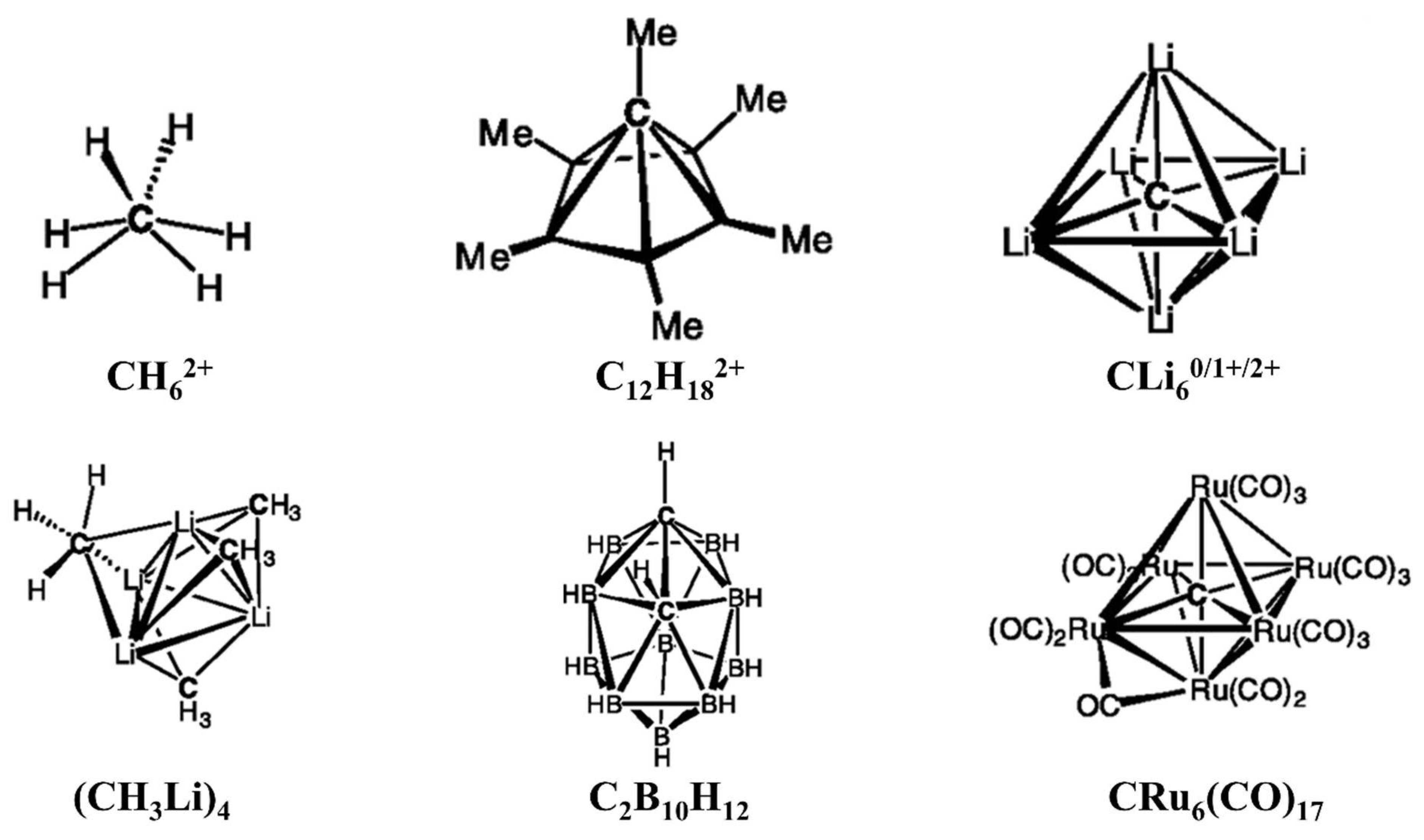
2. Planar Tetracoordinate Carbons (ptCs)
2.1. How to Achieve ptCs
2.2. Early Examples of ptCs
2.3. Recent Examples of ptCs with 18 Valence Electrons
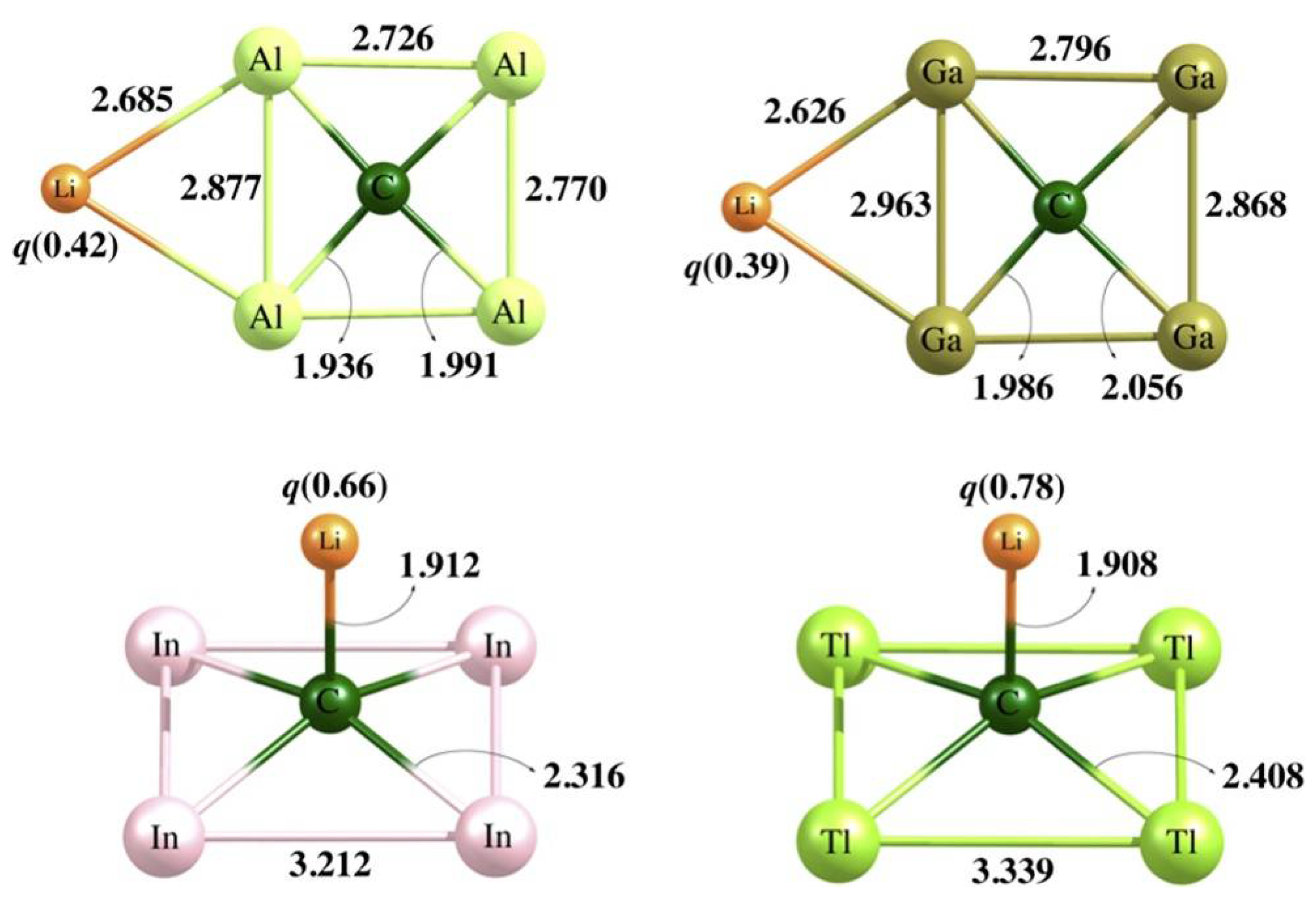
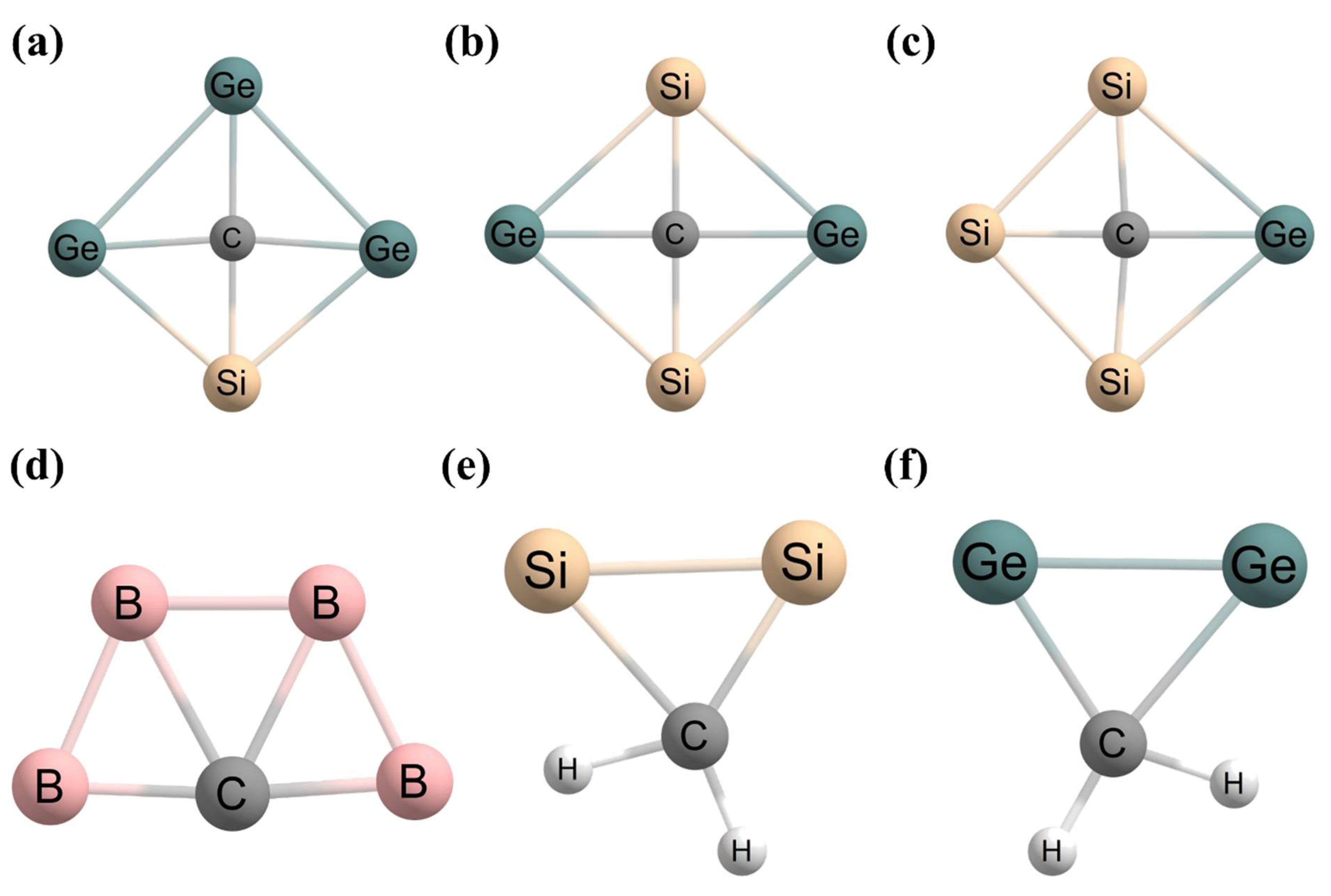
2.4. Examples of ptCs without 18 Valence Electrons
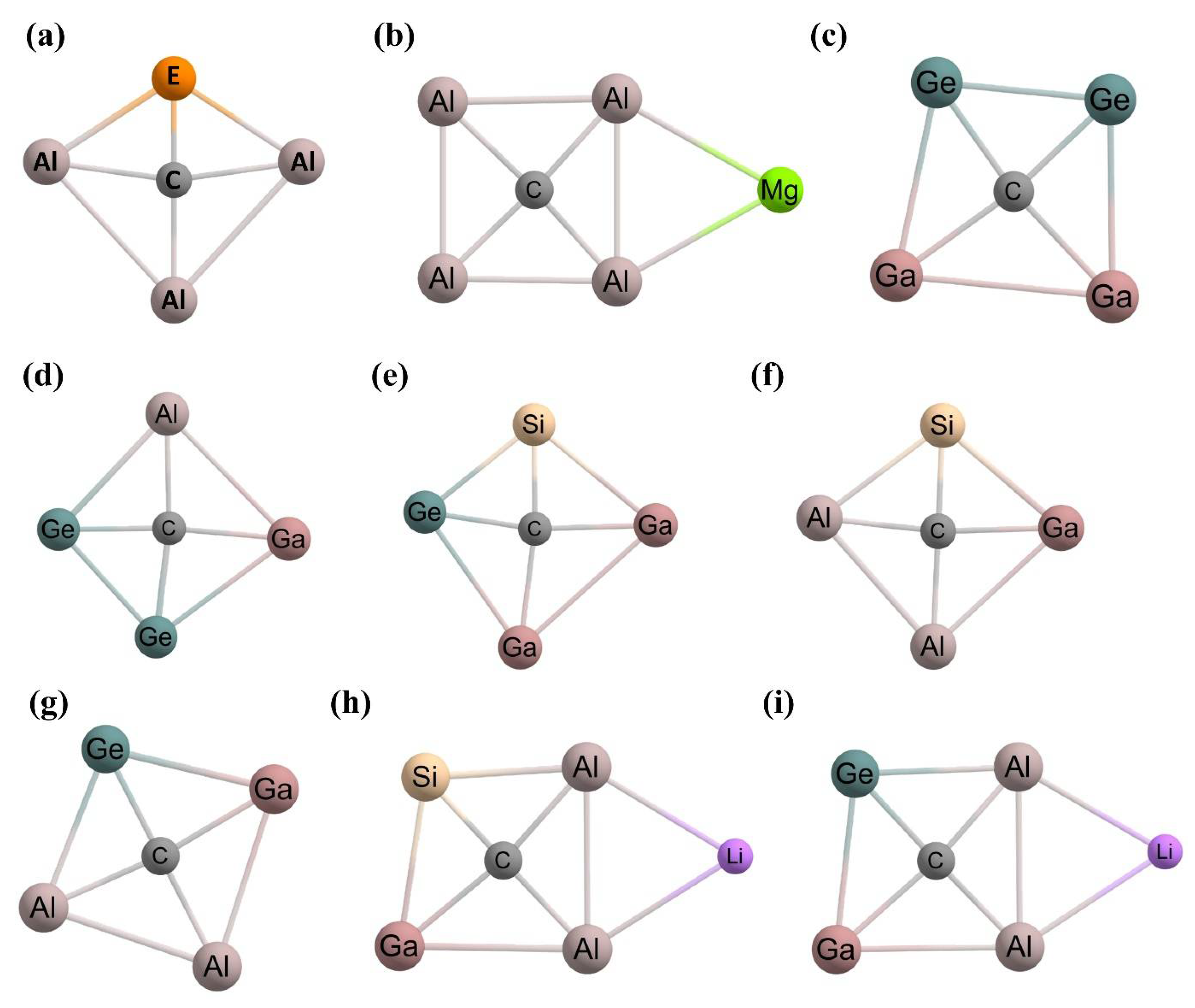
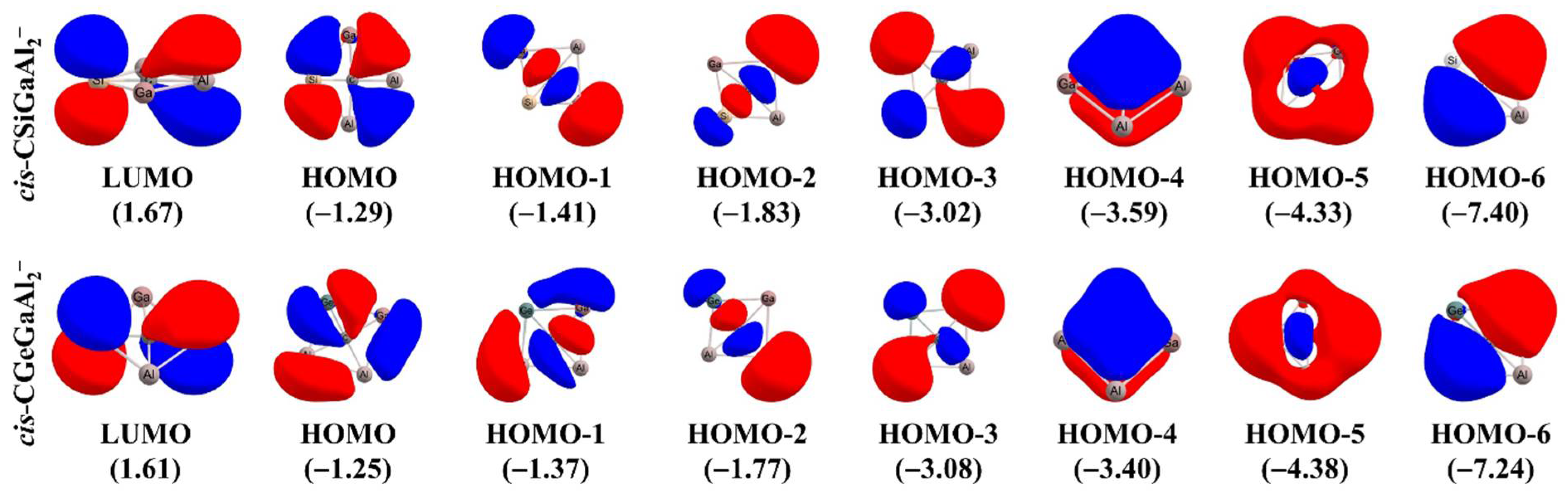
2.5. Examples of ptCs Using New Principles
2.6. Examples of Experimentally Predicted ptC Compounds
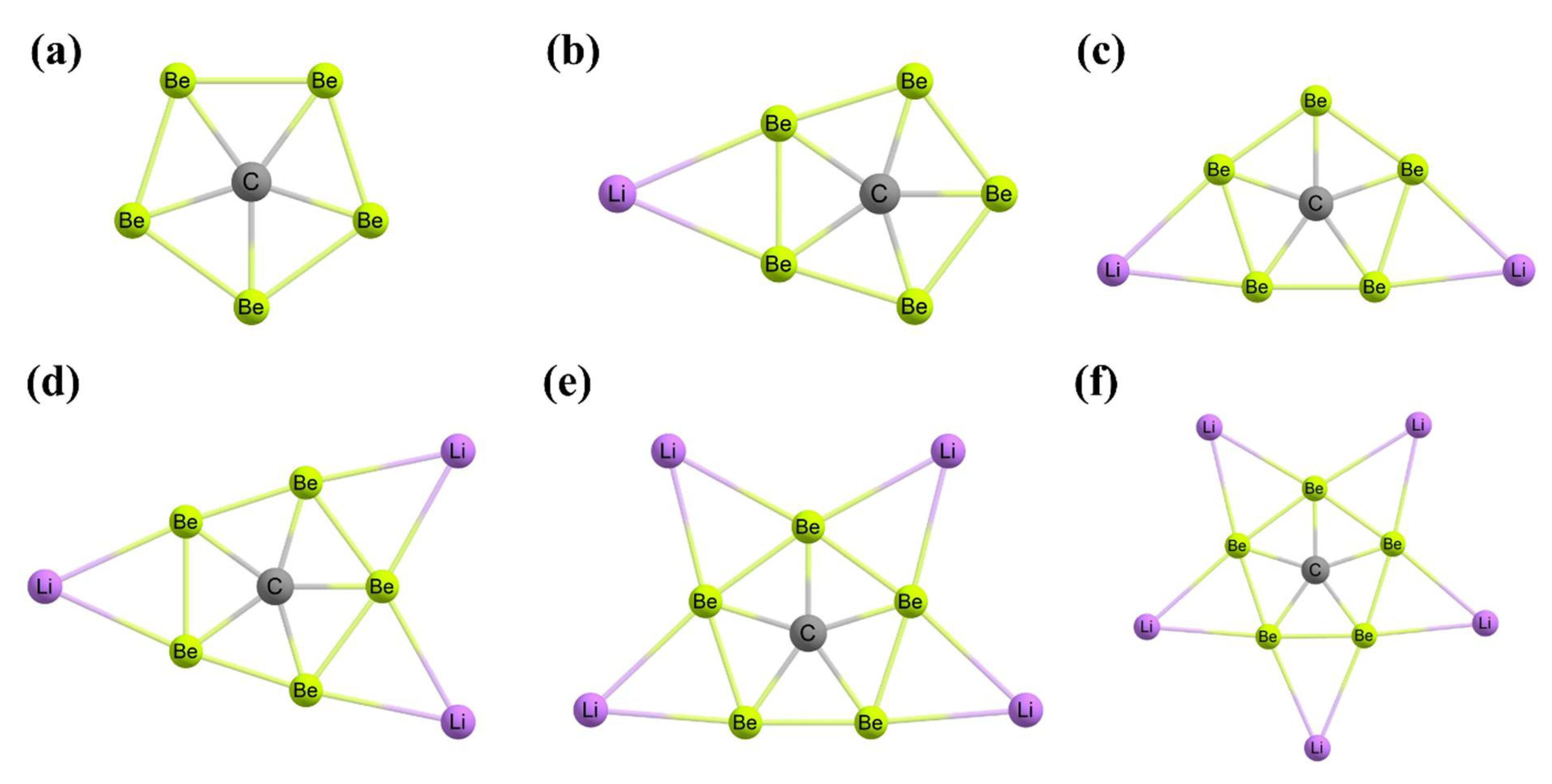
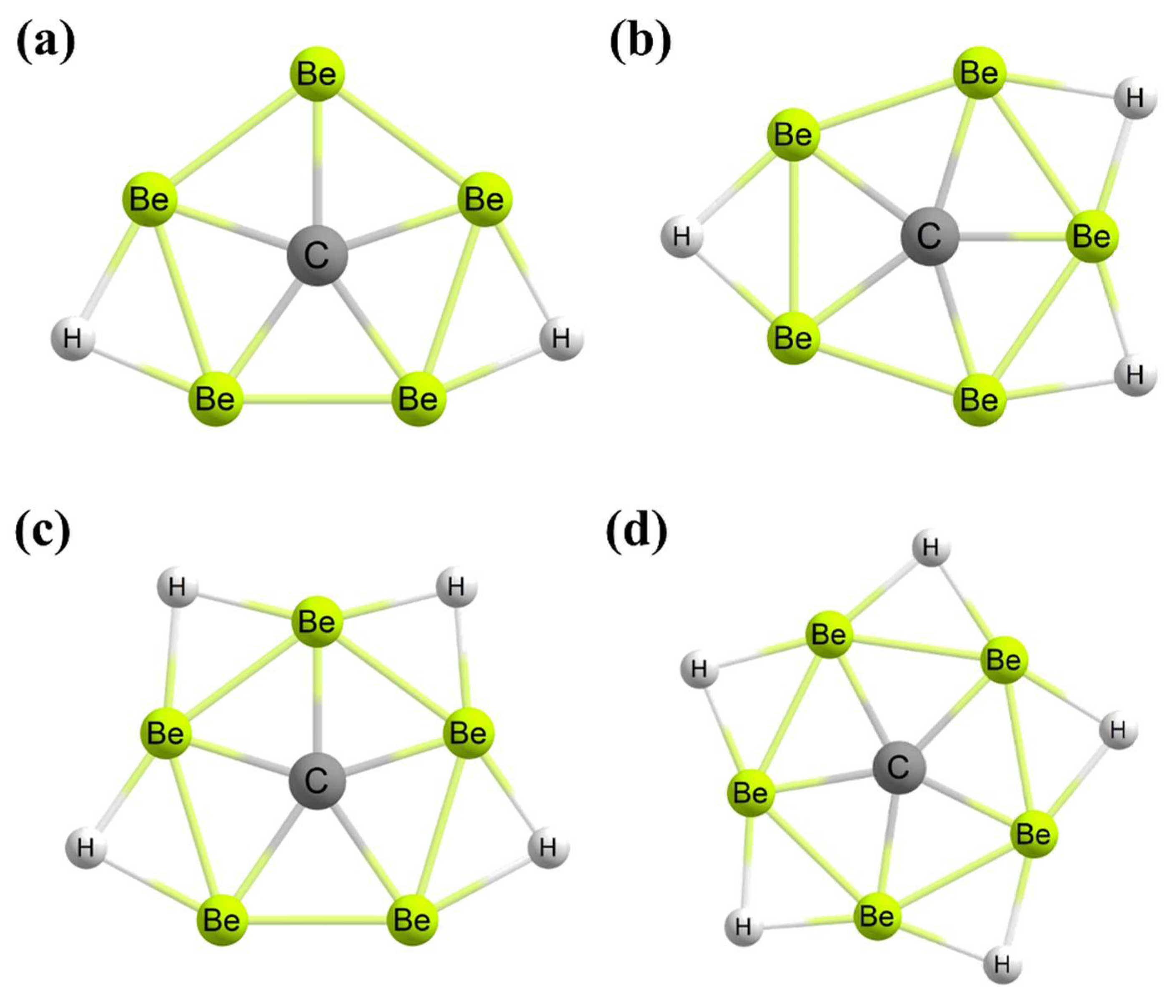
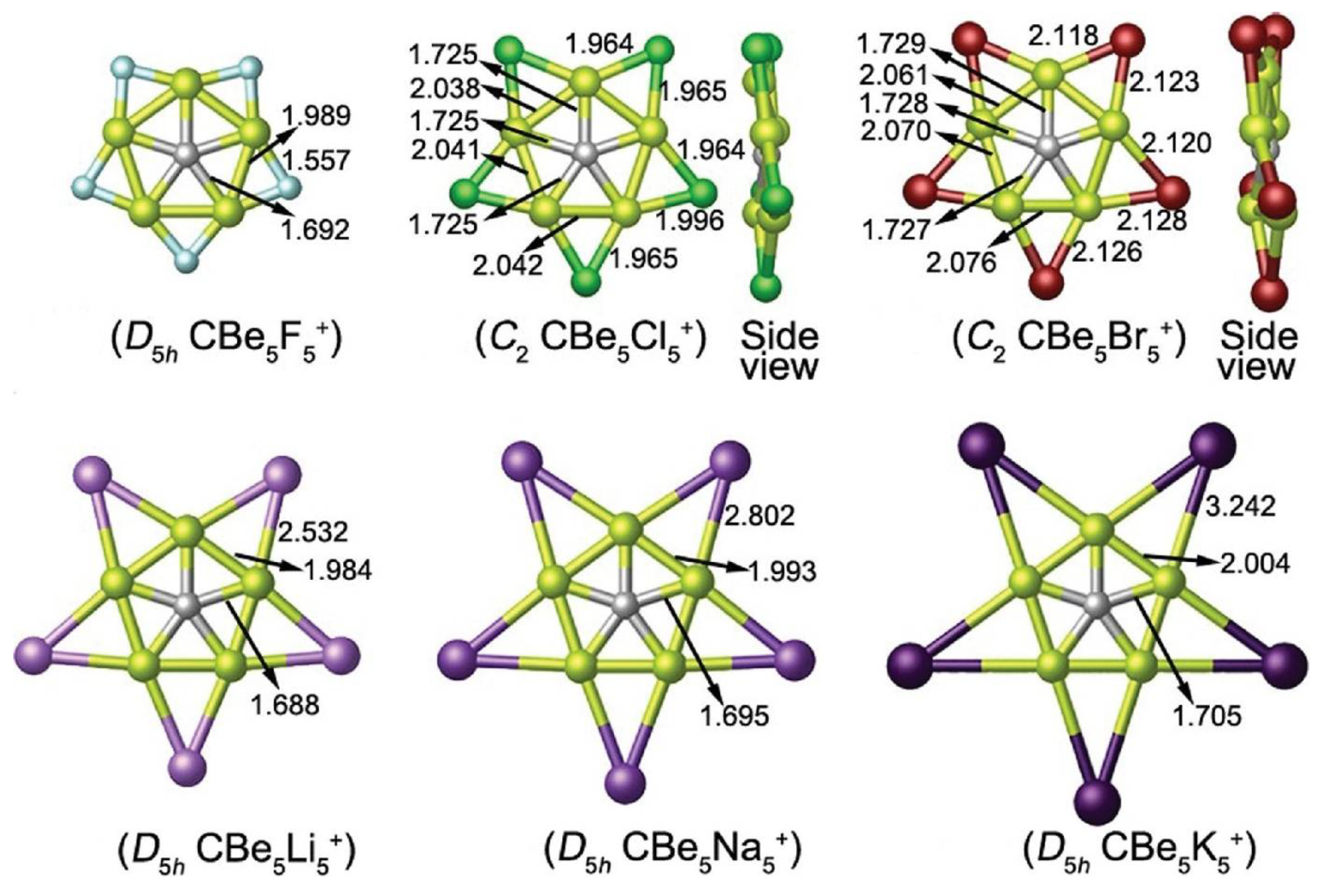
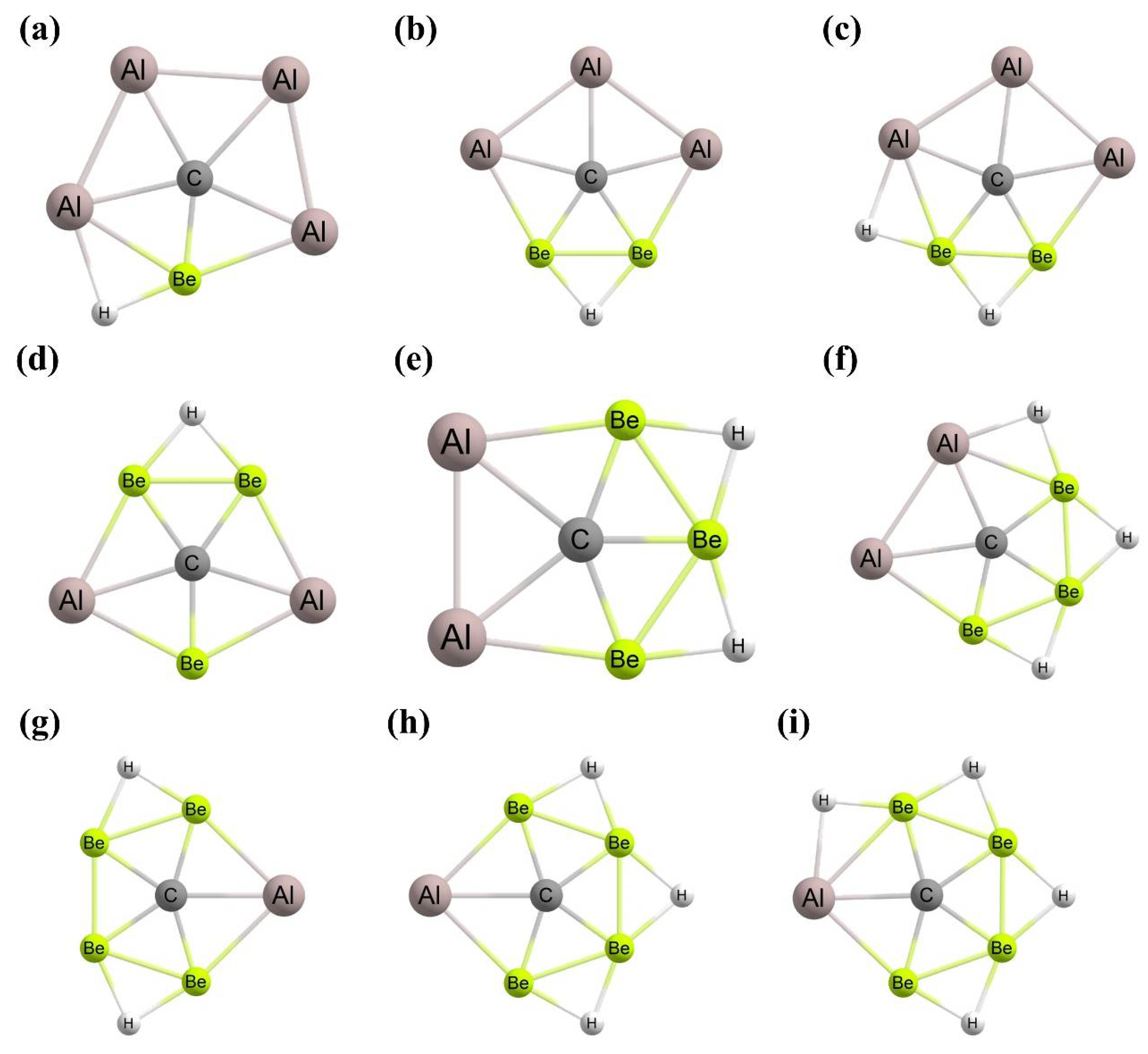
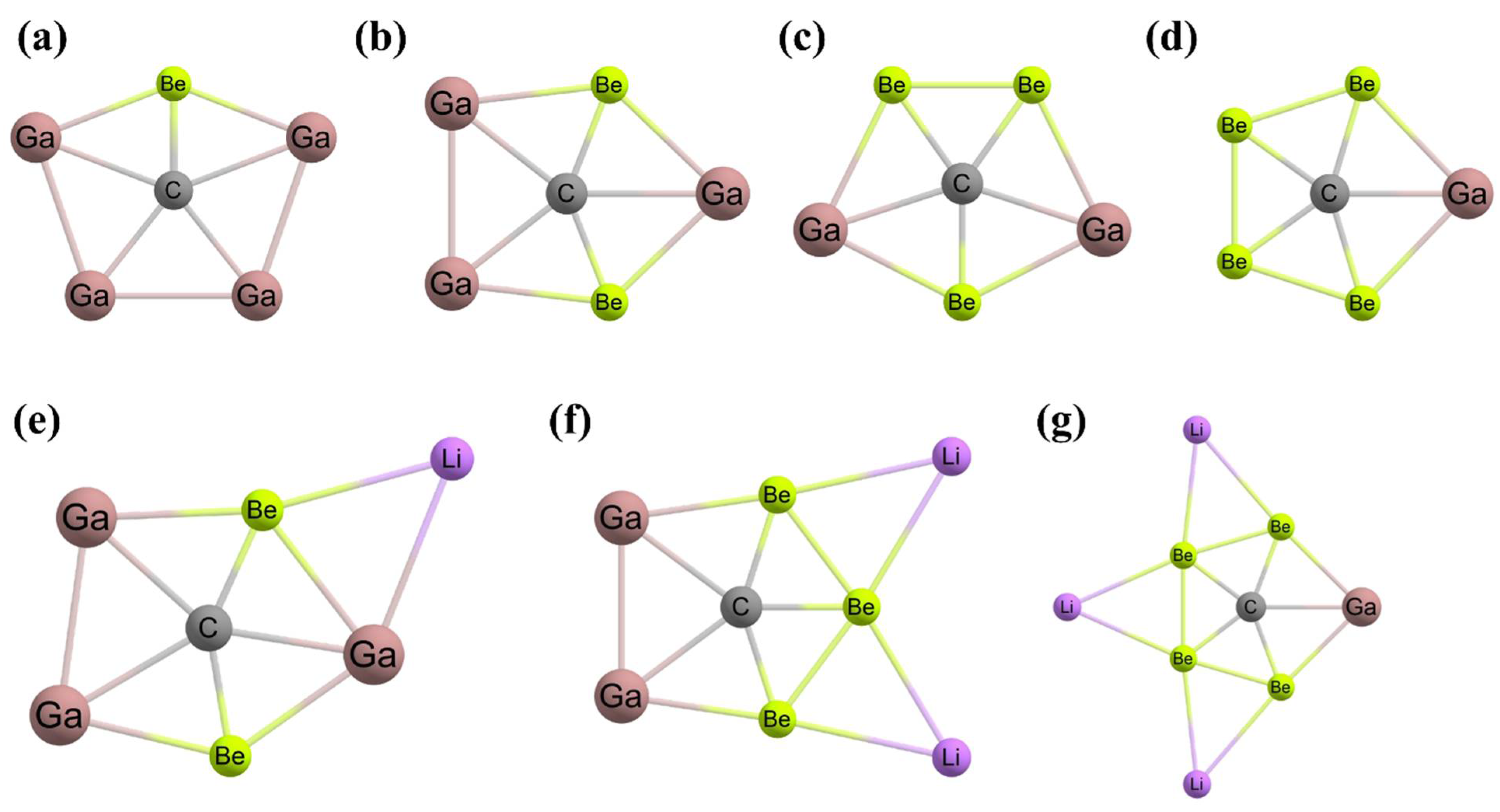
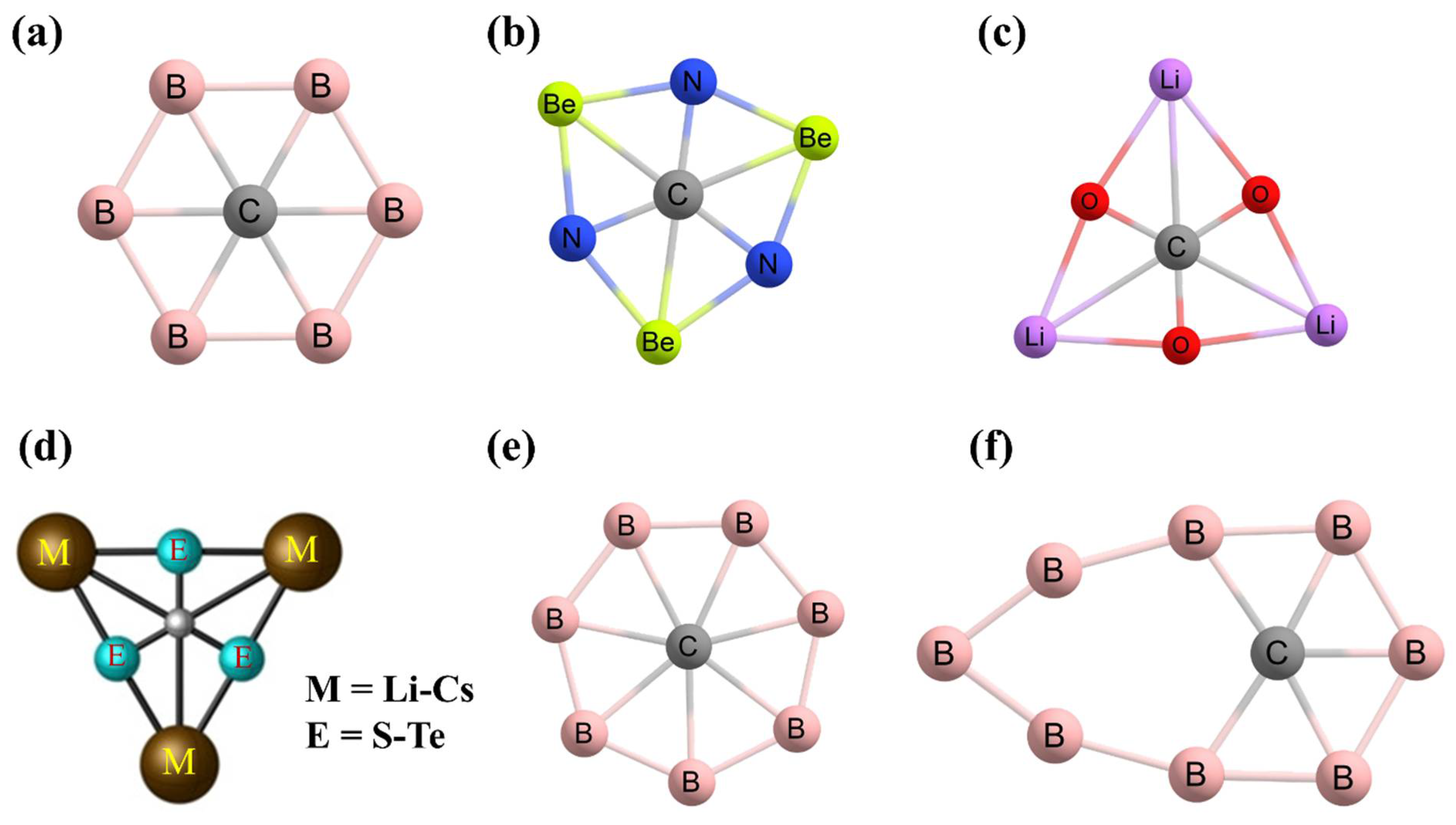
3. Planar Pentacoordinate Carbons (ppCs)
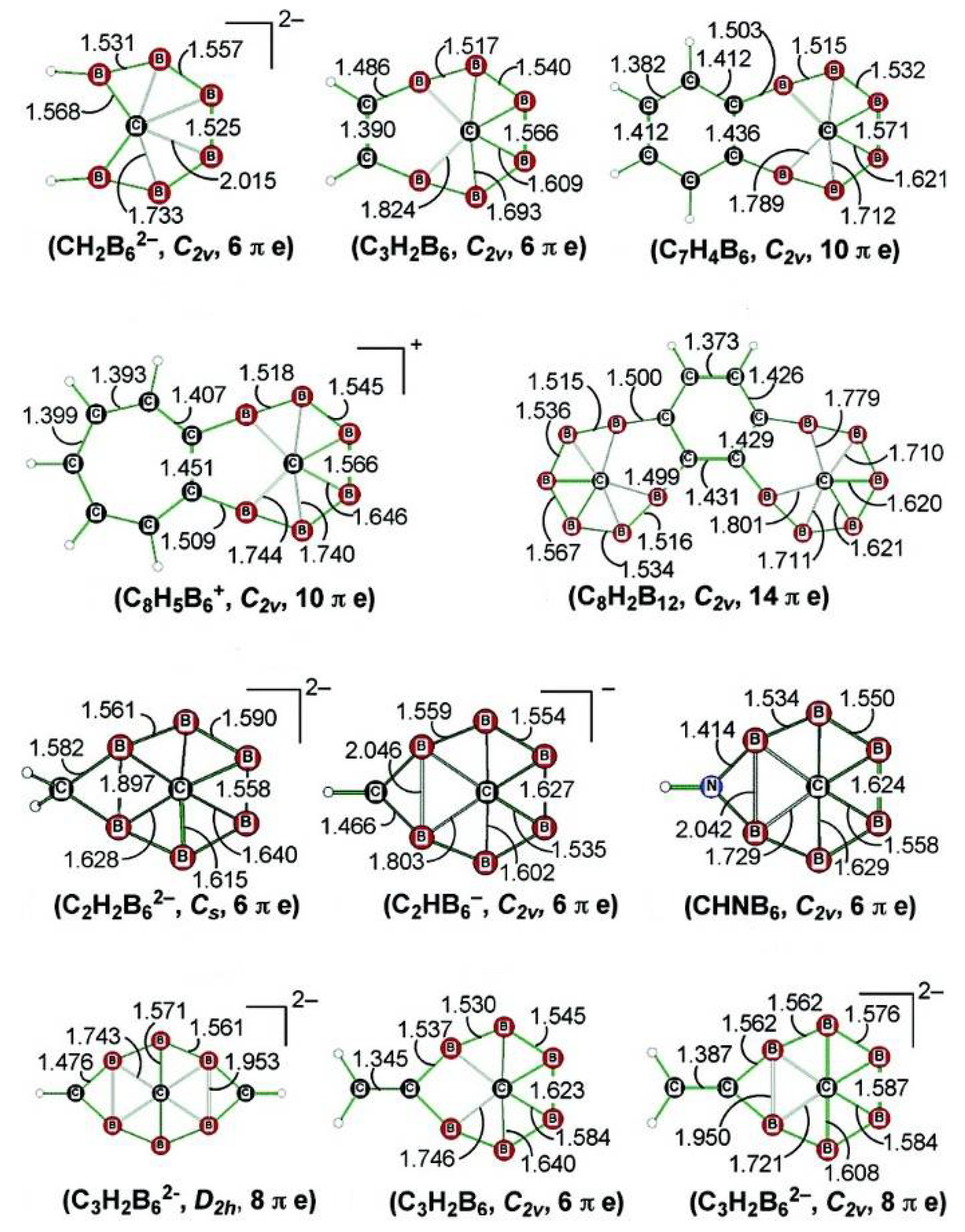
4. Planar Hexacoordinate Carbons (phCs)
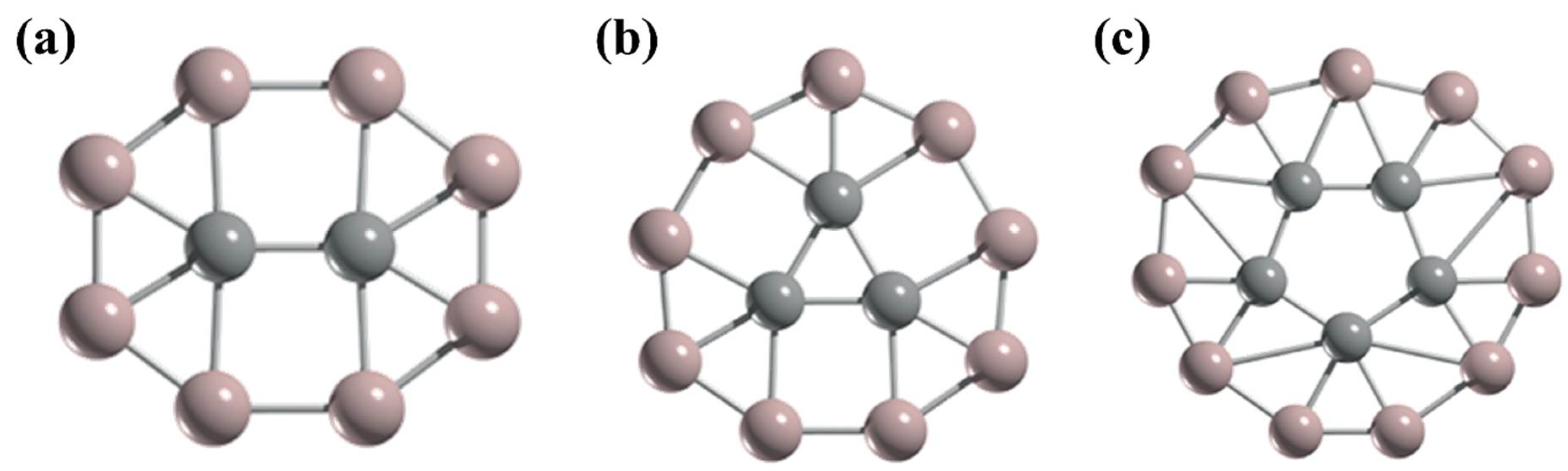
5. Higher Coordinate Carbons
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartlett, P.D. Nonclassical Ions; WA Benjamin: New York, NY, USA, 1965. [Google Scholar]
- Brown, H.C. The Nonclassical-Ion Problem; Plenum: New York, NY, USA, 1977. [Google Scholar]
- Le-Bel, J.A. On the Relations Which Exist between the Atomic Formulas of Organic Compounds and the Rotatory Power of Their Solutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Minkin, V.I.; Minyaev, R.M.; Hoffmann, R. Nonclassical structures of organic compounds: Non-standard stereochemistry and hypercoordination. Russ. Chem. Rev. 2002, 71, 869–892. [Google Scholar] [CrossRef] [Green Version]
- Minkin, V.I.; Minyaev, R.M.; Zdanov, J.A. Nonclassical Structures of Organic Compounds; Mir: Moscow, Russia, 1987. [Google Scholar]
- Das, P.; Pan, S.; Chattaraj, P.K. Planar Hypercoordinate Carbon. In Atomic Clusters with Unusual Structure, Bonding and Reactivity; Elsevier: Amsterdam, The Netherlands, 2023; pp. 357–372. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.; Wang, Z.X.; Schleyer, P.V.R. Four decades of the chemistry of planar hypercoordinate compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef] [PubMed]
- van’t Hoff, J.H. A Suggestion Looking to the Extension into Space of the Structural Formulas at Present Used in Chemistry, and a Note upon the Relation between the Optical Activity and the Chemical Constitution of Organic Compounds. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Monkhorst, H.J. Activation Energy for Interconversion of Enantiomers Containing an Asymmetric Carbon Atom without Breaking Bonds. Chem. Commun. 1968, 11, 1111–1112. [Google Scholar] [CrossRef]
- Wynberg, H.; Hekkert, G.L.; Houbiers, J.P.M.; Bosch, H.W. The Optical Activity of Butylethylhexylpropylmethane. J. Am. Chem. Soc. 1965, 87, 2635–2639. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Pepper, M.J.; Shavitt, I.; Schleyer, P.v.R.; Glukhovtsev, M.N.; Janoschek, R.; Quack, M. Is the stereomutation of methane possible? J. Comput. Chem. 1995, 16, 207–225. [Google Scholar] [CrossRef]
- Golden, D.M.; Walsh, R.; Benson, S.W. The Thermochemistry of the Gas Phase Equilibrium I2 + CH4 [UNK] CH3I + HI and the Heat of Formation of the Methyl Radical. J. Am. Chem. Soc. 1965, 87, 4053–4057. [Google Scholar] [CrossRef]
