
A Greener Technique for Microwave-Assisted O-Silylation and Silyl Ether Deprotection of Uridine and Other Substrates


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Instruments
2.2. General Procedure for the Synthesis of Silyl Ethers
2.3. General Procedure for the Synthesis of Alcohols via Deprotection of Silyl Ethers
3. Results and Discussion
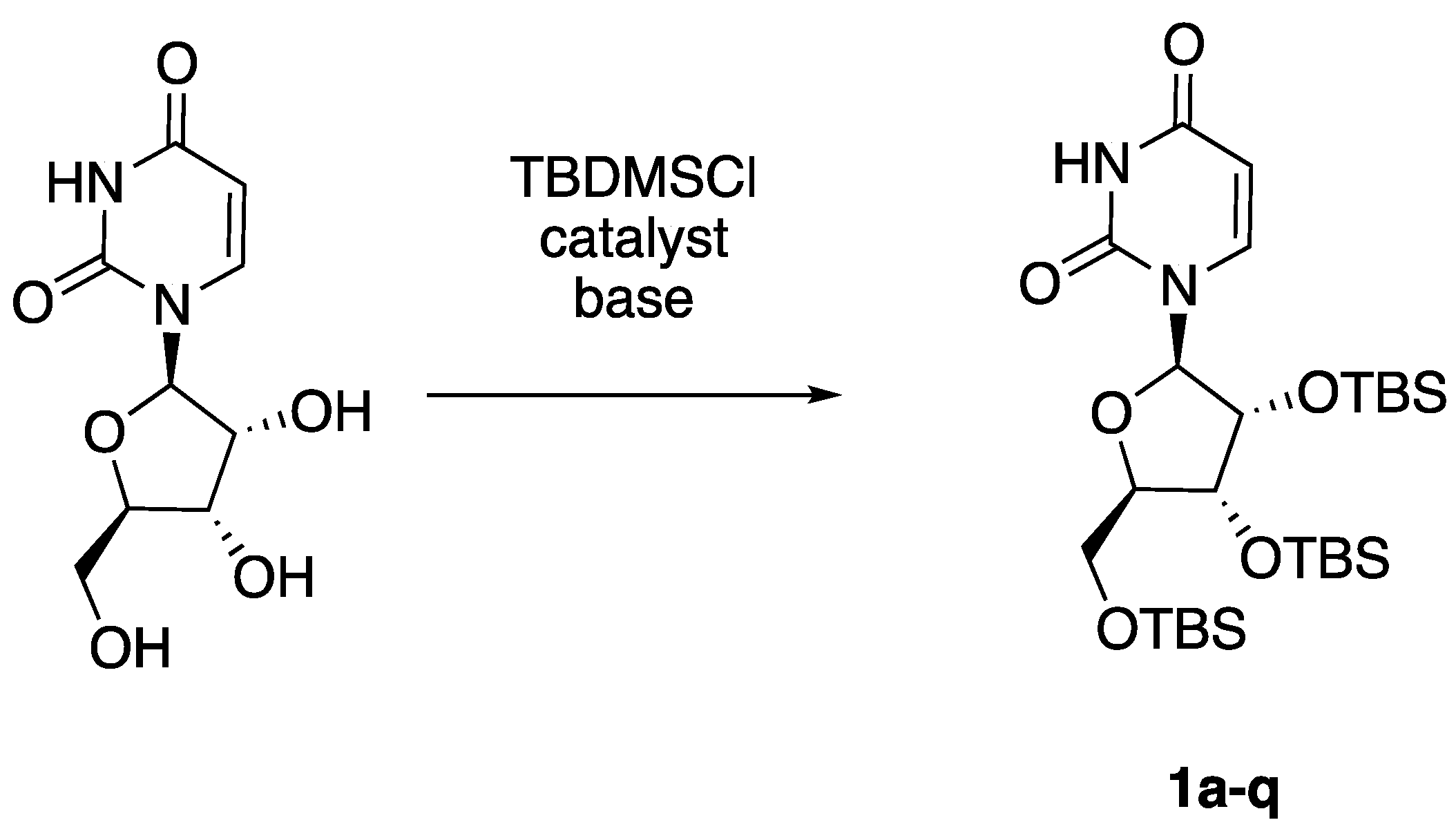
3.1. O-Silylation of Uridine
3.2. Scope Variation
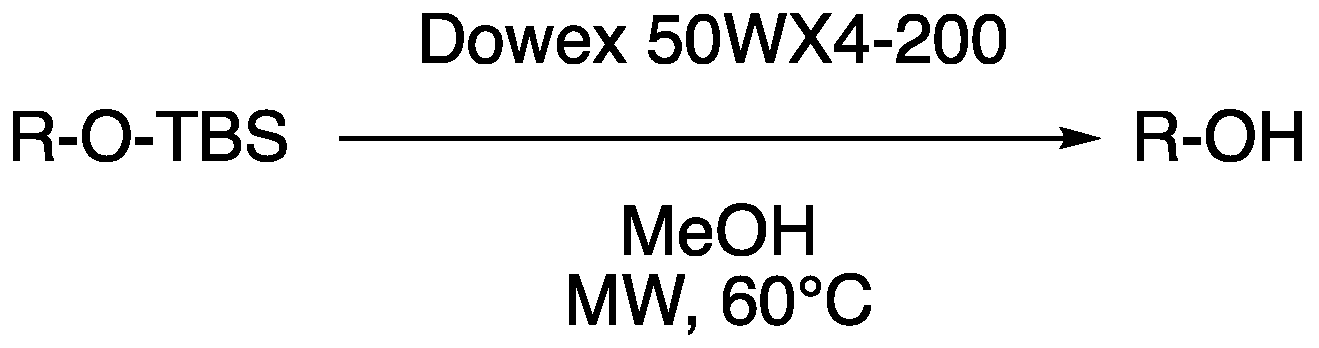
3.3. Microwave-Assisted Resin-Based Deprotection of a Panel of Silyl Ethers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cramer, J.; Sager, C.P.; Ernst, B. Hydroxyl Groups in Synthetic and Natural Product Derived Therapeutics: A Perspective on a Common Functional Group. J. Med. Chem. 2019, 62, 8915–8930. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewicz, A.; Kalek, M.; Stawinski, J. Iodine-Promoted Silylation of Alcohols with Silyl Chlorides. Synthetic and Mechanistic Studies. Tetrahedron 2008, 64, 8843–8850. [Google Scholar] [CrossRef]
- Wuts, P.G.M. Greene’s Protective Groups in Organic; John Wiley and Sons: Hoboken, NJ, USA, 2014; pp. 1–1360. [Google Scholar]
- Patschinski, P.; Zhang, C.; Zipse, H. The Lewis Base-Catalyzed Silylation of Alcohols-a Mechanistic Analysis. J. Org. Chem. 2014, 79, 8348–8357. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The Evolution of Nucleoside Analogue Antivirals: A Review for Chemists and Non-Chemists. Part 1: Early Structural Modifications to the Nucleoside Scaffold. Antiviral Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Somoza Àlvaro, S. Protecting Groups for RNA Synthesis: An Increasing Need for Selective Preparative Methods. Chem. Soc. Rev. 2008, 37, 2668–2675. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Venkateswarlu, A. Protection of Hydroxyl Groups as Tert-Butyldimethylsilyl Derivatives. J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar] [CrossRef]
- Jereb, M. Highly Atom Economical Uncatalysed and Iodine-Catalysed Silylation of Phenols, Alcohols and Carbohydrates, Using HMDS under Solvent-Free Reaction Conditions (SFRC). Tetrahedron 2012, 68, 3861–3867. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Liu, Z.; Li, C.; Zhang, D. Recent Advances in Catalytic Silylation of Hydroxyl-Bearing Compounds: A Green Technique for Protection of Alcohols Using Si–O Bond Formations. Appl. Organomet. Chem. 2021, 35, 1–24. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Hakimelahi, G.H.; Proba, Z.A.; Ogilvie, K.K. Nitrate Ion as Catalyst for Selective Silylations of Nucleosides. Tetrahedron Lett. 1981, 22, 4775–4778. [Google Scholar] [CrossRef]