- Crans, D.C.; Snyder, J.P. Tetracoordinate planar carbon: A singlet biradical. J. Am. Chem. Soc. 1980, 102, 7152–7154. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Boldyrev, A.I. A new, general strategy for achieving planar tetracoordinate geometries for carbon and other second row periodic elements. J. Chem. Soc. Chem. Commun. 1991, 1536–1538. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in Pentaatomic Molecules. J. Am. Chem. Soc. 1998, 120, 7967–7972. [Google Scholar] [CrossRef]
- Gribanova, T.N.; Minyaev, R.M.; Minkin, V.I. Planar Tetracoordinate Carbon in Organoboron Compounds: Ab initio Computational Study. Collect. Czech. Chem. Commun. 1999, 64, 1780–1789. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in the Al4C− Anion. A Combined Photoelectron Spectroscopy and ab Initio Study. J. Am. Chem. Soc. 1999, 121, 6033–6038. [Google Scholar] [CrossRef]
- Wang, Z.X.; Manojkumar, T.K.; Wannere, C.; Schleyer, P.V.R. A Theoretical Prediction of Potentially Observable Lithium Compounds with Planar Tetracoordinate Carbons. Org. Lett. 2001, 3, 1249–1252. [Google Scholar] [CrossRef]
- Wang, Z.X.; Schleyer, P.v.R. A New Strategy To Achieve Perfectly Planar Carbon Tetracoordination. J. Am. Chem. Soc. 2001, 123, 994–995. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Vela, A. (C5M2-n)n- (M = Li, Na, K, and n = 0, 1, 2). A New Family of Molecules Containing Planar Tetracoordinate Carbons. J. Am. Chem. Soc. 2003, 125, 6026–6027. [Google Scholar] [CrossRef]
- Sahin, Y.; Prasang, C.; Hofmann, M.; Subramanian, G.; Geiseler, G.; Massa, W.; Berndt, A. A Diboracyclopropane with a Planar-Tetracoordinate Carbon Atom and a Triborabicyclobutane. Angew. Chem. Int. Ed. 2003, 42, 671–674. [Google Scholar] [CrossRef]
- Li, S.D.; Ren, G.M.; Miao, C.Q.; Jin, Z.H. M4H4X: Hydrometals (M=Cu, Ni) Containing Tetracoordinate Planar Nonmetals (X = B, C, N, O). Angew. Chem. Int. Ed. 2004, 43, 1371–1373. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Beltran, H.I.; Corminboeuf, C.; Heine, T.; Vela, A. Theoretical Analysis of the Smallest Carbon Cluster Containing a Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 2004, 126, 16160–16169. [Google Scholar] [CrossRef]
- Pancharatna, P.D.; Méndez-Rojas, M.A.; Merino, G.; Vela, A.; Hoffmann, R. Planar Tetracoordinate Carbon in Extended Systems. J. Am. Chem. Soc. 2004, 126, 15309–15315. [Google Scholar] [CrossRef] [PubMed]
- Priyakumar, U.D.; Reddy, A.S.; Sastry, G.N. The design of molecules containing planar tetracoordinate carbon. Tetrahedron Lett. 2004, 45, 2495–2498. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Vela, A.; Heine, T. Recent advances in planar tetracoordinate carbon chemistry. J. Comput. Chem. 2007, 28, 362–372. [Google Scholar] [CrossRef]
- Minkin, V.I.; Gribanova, T.N.; Minkin, V.I.; Starikov, A.G.; Hoffmann, R. Planar and Pyramidal Tetracoordinate Carbon in Organoboron Compounds. J. Org. Chem. 2005, 70, 6693–6704. [Google Scholar]
- Su, M.D. Theoretical Designs for Planar Tetracoordinated Carbon in Cu, Ag, and Au Organometallic Chemistry: A New Target for Synthesis. Inorg. Chem. 2005, 44, 4829–4833. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Ren, G.M.; Miao, C.Q. (M4H3X)2B2O2: Hydrometal Complexes (M = Ni, Mg) Containing Double Tetracoordinate Planar Nonmetal Centers (X = C, N). J. Phys. Chem. A 2005, 109, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Esteves, P.M.; Ferreira, N.B.P.; Corrêa, R.J. Neutral Structures with a Planar Tetracoordinated Carbon Based on Spiropentadiene Analogues. J. Am. Chem. Soc. 2005, 127, 8680–8685. [Google Scholar] [CrossRef]
- Erker, G. Stereochemistry and catalysis with zirconium complexes. Pure Appl. Chem. 1991, 63, 797–806. [Google Scholar] [CrossRef]
- Erker, G.; Albrecht, M.; Kruger, C.; Werner, S. Novel synthetic route to hydrocarbyl-bridged dinuclear zirconium/aluminum complexes exhibiting a planar tetracoordinate carbon center. Organometallics 1991, 10, 3791–3793. [Google Scholar] [CrossRef]
- Albrecht, M.; Erker, G.; Nolte, M.; Kruger, C. Planar tetracoordinate carbon stabilized in a dimetallic hafnium/aluminium compound: Formation and crystal structure of Cp2Hf[μ-η1: η2-MeCC(C6H11)][μ-CC(C6H11)]A1Me2. J. Organomet. Chem. 1992, 427, C21–C25. [Google Scholar] [CrossRef]
- Rottger, D.; Erker, G.; Frohlich, R.; Grehl, M.; Silverio, S.J.; Hylakryspin, I.; Gleiter, R. Determination of the Stabilization Energy of Planar-Tetracoordinate Carbon in Dynamic Dinuclear (μ-Hydrocarbyl)bis(zirconocene) Cation Complexes and Detection of an Organometallic Memory Effect in Their Formation. J. Am. Chem. Soc. 1995, 117, 10503–10512. [Google Scholar] [CrossRef]
- Rottger, D.; Erker, G.; Frohlich, R. Formation of stable organometallic planar-tetracoordinate carbon compounds containing a cationic (μ-R1CCR2) [μ-chloro(ZrCp2)2] framework. J. Organomet. Chem. 1996, 518, 221–225. [Google Scholar] [CrossRef]
- Rottger, D.; Erker, G.; Frohlich, R.; Kotila, S. Stabilization of a Planar-tetracoordinate Carbon Center in an Organometallic Complex Containing Both a Zirconocene and a Hafnocene Moiety. Chem. Ber. 1996, 129, 1–3. [Google Scholar] [CrossRef]
- Schottek, J.; Erker, G.; Frohlich, R. Formation of Metallocene-Stabilized Planar-Tetracoordinate Carbon Compounds by a Protonation Route. Eur. J. Inorg. Chem. 1998, 1998, 551–558. [Google Scholar] [CrossRef]
- Choukroun, R.; Donnadieu, B.; Zhao, J.S.; Cassoux, P.; Lepetit, C.; Silvi, B. Synthesis and Characterization of [Cp2V(μ-η2:η4-butadiyne)ZrCp’2] Heterodimetallic Complexes (Cp’ = C5H4t-Bu, C5H4Me). Formation Mechanism and Theoretical (ELF) Evidence for the Existence of Planar Tetracoordinate Carbon (ptC). Organometallics 2000, 19, 1901–1911. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.F.; Wang, L.S.; Geske, G.D.; Boldyrev, A.I. Pentaatomic Tetracoordinate Planar Carbon, [CAl4]2−: A New Structural Unit and Its Salt Complexes. Angew. Chem. Int. Ed. 2000, 39, 3630–3632. [Google Scholar] [CrossRef]
- Hoffmann, R. The theoretical design of novel stabilized systems. Pure Appl. Chem. 1971, 28, 181–194. [Google Scholar] [CrossRef]
- Sorger, K.; Schleyer, P.v.R. Planar and inherently non-tetrahedral tetracoordinate carbon: A status report. J. Mol. Struct. THEOCHEM 1995, 338, 317–346. [Google Scholar] [CrossRef]
- Röttger, D.; Erker, G. Compounds containing planar-tetracoordinate carbon. Angew. Chem. Int. Ed. 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Radom, L.; Rasmussen, D.R. The planar carbon story. Pure Appl. Chem. 1998, 70, 1977–1984. [Google Scholar] [CrossRef]
- Erker, G. Using bent metallocenes for stabilizing unusual coordination geometries at carbon. Chem. Soc. Rev. 1999, 28, 307–314. [Google Scholar] [CrossRef]
- Siebert, W.; Gunale, A. Compounds containing a planar-tetracoordinate carbon atom as analogues of planar methane. Chem. Soc. Rev. 1999, 28, 367–371. [Google Scholar] [CrossRef]
- Keese, R. Carbon flatland: planar tetracoordinate carbon and fenestrenes. Chem. Rev. 2006, 106, 4787–4808. [Google Scholar] [CrossRef]