- Giri, N.; Bowen, C.; Vyle, J.S.; James, S.L. Fast, Quantitative Nucleoside Protection under Solvent-Free Conditions. Green Chem. 2008, 10, 627–662. [Google Scholar] [CrossRef]
- Karimi, B.; Golshani, B. Mild and Highly Efficient Method for the Silylation of Alcohols Using Hexamethyldisilazane Catalyzed by Iodine under Nearly Neutral Reaction Conditions. J. Org. Chem. 2000, 65, 7228–7230. [Google Scholar] [CrossRef] [PubMed]
- Saxena, I.; Deka, N.; Sarma, J.C.; Tsuboi, S. A Convenient Method for Protection and Deprotection of Alcohols and Phenols as Alkylsilyl Ethers Catalyzed by Iodine under Microwave Irradiation. Synth. Commun. 2003, 33, 4185–4191. [Google Scholar] [CrossRef]
- Loupy, A. Solvent-Free Reactions. In Modern Solvents in Organic Synthesis; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Painter, G.R.; Guthrie, D.B.; Bluemling, G.R.; Natchus, M.G. N4-Hydroxycytidine and Derivatives and Anti-Viral Uses Related Thereto. WO2016106050A1, 30 June 2016. [Google Scholar]
- Gadakh, B.; Vondenhoff, G.; Lescrinier, E.; Rozenski, J.; Froeyen, M.; Van Aerschot, A. Base Substituted 5′-O-(N-Isoleucyl)Sulfamoyl Nucleoside Analogues as Potential Antibacterial Agents. Bioorganic Med. Chem. 2014, 22, 2875–2886. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, K.K.; McGee, D.P.C.; Boisvert, S.M.; Hakimelahi, G.H.; Proba, Z.A. The Preparation of Protected Arabinonucleosides. Can. J. Chem. 1983, 61, 1204–1212. [Google Scholar] [CrossRef]
- Hadrup, N.; Sharma, A.K.; Loeschner, K. Toxicity of Silver Ions, Metallic Silver, and Silver Nanoparticle Materials after in Vivo Dermal and Mucosal Surface Exposure: A Review. Regul. Toxicol. Pharmacol. 2018, 98, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teran, C.G.; Sura, S.; Cabandugama, P.; Berson, C. Silver Nitrate Ingestion: Report of a Case with an Uneventful Course and Review of the Literature. Clin. Pract. 2011, 1, e43. [Google Scholar] [CrossRef]