- McGrath, M.P.; Radom, L. Alkaplanes: A class of neutral hydrocarbons containing a potentially planar tetracoordinate carbon. J. Am. Chem. Soc. 1993, 115, 3320–3321. [Google Scholar] [CrossRef]
- Lyons, J.E.; Rasmussen, D.R.; McGrath, M.P.; Nobes, R.H.; Radom, L. Octaplan: Ein gesättigter Kohlenwasserstoff mit ungewöhnlich niedriger Ionisierungsenergie und einem planar-tetrakoordinierten Kohlenstoffatom im Radikalkation. Angew. Chem. Int. Ed. Engl. 1994, 33, 1667–1668. [Google Scholar] [CrossRef]
- Ding, B.W.; Keese, R.; Stoeckli-Evans, H. First Synthesis and Structure of a Tetraazasilafenestrane. Angew. Chem. Int. Ed. 1999, 38, 375–376. [Google Scholar] [CrossRef]
- Rasmussen, D.R.; Radom, L. Planar tetrakoordinierter Kohlenstoff in einem neutralen gesättigten Kohlenwasserstoff: Theoretischer Entwurf und Charakterisierung. Angew. Chem. Int. Ed. 1999, 38, 2875–2878. [Google Scholar] [CrossRef]
- Wang, Z.X.; Schleyer, P.v.R. The Theoretical Design of Neutral Planar Tetracoordinate Carbon Molecules with C(C)4 Substructures. J. Am. Chem. Soc. 2002, 124, 11979–11982. [Google Scholar] [CrossRef]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; Schleyer, P.v.R.; Seeger, R.; Pople, J.A. Stabilization of Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Cotton, F.A.; Millar, M. The probable existence of a triple bond between two vanadium atoms. J. Am. Chem. Soc. 1977, 99, 7886–7891. [Google Scholar] [CrossRef]
- Xie, Y.; Schaefer, H.F. Hexalithiobenzene: A D6h equilibrium geometry with six lithium atoms in bridging positions. Chem. Phys. Lett. 1991, 179, 563–567. [Google Scholar] [CrossRef]
- Wang, L.S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Castro, A.C.; Audiffred, M.; Mercero, J.M.; Ugalde, J.M.; Méndez-Rojas, M.A.; Merino, G. Planar tetracoordinate carbon in CE42− (E = Al–Tl) clusters. Chem. Phys. Lett. 2012, 519–520, 29–33. [Google Scholar] [CrossRef]
- Cui, Z.H.; Ding, Y.H.; Cabellos, J.L.; Osorio, E.; Islas, R.; Restrepoe, A.; Merino, G. Planar tetracoordinate carbons with a double bond in CAl3E clusters. Phys. Chem. Chem. Phys. 2015, 17, 8769–8775. [Google Scholar] [CrossRef]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory based Electronic Structure Principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Cedillo, A.; Parr, R.G.; Arnett, E.M. Appraisal of Chemical Bond Making, Bond Breaking, and Electron Transfer in Solution in the Light of the Principle of Maximum Hardness. J. Org. Chem. 1995, 60, 4707–4714. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing Paradigms of Chemical Bonding: Adaptive Natural Density Partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Revealing Intuitively Assessable Chemical Bonding Patterns in Organic Aromatic Molecules via Adaptive Natural Density Partitioning. J. Org. Chem. 2008, 73, 9251–9258. [Google Scholar] [CrossRef]
- Millam, J.M.; Bakken, V.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab initio classical trajectories on the Born–Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Job, N.; Khatun, M.; Thirumoorthy, K.; CH, S.S.R.; Chandrasekaran, V.; Anoop, A.; Thimmakondu, V.S. CAl4Mg0/−: Global Minima with a Planar Tetracoordinate Carbon Atom. Atoms 2021, 9, 24. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, M.J.R.V.E. Nucleus- Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Patra, S.G.; Chattaraj, P.K. CB6Al0/+: Planar hexacoordinate boron (phB) in the global minimum structure. Phys. Chem. Chem. Phys. 2022, 24, 22634–22644. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Chattaraj, P.K. Electride characteristics of some binuclear sandwich complexes of alkaline earth metals, M2(η5-L)2 (M = Be, Mg; L = C5H5–, N5–, P5–, As5–). J. Phys. Chem. A 2020, 124, 9801–9810. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Chattaraj, P.K. Substituent effects on electride characteristics of Mg2(η5-C5H5)2: A theoretical study. J. Phys. Chem. A 2021, 125, 6207–6220. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, H.B.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency, energy conservation and choice of initial conditions. J. Chem. Phys. 2001, 115, 10291–10302. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Iyengar, S.S.; Li, X.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer dynamics. J. Chem. Phys. 2002, 117, 8694–8704. [Google Scholar] [CrossRef] [Green Version]
- Das, P.; Saha, R.; Chattaraj, P.K. Encapsulation of Mg2 inside a C60 cage forms an electride. J. Comput. Chem. 2020, 41, 1645–1653. [Google Scholar] [CrossRef]
- Das, P.; Chattaraj, P.K. Comparison Between Electride Characteristics of Li3@B40 and Li3@C60. Front. Chem. 2021, 9, 638581. [Google Scholar] [CrossRef] [PubMed]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Saha, R.; Das, P.; Chattaraj, P.K. Molecular Electrides: An In Silico Perspective. ChemPhysChem 2022, 23, e202200329. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Chattaraj, P.K. In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon. Atoms 2021, 9, 65. [Google Scholar] [CrossRef]
- Das, P.; Chattaraj, P.K. CSiGaAl2−/0 and CGeGaAl2−/0 having planar tetracoordinate carbon atoms in their global minimum energy structures. J. Comput. Chem. 2022, 43, 894–905. [Google Scholar] [CrossRef]
- Das, P.; Khatun, M.; Anoop, A.; Chattaraj, P.K. CSinGe4−n2+ (n = 1–3): Prospective systems containing planar tetracoordinate carbon (ptC). Phys. Chem. Chem. Phys. 2022, 24, 16701–16711. [Google Scholar] [CrossRef]
- Zhang, J.; Dolg, M. ABCluster: The artificial bee colony algorithm for cluster global optimization. Phys. Chem. Chem. Phys. 2015, 17, 24173–24181. [Google Scholar] [CrossRef]
- Zhang, J.; Dolg, M. Global optimization of clusters of rigid molecules using the artificial bee colony algorithm. Phys. Chem. Chem. Phys. 2016, 18, 3003–3010. [Google Scholar] [CrossRef]
- Karaboga, D. An Idea Based on Honey Bee Swarm for Numerical Optimization, Technical Report-TR06; Erciyes University, Engineering Faculty, Computer Engineering Department: Kayseri, Türkiye, 2005. [Google Scholar]
- Sarkar, K.; Bhattacharyya, S.P. Soft Computing in Chemical and Physical Sciences: A Shift in Computing Paradigm, 1st ed.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar] [CrossRef]
- Cui, Z.H.; Contreras, M.; Ding, Y.H.; Merino, G. Planar Tetracoordinate Carbon versus Planar Tetracoordinate Boron: The Case of CB4 and Its Cation. J. Am. Chem. Soc. 2011, 133, 13228–13231. [Google Scholar] [CrossRef]
- Becker, S.; Dietze, H.J. Cluster ions in the laser mass spectra of boron carbide. Int. J. Mass Spectrom. Ion Process. 1988, 82, 287–298. [Google Scholar] [CrossRef]
- Vogt-Geisse, S.; Wu, J.I.C.; Schleyer, P.v.R.; Schaefer, H.F. Bonding, aromaticity, and planar tetracoordinated carbon in Si2CH2 and Ge2CH2. J. Mol. Model 2015, 21, 217. [Google Scholar] [CrossRef] [PubMed]
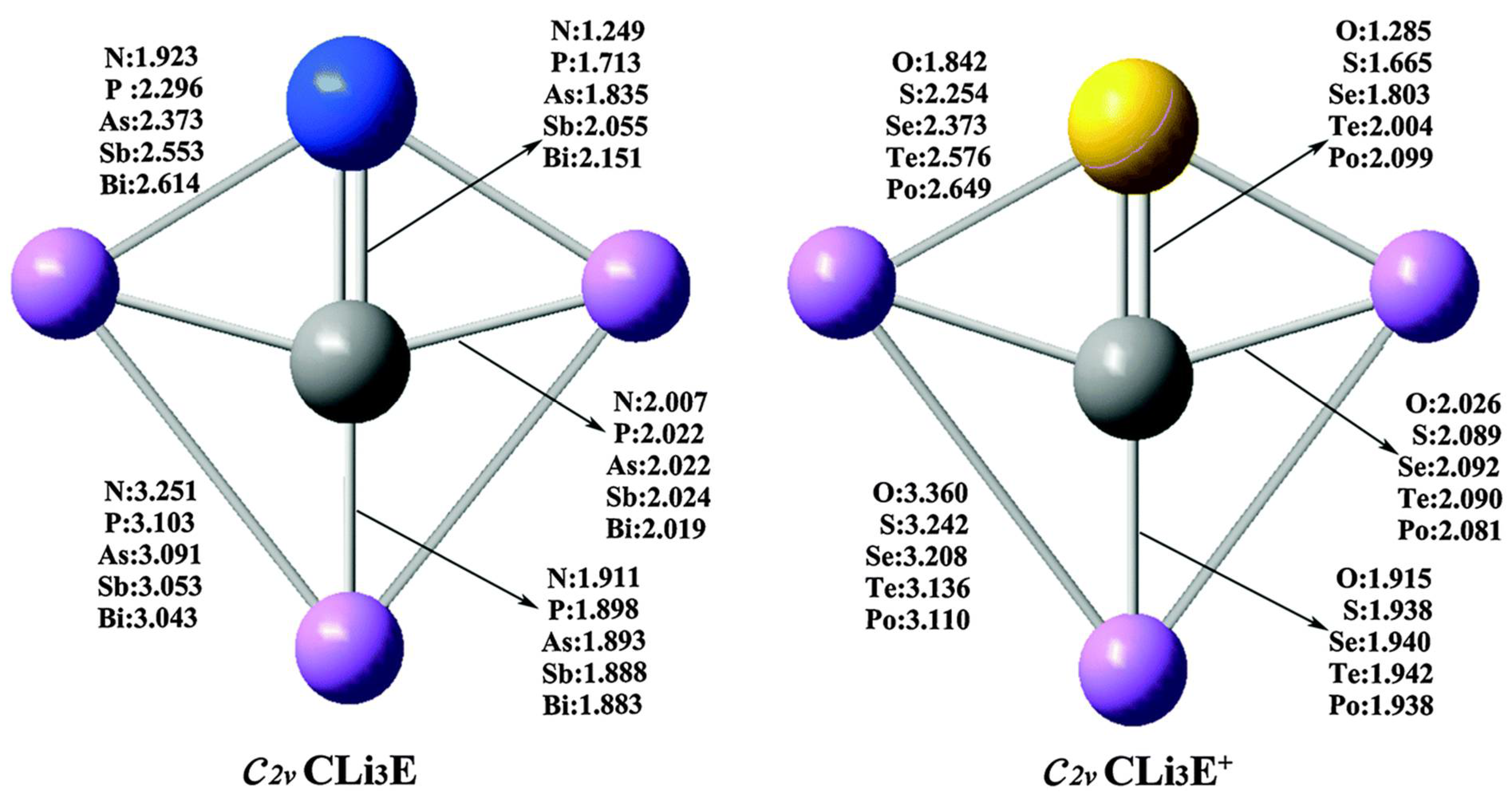
- Guo, J.Y.; Chai, H.Y.; Duan, Q.; Qin, J.M.; Shen, X.D.; Jiang, D.Y.; Hou, J.H.; Yan, B.; Li, Z.R.; Gu, F.L.; et al. Planar tetracoordinate carbon species CLi3E with 12-valence-electrons. Phys. Chem. Chem. Phys. 2016, 18, 4589–4593. [Google Scholar] [CrossRef]
- Das, P.; Chattaraj, P.K. Electride characteristics of M2(η5-E5)2 (M = Be, Mg; E = Sb5-). Struct. Chem. 2021, 32, 2107–2114. [Google Scholar] [CrossRef]
- Das, P.; Chattaraj, P.K. Stabilisation of Li(0)-Li(0) bond by normal and mesoionic carbenes and electride characteristics of the complexes. Mol. Phys. 2022, 120, e2026512. [Google Scholar] [CrossRef]
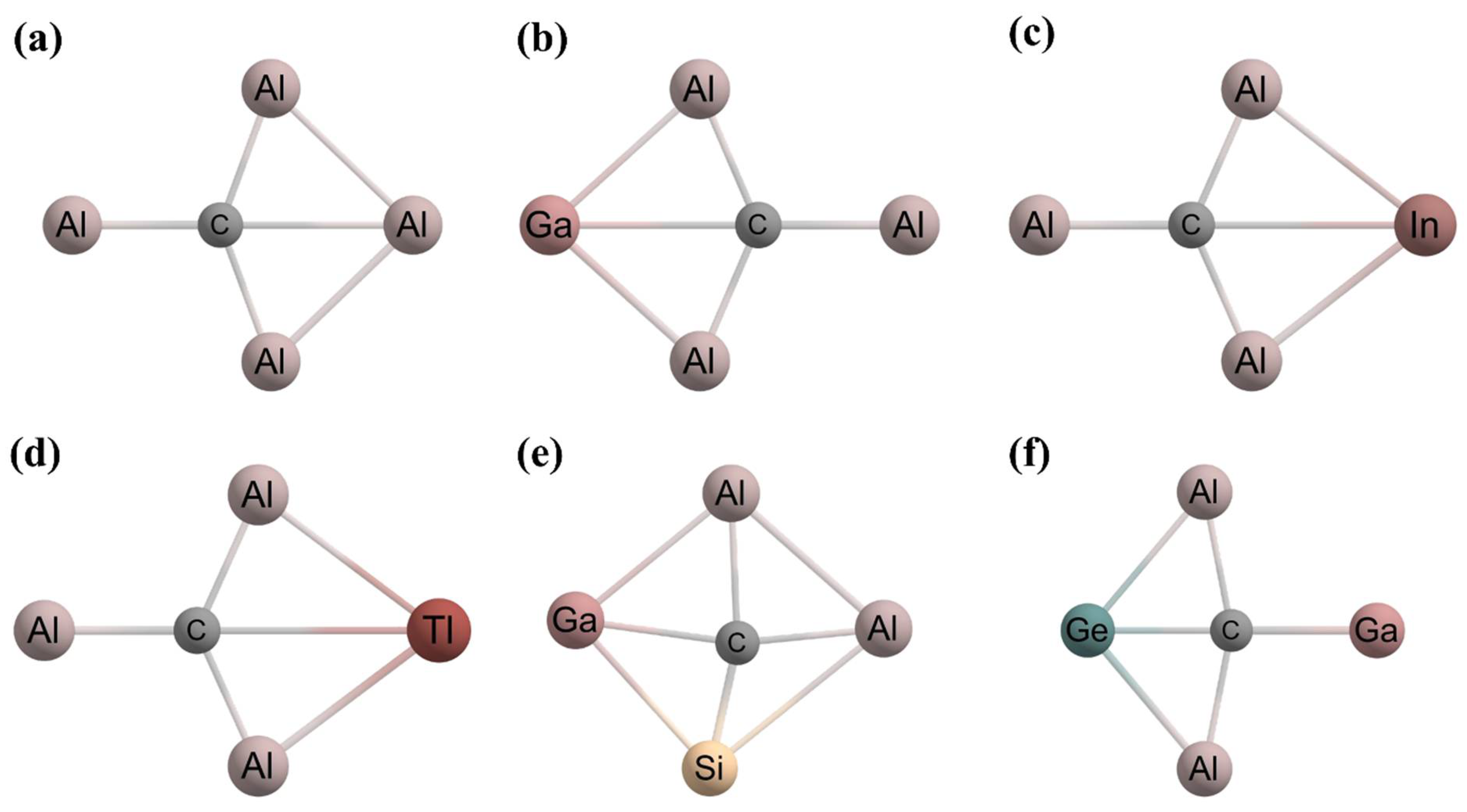
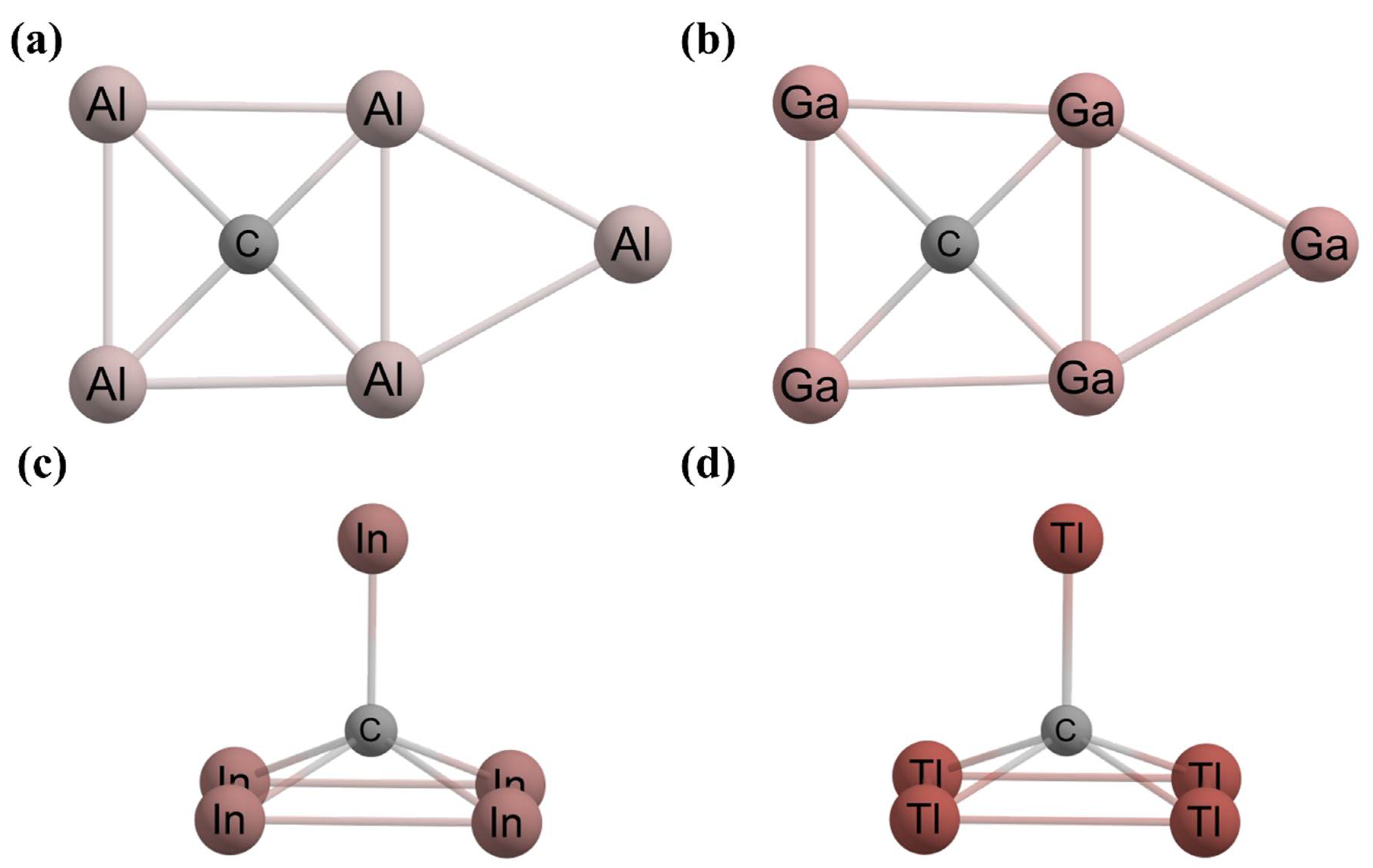
- Zheng, H.F.; Yu, S.; Hu, T.D.; Xu, J.; Ding, Y.H. CAl3X (X = B/Al/Ga/In/Tl) with 16 Valence Electrons: Can Planar Tetracoordinate Carbon Be Stable? Phys. Chem. Chem. Phys. 2018, 20, 26266–26272. [Google Scholar] [CrossRef]
- Ravell, E.; Jalife, S.; Barroso, J.; Orozco-Ic, M.; Hernandez-Juarez, G.; Ortiz-Chi, F.; Pan, S.; Cabellos, J.L.; Merino, G. Structure and Bonding in CE5− (E = Al − Tl) Clusters: Planar Tetracoordinate Carbon vs Pentacoordinate Carbon. Chem.-Asian J. 2018, 13, 1467–1473. [Google Scholar] [CrossRef]
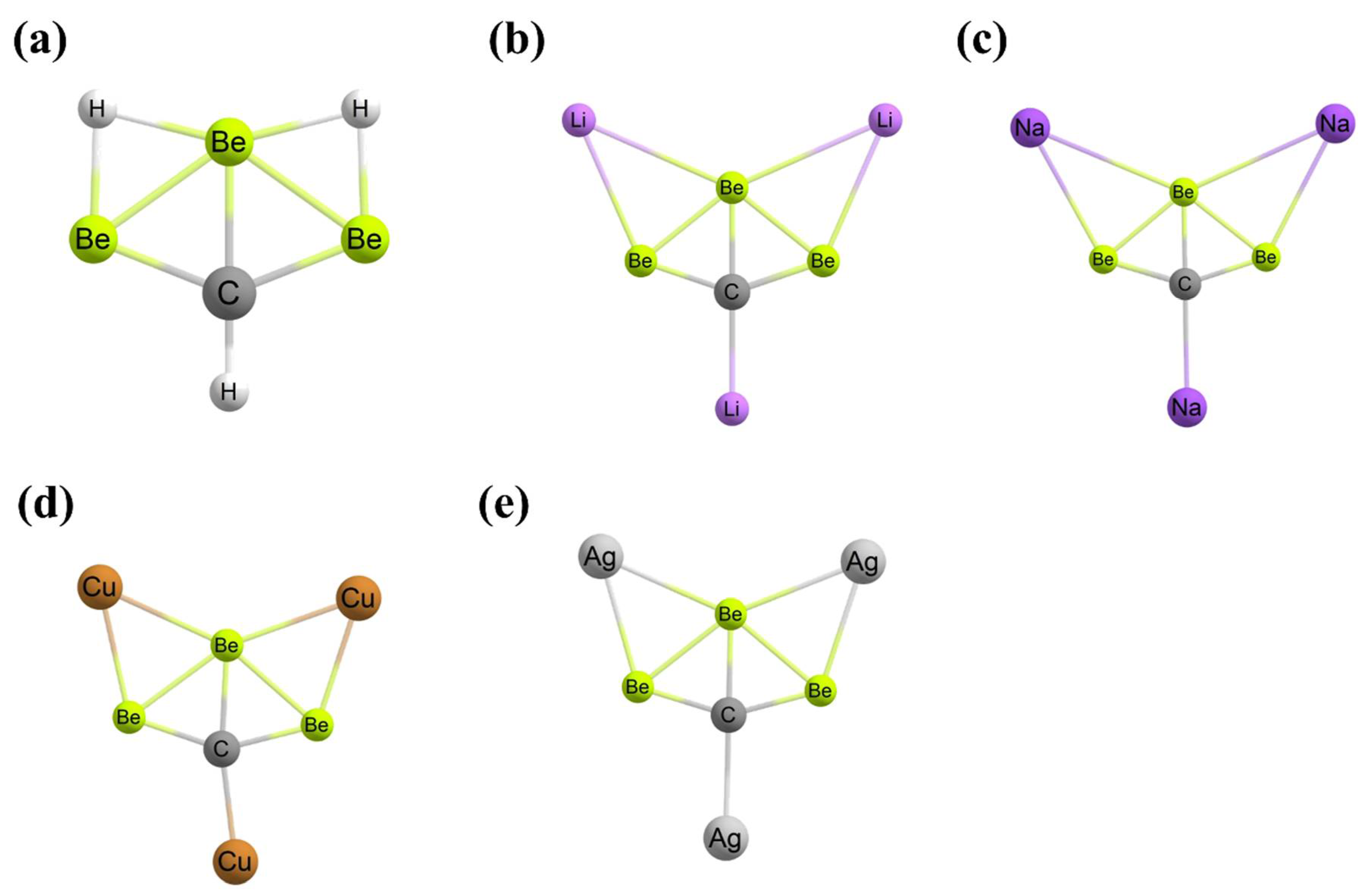
- Guo, J.C.; Feng, L.Y.; Dong, C.; Zhai, H.J. Ternary 12-electron CBe3X3+ (X = H, Li, Na, Cu, Ag) Clusters: Planar Tetracoordinate Carbons and Superalkali Cations. Phys. Chem. Chem. Phys. 2019, 21, 22048–22056. [Google Scholar] [CrossRef]
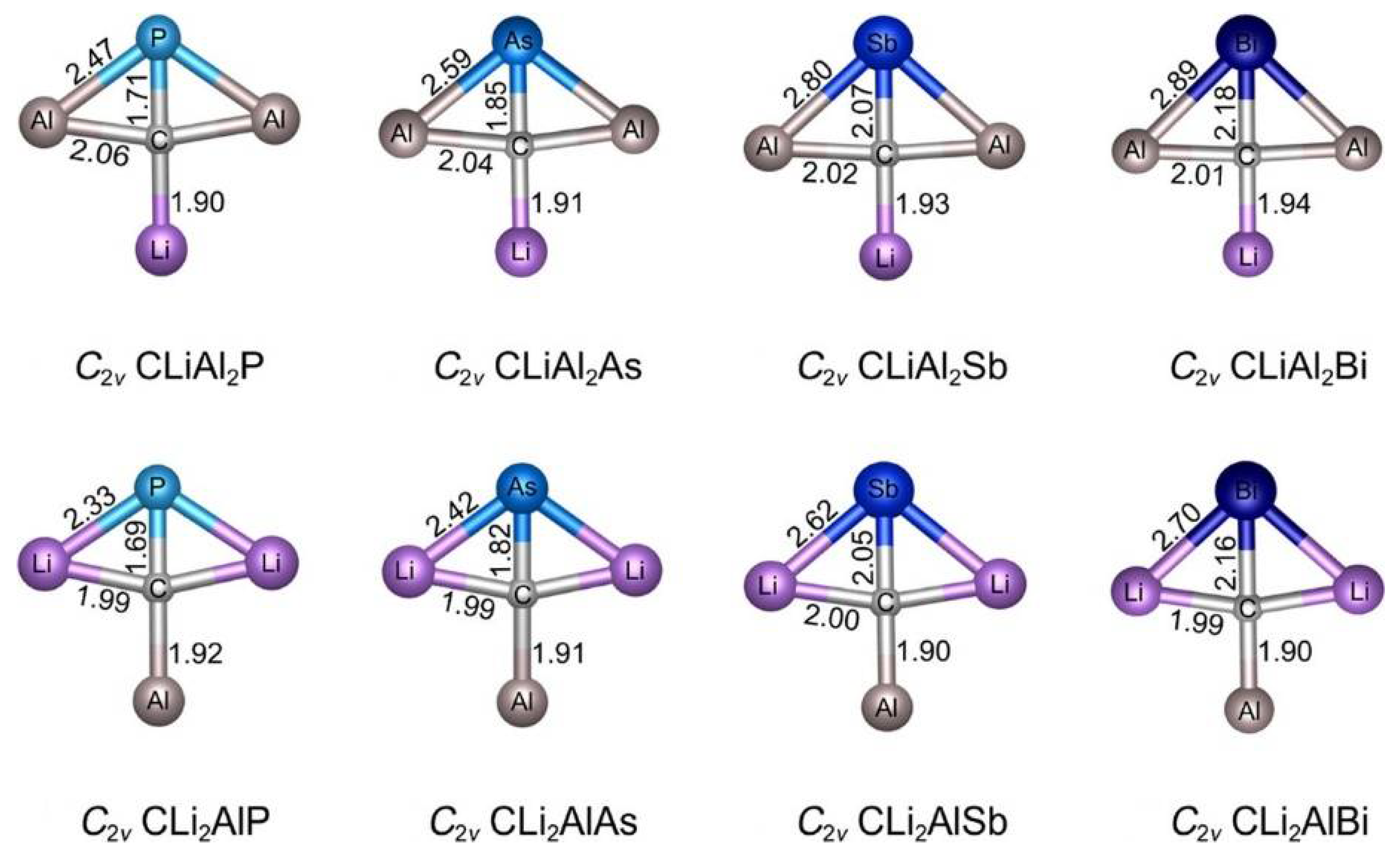
- Wu, X.F.; Cheng, Y.X.; Guo, J.C. CLiAl2E and CLi2AlE (E = P, As, Sb, Bi): Planar Tetracoordinate Carbon Clusters with 16 and 14 Valence Electrons. ACS Omega 2019, 4, 21311–21318. [Google Scholar] [CrossRef] [Green Version]
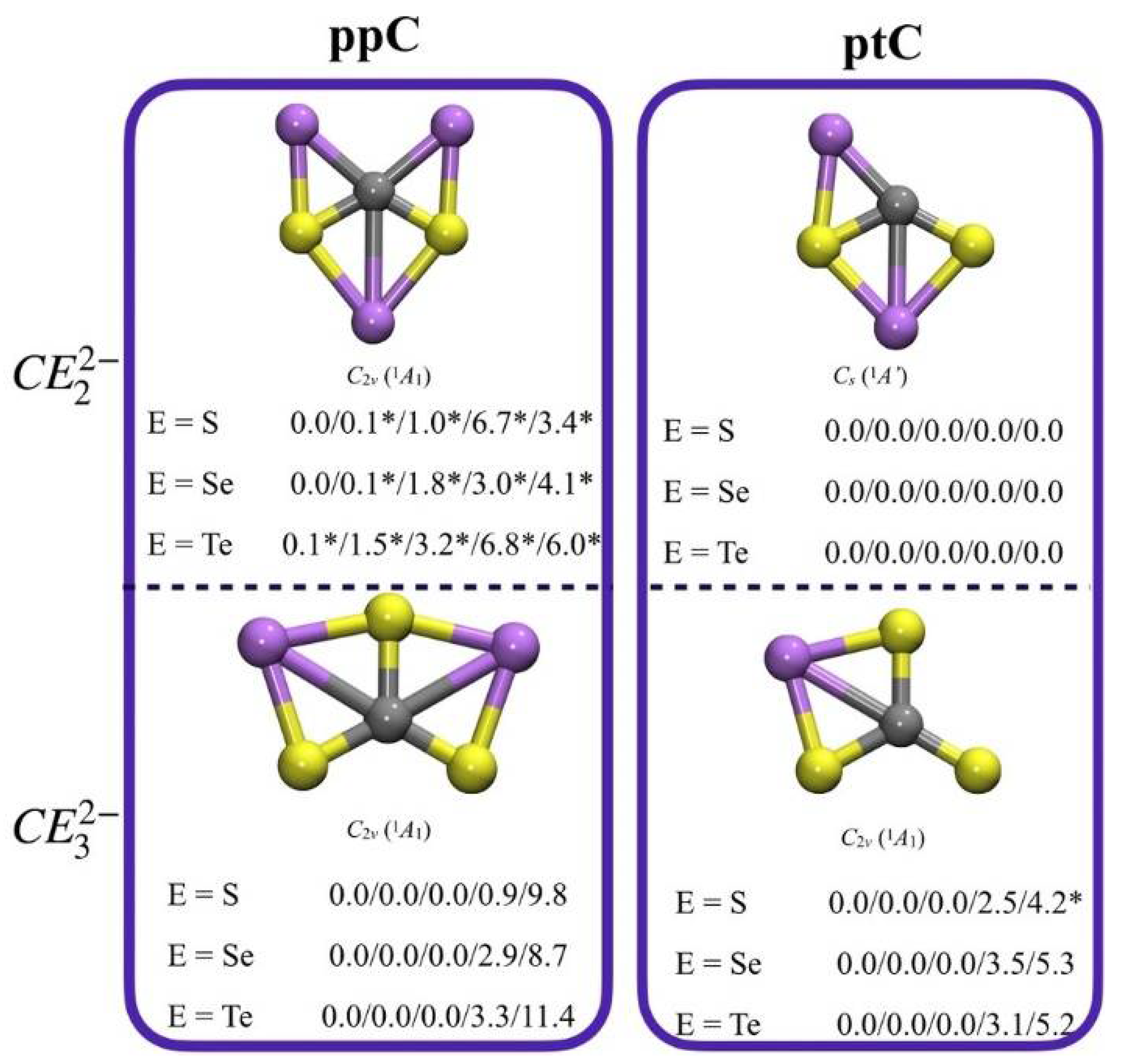
- Leyva-Parra, L.; Diego, L.; Inostroza, D.; Yañez, O.; Pumachagua-Huertas, R.; Barroso, J.; Vásquez-Espinal, A.; Merino, G.; Tiznado, W. Planar Hypercoordinate Carbons in Alkali Metal Decorated CE32− and CE22− Dianions. Chem. Eur. J. 2021, 27, 16701–16706. [Google Scholar] [CrossRef]
- Yañez, O.; Vásquez-Espinal, A.; Pino-Rios, R.; Ferraro, F.; Pan, S.; Osorio, E.; Merino, G.; Tiznado, W. Exploiting electronic strategies to stabilize a planar tetracoordinate carbon in cyclic aromatic hydrocarbons. Chem. Commun. 2017, 53, 12112–12115. [Google Scholar] [CrossRef]
- Wang, M.; Orozco-Ic, M.; Leyva-Parra, L.; Tiznado, W.; Barroso, J.; Ding, Y.; Cui, Z.H.; Merino, G. Planar Tetracoordinate Carbons in Allene-Type Structures. J. Phys. Chem. A 2021, 125, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
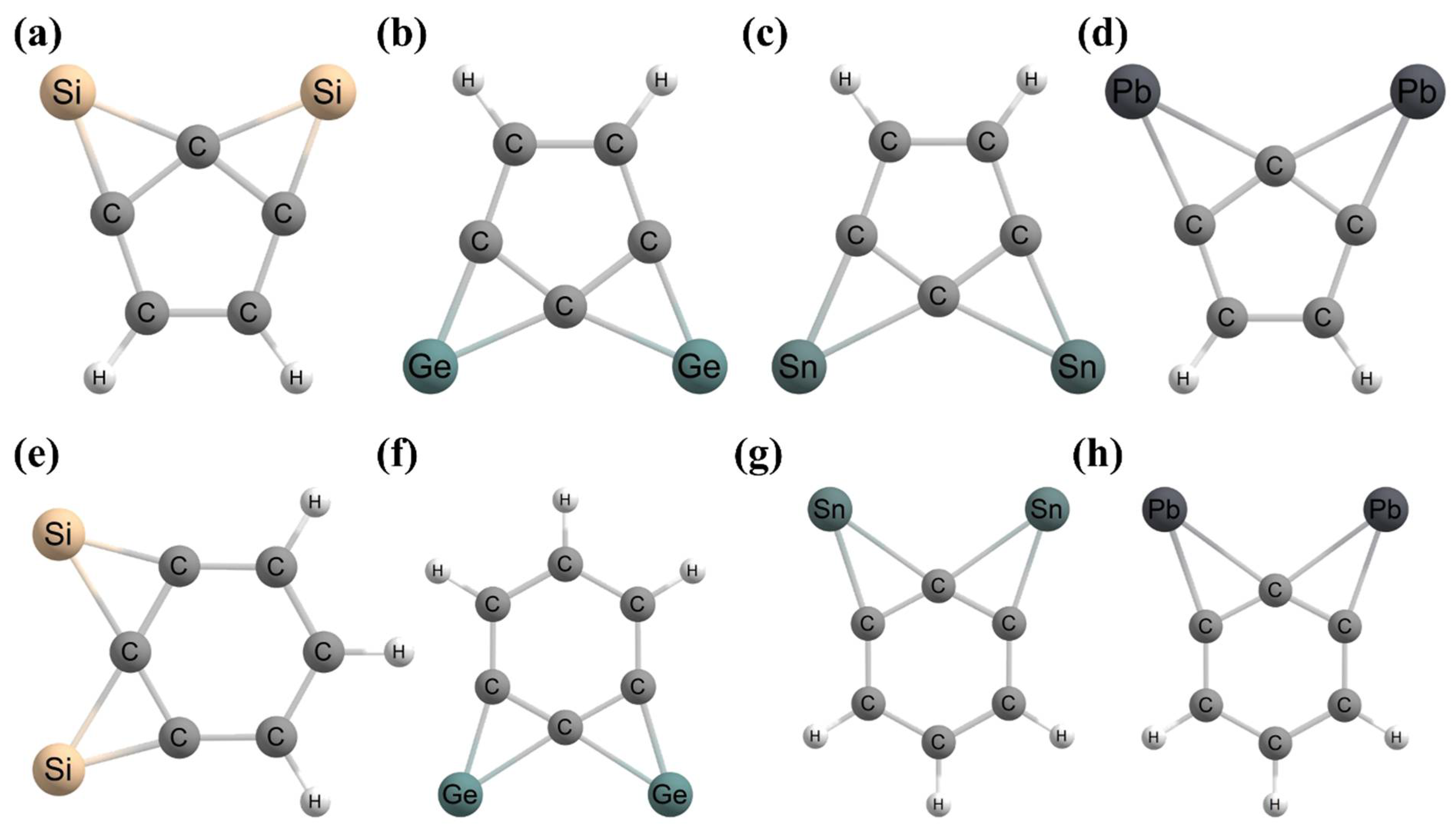
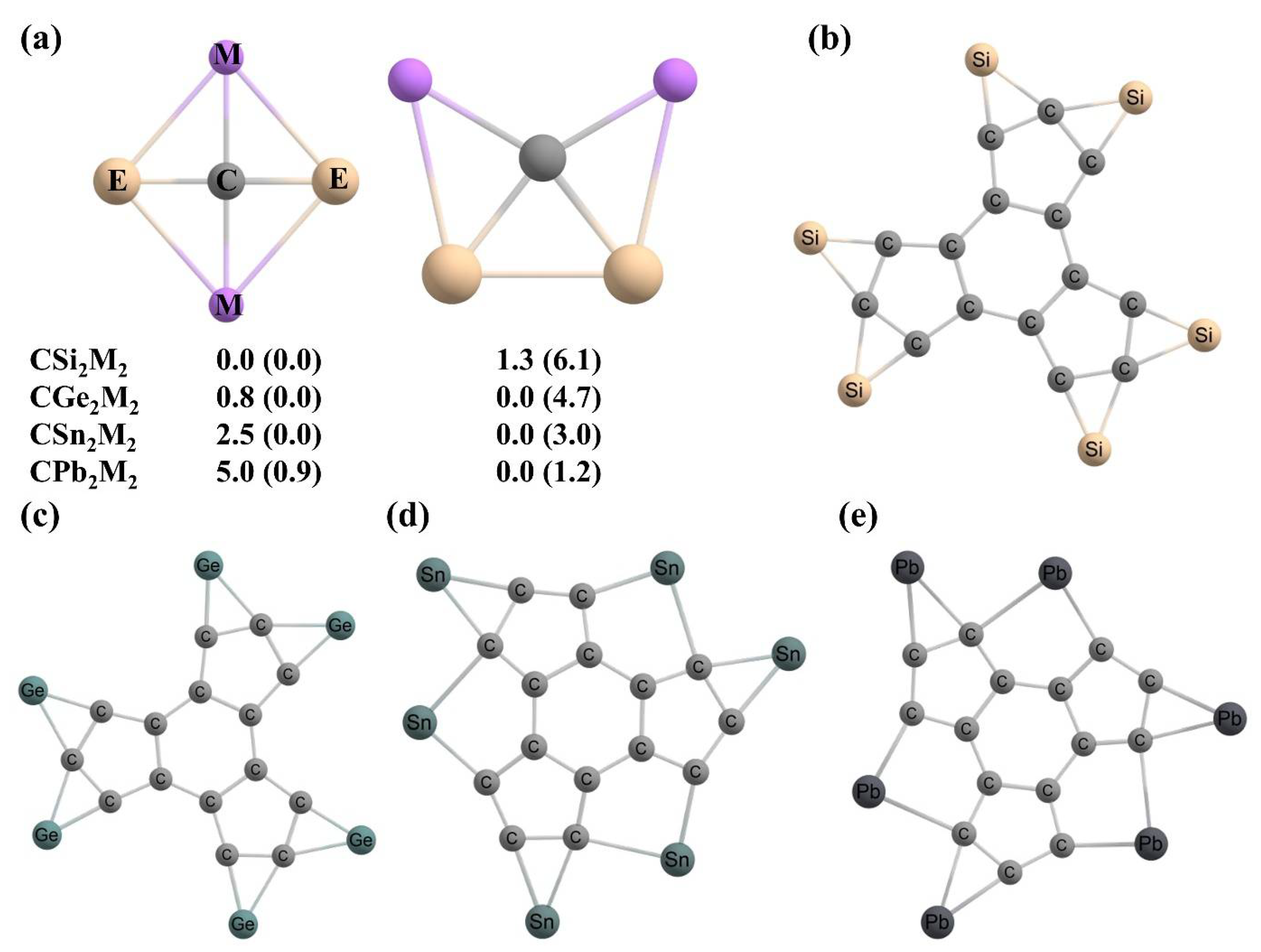
- Inostroza, D.; Leyva-Parra, L.; Vásquez-Espinal, A.; Contreras-García, J.; Cui, Z.H.; Pan, S.; Thimmakondu, V.S.; Tiznado, W. E6C15 (E = Si–Pb): Polycyclic aromatic compounds with three planar tetracoordinate carbons. Chem. Commun. 2022, 58, 13075–13078. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Yu, S.; Ding, Y.H.; Bowen, K.H. Identifying the Hydrogenated Planar Tetracoordinate Carbon: A Combined Experimental and Theoretical Study of CAl4H and CAl4H–. J. Phys. Chem. Lett. 2017, 8, 2263–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev-Galindo, V.; Pan, S.; Donald, K.J.; Merino, G. Planar pentacoordinate carbons. Nat. Rev. Chem. 2018, 2, 0114. [Google Scholar] [CrossRef]