- Pace, V.; Hoyos, P.; Castoldi, L.; Domínguez De María, P.; Alcántara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A Biomass-Derived Solvent with Broad Application in Organic Chemistry. Chem. Sus. Chem. 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- Cai, C.M.; Zhang, T.; Kumar, R.; Wyman, C.E. Integrated Furfural Production as a Renewable Fuel and Chemical Platform from Lignocellulosic Biomass. J. Chem. Technol. Biotechnol. 2014, 89, 2–10. [Google Scholar] [CrossRef]
- Shieh, W.C.; Dell, S.; Repič, O. 1,8-Diazabicyclo[5.4.0]Undec-7-Ene (DBU) and Microwave-Accelerated Green Chemistry in Methylation of Phenols, Indoles, and Benzimidazoles with Dimethyl Carbonate. Org. Lett. 2001, 3, 4279–4281. [Google Scholar] [CrossRef]
- Hermecz, I. Chemistry of Diazabicycloundecene (DBU) and Other Pyrimidoazepines. Adv. Heterocycl. Chem. 1987, 42, 83–202. [Google Scholar] [CrossRef]
- Nand, B.; Khanna, G.; Chaudhary, A.; Lumb, A.; Khurana, J.M. 1,8-Diazabicyclo[5.4.0]Undec-7-Ene (DBU): A Versatile Reagent in Organic Synthesis. Curr. Org. Chem. 2015, 19, 790–812. [Google Scholar] [CrossRef]
- Henderson, R.K.; Hill, A.P.; Redman, A.M.; Sneddon, H.F. Development of GSK’s Acid and Base Selection Guides. Green Chem. 2015, 17, 945–949. [Google Scholar] [CrossRef]
- Hatano, B.; Toyota, S.; Toda, F. Efficient Solvent-Free O-Silylation of Alcohols with R3SiCl. Green Chem. 2001, 3, 140–142. [Google Scholar] [CrossRef]
- Crouch, R.D. Selective Deprotection of Silyl Ethers. Tetrahedron 2013, 69, 2383–2417. [Google Scholar] [CrossRef]
- Wang, B.; Sun, H.X.; Chen, B.; Sun, Z.H. Practical, Environment-Benign and Atom Economic KOAc-Catalysed Deprotection of Aryl TIPS Ethers under Mild Fluoride-Free Conditions. Green Chem. 2009, 11, 1112–1114. [Google Scholar] [CrossRef]
- Crouch, R.; Williams, A. Rapid, Acid-Mediated Deprotection of Silyl Ethers Using Microwave Heating. Synth. Commun. 2006, 36, 959–964. [Google Scholar] [CrossRef]
- Tan, Z.P.; Wang, L.; Wang, J.B. Deprotection of T-Butyldimethylsiloxy (TBDMS) Protecting Group with Catalytic Copper (II) Chloride Dihydrate. Chin. Chem. Lett. 2000, 11, 753–756. [Google Scholar]
- González-Calderón, D.; Benítez-Puebla, L.J.; González-González, C.A.; Assad-Hernández, S.; Fuentes-Benítez, A.; Cuevas-Yáñez, E.; Corona-Becerril, D.; González-Romero, C. Selective Deprotection of TBDMS Alkyl Ethers in the Presence of TIPS or TBDPS Phenyl Ethers by Catalytic CuSO4·5H2O in Methanol. Tetrahedron Lett. 2013, 54, 5130–5132. [Google Scholar] [CrossRef]