- Bolton, E.E.; Laidig, W.D.; Schleyer, P.v.R.; Schaefer, H.F. Does singlet 1,1-dilithioethene really prefer a perpendicular structure? J. Phys. Chem. 1995, 99, 17551–17557. [Google Scholar] [CrossRef]
- Wang, Z.X.; Schleyer, P.v.R. Construction principles of “hyparenes”: Families of molecules with planar pentacoordinate carbons. Science 2001, 292, 2465–2469. [Google Scholar] [CrossRef]
- Tsipis, C.A.; Karagiannis, E.E.; Kladou, P.F.; Tsipis, A.C. Aromatic Gold and Silver ‘Rings’: Hydrosilver(I) and Hydrogold(I) Analogues of Aromatic Hydrocarbons. J. Am. Chem. Soc. 2004, 126, 12916–12929. [Google Scholar] [CrossRef]
- Tsipis, A.C.; Tsipis, C.A. Hydrometal Analogues of Aromatic Hydrocarbons: A New Class of Cyclic Hydrocoppers(I). J. Am. Chem. Soc. 2003, 125, 1136–1137. [Google Scholar] [CrossRef]
- Li, S.D.; Miao, C.Q.; Ren, G.M. D5h Cu5H5X: Pentagonal hydrocopper Cu5H5 containing pentacoordinate planar nonmetal centers (X = B, C, N, O). Eur. J. Inorg. Chem. 2004, 2004, 2232–2234. [Google Scholar] [CrossRef]
- Li, S.D.; Guo, Q.L.; Miao, C.Q.; Ren, G.M. Investigation on transition-metal hydrometal complexes MnHnC with planar coordinate carbon centers by density functional theory. Acta Phys. Chim. Sin. 2007, 23, 743–745. [Google Scholar]
- Pei, Y.; An, W.; Ito, K.; Schleyer, P.v.R.; Zeng, X.C. Planar pentacoordinate carbon in CAl5+: A global minimum. J. Am. Chem. Soc. 2008, 130, 10394–10400. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Halla, J.O.C.; Wu, Y.B.; Wang, Z.X.; Islas, R.; Heine, T.; Merino, G. CAl4Be and CAl3Be2−: Global Minima with a Planar Pentacoordinate Carbon Atom. Chem. Commun. 2010, 46, 8776–8778. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.B.; Duan, Y.; Lu, H.G.; Li, S.D. CAl2Be32– and its salt complex LiCAl2Be3–: Anionic global minimawith planar pentacoordinate carbon. J. Phys. Chem. A 2012, 116, 3290–3294. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.C.; Martínez-Guajardo, G.; Johnson, T.; Ugalde, J.M.; Wu, Y.B.; Mercero, J.M.; Heine, T.; Donald, K.J.; Merino, G. CBe5E– (E = Al, Ga, In, Tl): Planar pentacoordinate carbon in heptaatomic clusters. Phys. Chem. Chem. Phys. 2012, 14, 14764–14768. [Google Scholar] [CrossRef]
- Luo, Q. Theoretical observation of hexaatomic molecules containing pentacoordinate planar carbon. Sci. China, Ser. B Chem. 2008, 51, 1030–1035. [Google Scholar] [CrossRef]
- Grande-Aztatzi, R.; Cabellos, J.L.; Islas, R.; Infante, I.; Mercero, J.M.; Restrepo, A.; Merino, G. Planar pentacoordinate carbons in CBe54– derivatives. Phys. Chem. Chem. Phys. 2015, 17, 4620–4624. [Google Scholar] [CrossRef]
- Guo, J.C.; Ren, G.M.; Miao, C.Q.; Tian, W.J.; Wu, Y.B.; Wang, X. CBe5Hnn−4 (n = 2–5): Hydrogen-stabilized CBe5 pentagons containing planar or quasi-planar pentacoordinate carbons. J. Phys. Chem. A 2015, 119, 13101–13106. [Google Scholar] [CrossRef]
- Guo, J.C.; Tian, W.J.; Wang, Y.J.; Zhao, X.F.; Wu, Y.B.; Zhai, H.J.; Li, S.D. Star-like superalkali cations featuring planar pentacoordinate carbon. J. Chem. Phys. 2016, 144, 244303. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.F.; Bian, J.H.; Huang, F.; Yuan, C.; Wang, Q.; Liu, P.; Li, D.; Wang, X.; Wu, Y.B. Stabilization of beryllium-containing planar pentacoordinate carbon species through attaching hydrogen atoms. RSC Adv. 2018, 8, 36521–36526. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Cabellos, J.L.; Orozco-Ic, M.; Chattaraj, P.K.; Zhao, L.; Merino, G. Planar pentacoordinate carbon in CGa5+ derivatives. Phys. Chem. Chem. Phys. 2018, 20, 12350–12355. [Google Scholar] [CrossRef]
- Zdetsis, A.D. Novel pentagonal silicon rings and nanowheels stabilized by flat pentacoordinate carbon(s). J. Chem. Phys. 2011, 134, 094312. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.H.; Vassilev-Galindo, V.; Cabellos, J.L.; Osorio, E.; Orozco, M.; Pan, S.; Ding, Y.H.; Merino, G. Planar pentacoordinate carbon atoms embedded in a metallocene framework. Chem. Commun. 2017, 53, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Jin, B.; Huo, B.; Yuan, C.; Zhai, H.J.; Wu, Y.B. Planar pentacoordinate carbon in a sulphur-surrounded boron wheel: The global minimum of CB5S5+. Chem. Commun. 2022, 58, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Frenking, G.; Chen, Z.F.; Schleyer, P.v.R. Aromatic boron wheels with more than one carbon atom in the center: C2B8, C3B93+, and C5B11+. Angew. Chem. Int. Ed. 2005, 44, 1078–1082. [Google Scholar] [CrossRef]
- Lammertsma, K.; Barzaghi, M.; Olah, G.A.; Pople, J.A.; Schleyer, P.v.R.; Simonetta, M. Carbodications. 7. Structure and stability of diprotonated methane, CH62+. J. Am. Chem. Soc. 1983, 105, 5258–5263. [Google Scholar] [CrossRef]
- Sirigu, A.; Bianchi, M.; Benedetti, E. The crystal structure of Ru6C(CO)17. J. Chem. Soc. D 1969, 596a. [Google Scholar] [CrossRef]
- Hogeveen, H.; Kwant, P.W. Pyramidal mono- and dications. Bridge between organic and organometallic chemistry. Acc. Chem. Res. 1975, 8, 413–420. [Google Scholar] [CrossRef]
- Exner, K.; Schleyer, P.v.R. Planar Hexacoordinate Carbon: A Viable Possibility. Science 2000, 290, 1937–1940. [Google Scholar] [CrossRef]
- Ito, K.; Chen, Z.; Corminboeuf, C.; Wannere, C.S.; Zhang, X.H.; Li, Q.S.; Schleyer, P.v.R. Myriad planar hexacoordinate carbon molecules inviting synthesis. J. Am. Chem. Soc. 2007, 129, 1510–1511. [Google Scholar] [CrossRef]
- Wu, Y.B.; Duan, Y.; Lu, G.; Lu, H.G.; Yang, P.; Schleyer, P.v.R.; Merino, G.; Islas, R.; Wang, Z.X. D3h CN3Be3+ and CO3Li3+: Viable planar hexacoordinate carbon prototypes. Phys. Chem. Chem. Phys. 2012, 14, 14760–14763. [Google Scholar] [CrossRef]
- Leyva-Parra, L.; Diego, L.; Yañez, O.; Inostroza, D.; Barroso, J.; Vásquez-Espinal, A.; Merino, G.; Tiznado, W. Planar hexacoordinate carbons: Half covalent, half ionic. Angew. Chem. Int. Ed. 2021, 60, 8700–8704. [Google Scholar] [CrossRef] [PubMed]
- Minyaev, R.M.; Gribanova, T.N.; Starikov, A.G.; Minkin, V.I. Heptacoordinated carbon and nitrogen in a planar boron ring. Dokl. Chem. 2002, 382, 41–45. [Google Scholar] [CrossRef]
- Zhai, H.J.; Alexandrova, A.N.; Birch, K.A.; Boldyrev, A.I.; Wang, L.S. Hepta- and Octacoordinate Boron in Molecular Wheels of Eight- and Nine-Atom Boron Clusters: Observation and Confirmation. Angew. Chem. Int. Ed. 2003, 42, 6004–6008. [Google Scholar] [CrossRef] [PubMed]






























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, P.; Chattaraj, P.K. Structure and Bonding in Planar Hypercoordinate Carbon Compounds. Chemistry 2022, 4, 1723-1756. https://doi.org/10.3390/chemistry4040113
Das P, Chattaraj PK. Structure and Bonding in Planar Hypercoordinate Carbon Compounds. Chemistry. 2022; 4(4):1723-1756. https://doi.org/10.3390/chemistry4040113
Chicago/Turabian StyleDas, Prasenjit, and Pratim Kumar Chattaraj. 2022. "Structure and Bonding in Planar Hypercoordinate Carbon Compounds" Chemistry 4, no. 4: 1723-1756. https://doi.org/10.3390/chemistry4040113