- Vaino, A.R.; Szarek, W.A. A Mild and Efficient Method for the Deprotection of Tert-Butyldimethylsilyl Ethers Using Iodine in Methanol. Chem. Commun. 1996, 2351–2352. [Google Scholar] [CrossRef]
- Fujii, H.; Shimada, N.; Ohtawa, M.; Karaki, F.; Koshizuka, M.; Hayashida, K.; Kamimura, M.; Makino, K.; Nagamitsu, T.; Nagase, H. Deprotection of Silyl Ethers by Using SO3H Silica Gel: Application to Sugar, Nucleoside, and Alkaloid Derivatives. Tetrahedron 2017, 73, 5425–5429. [Google Scholar] [CrossRef]
- Varma, R.S.; Lamture, J.B.; Varma, M. Alumina-Mediated Cleavage of t-Butyldimethylsilyl Ethers. Tetrahedron Lett. 1993, 34, 3029–3032. [Google Scholar] [CrossRef]
- Feixas, J.; Capdevila, A.; Guerrero, A. Utilization of Neutral Alumina as a Mild Reagent for the Selective Cleavage of Primary and Secondary Silyl Ethers. Tetrahedron 1994, 50, 8539–8550. [Google Scholar] [CrossRef]
- Upadhya, T.T.; Daniel, T.; Sudalai, A.; Ravindranathan, T.; Sabu, K.R. Natural Kaolinitic Clay: A Mild and Efficient Catalyst for the Tetrahydropyranylation and Trimethylsilylation of Alcohols. Synth. Commun. 1996, 26, 4539–4544. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Li, T.S.; Yang, F.; Fu, C.G. Montmorillonite Clay Catalysis XI1: Protection and Deprotection of Hydroxyl Group by Formation and Cleavage of Trimethylsilyl Ethers Catalysed by Catalysed by Montmorillonite K-10. Synth. Commun. 1998, 28, 3105–3114. [Google Scholar] [CrossRef]
- Ghanei, M.; Khalilzadeh, M.A.; Hashemi, M.M. Potassium Fluoride/K10-Montmorillonite Nanostructure as a Green and Reusable Catalyst under Mild Reaction Conditions for the Knovenagel Condensation. Orient. J. Chem. 2016, 32, 665–669. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Entry | TBDMSCl (mmol) | Catalyst (mmol) | Base/Nucleophile (mmol) | Solvent | Heating Method | Time | Yield |
|---|---|---|---|---|---|---|---|
| 1a | 4.0 | DMAP (0.1) | Imidazole (4.0) | DCM | None | 3 d | 11% |
| 1b | 6.0 | / | Imidazole (10.0) | DMF | Conventional | 48 h | 27% |
| 1c | 12.0 | AgNO3 (11.0) | Imidazole (26.0) | DMF | Conventional | 3 h | 86% |
| 1d | 12.0 | KNO3 (11.0) | Imidazole (26.0) | DMF | Conventional | 3 h | 97% |
| 1e | 12.0 | KNO3 (11.0) | Imidazole (26.0) | DMF | None | 3 h | 47% |
| 1f | 12.0 | KNO3 (11.0) | Imidazole (26.0) | DMF | MW | 5 min | 99% |
| 1g | 12.0 | KNO3 (11.0) | CsCO3 (26.0) | DMF | MW | 5 min | / |
| 1h | 12.0 | KNO3 (11.0) | Na2CO3 (26.0) | DMF | MW | 5 min | / |
| 1i | 12.0 | KNO3 (11.0) | Imidazole (26.0) | 2-Me-THF | MW | 5 min | 99% |
| 1j | 6.0 | KNO3 (6.0) | Imidazole (26.0) | 2-Me-THF | MW | 10 min | 19% |
| 1k | 9.0 | KNO3 (8.2) | Imidazole (19.5) | 2-Me-THF | MW | 50 min | 88% |
| 1l | 9.0 | KNO3 (8.2) | DBU (19.5) | 2-Me-THF | MW | 5 min | 90% |
| 1m | 9.0 | KNO3 (3.0) | DBU (19.5) | 2-Me-THF | MW | 10 min | 89% |
| 1n | 9.0 | KNO3 (1.0) | DBU (19.5) | 2-Me-THF | MW | 10 min | 99% |
| 1o | 6.0 | KNO3 (1.0) | DBU (13.0) | 2-Me-THF | MW | 10 min | 90% |
| 1p | 6.0 | KNO3 (1.0) | 2,6-lutidine (13.0) | 2-Me-THF | MW | 10 min | 21% |
| 1q | 4.5 | KNO3 (1.0) | DBU (8.0) | Solvent-free | MW | 10 min | 98% |
| Entry | Starting Alcohol | Product | t (min) | Yield a |
|---|---|---|---|---|
| 1 |  |  | 10 | 98% |
| 2 |  |  | 5 | 82% |
| 3 |  |  | 40 | 88% |
| 4 |  |  | 30 | 86% |
| 5 |  |  | 20 | 93% |
| 6 |  |  | 5 | 84% |
| 7 |  |  | 5 | 50% |
| 8 |  |  | 10 | 81% |
| 9 |  |  | 20 | 43% |
| 10 |  |  | 5 | 62% b |
| Entry | Starting Alcohol | Product | t (min) | Yield a |
|---|---|---|---|---|
| D1 |  1 |  | 100 | 83% |
| D2 |  2 |  | 20 | 56% |
| D3 |  3 3 |  | 20 | 64% |
| D4 |  4 |  | 60 | 40% |
| D5 |  5 |  | 40 | / a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasqualini, C.; Poggialini, F.; Vagaggini, C.; Brai, A.; Dreassi, E. A Greener Technique for Microwave-Assisted O-Silylation and Silyl Ether Deprotection of Uridine and Other Substrates. Chemistry 2022, 4, 1714-1722. https://doi.org/10.3390/chemistry4040112
Pasqualini C, Poggialini F, Vagaggini C, Brai A, Dreassi E. A Greener Technique for Microwave-Assisted O-Silylation and Silyl Ether Deprotection of Uridine and Other Substrates. Chemistry. 2022; 4(4):1714-1722. https://doi.org/10.3390/chemistry4040112
Chicago/Turabian StylePasqualini, Claudia, Federica Poggialini, Chiara Vagaggini, Annalaura Brai, and Elena Dreassi. 2022. "A Greener Technique for Microwave-Assisted O-Silylation and Silyl Ether Deprotection of Uridine and Other Substrates" Chemistry 4, no. 4: 1714-1722. https://doi.org/10.3390/chemistry4040112