
Dissociative Electron Attachment Cross Sections for Ni(CO)4, Co(CO)3NO, Cr(CO)6



Abstract
:1. Introduction
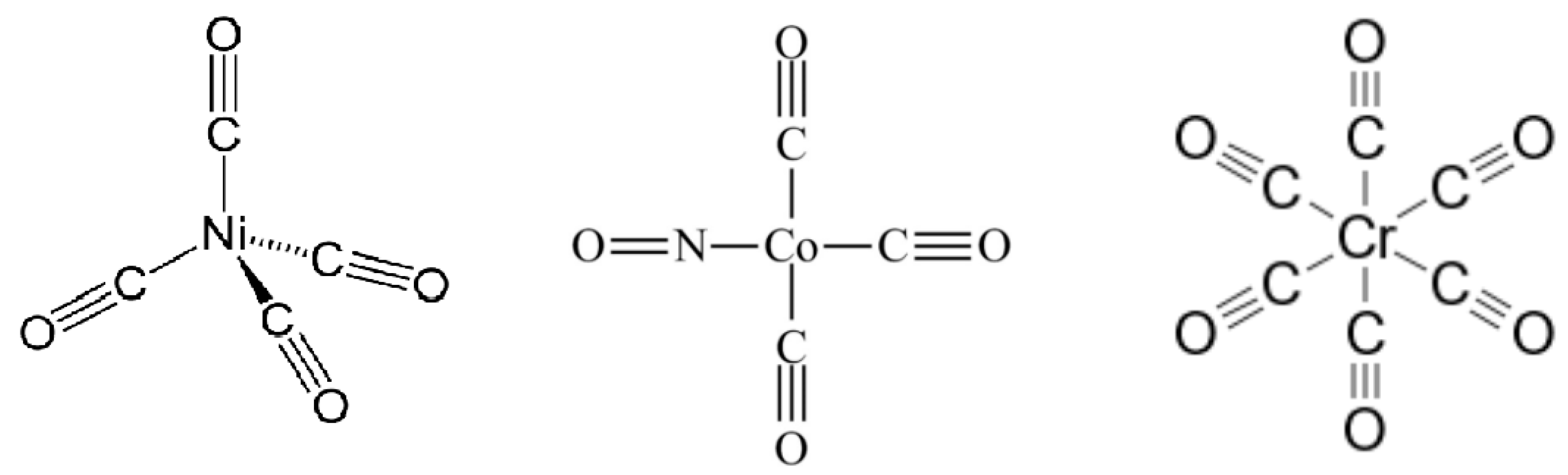
Molecular Complexes Used for Calculations
2. R-Matrix Theory and Method of Calculation
3. Computational Details
4. Cross Sections
→ Co(CO)3− + NO → Co(CO)2− + NO + CO
→ Co(CO)− + NO + 2CO
→ Co− + NO + 3(CO)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Negative Ion | Incident Electron Energy (eV) [10] | Ion Formation | Incident Electron Energy (eV) [21] | Negative Ion | Incident Electron Energy (eV) [22] |
|---|---|---|---|---|---|
| Co(CO)2NO− | 0.9 | Ni(CO)3− | 0.8 | Cr(CO)5− | 0.1 |
| CoCONO− | 2 | Ni(CO)2− | 1.7 | Cr(CO)4− | 1.5 |
| CoNO− | 5 | Ni(CO)− | 4.6 | Cr(CO)3− | 4.7 |
| Co(CO)−3 | 1.8 | Ni- | 5.4 | Cr(CO)2− | 5.9 |
| Co(CO)−2 | 3 | CrCO− | 8.5 | ||
| CoCO− | 6.4 | Cr− | 8.8 | ||
| Co− | 7.1 |
| Molecule | Bond Distances (Å) CCSD (Cr-C) [27] | Bond Distances (Å) CCSD (C-O) [27] | Bond Distances (Å) CCSD (Cr-C)eq [25] | Bond Distances (Å) CCSD (C-O)eq [25] |
|---|---|---|---|---|
| Cr(CO)6 | 3.684 | 2.207 | 1.918 | 1.141 |
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
| Atom Label | X [Å] | Y [Å] | Z [Å] |
|---|---|---|---|
| C1 | 1.34 | 1.34 | 0 |
| C2 | −1.34 | −1.34 | 0 |
| C3 | 0 | 0 | 1.9 |
| C4 | −1.34 | 1.34 | 0 |
| C5 | 0 | 0 | 0 |
| C6 | 2.15 | 2.15 | 0 |
| Cr7 | −2.15 | −2.15 | 0 |
| O8 | 0 | 0 | 3.04 |
| O9 | −2.15 | 2.15 | 0 |
| O10 | 0 | 0 | −3.04 |
| O11 | 2.15 | −2.15 | 0 |
| O12 | 0 | 0 | −3.04 |
| O13 | 0 | −3.04 | 0 |
| Atom Label | X [Å] | Y [Å] | Z [Å] |
|---|---|---|---|
| Co1 | −0.1 | 0 | 0 |
| C2 | 0.66 | −0.81 | −1.4 |
| C3 | 0.66 | −0.81 | 1.4 |
| C4 | 0.66 | 1.62 | 0 |
| O5 | 1.12 | −1.34 | 2.32 |
| O6 | 1.12 | −1.34 | −2.32 |
| O7 | 1.11 | 2.68 | 0 |
| O8 | −2.92 | 0 | 0 |
| N9 | −1.76 | 0 | 0 |
| Atom Label | X [Å] | Y [Å] | Z [Å] |
|---|---|---|---|
| Ni1 | 0 | 0 | 0 |
| C2 | −0.09 | −1.8 | 0.18 |
| C3 | 1.73 | 0.52 | 0.04 |
| C4 | −0.74 | 0.48 | −1.58 |
| C5 | −0.9 | 0.79 | 1.35 |
| O6 | −0.15 | −2.94 | 0.3 |
| O7 | 2.83 | 0.85 | 0.07 |
| O8 | −1.2 | 0.79 | −2.58 |
| O9 | −1.47 | 1.29 | 2.21 |
References
- Huth, M.; Porrati, F.; Schwalb, C.; Winhold, M.; Sachser, R.; Dukic, M.; Adams, J.; Fantner, G. Focused electron beam induced deposition: A perspective. Beilstein J. Nanotechnol. 2012, 3, 597–619. [Google Scholar] [CrossRef]
- Thorman, R.M.; Kumar RT, P.; Fairbrother, H.D.; Ingólfsson, O. The role of low-energy electrons in focused electron beam induced deposition: Four case studies of representative precursors. Beilstein J. Nanotechnol. 2015, 6, 1904–1926. [Google Scholar] [CrossRef]
- Van Dorp, W.F.; Hagen, C.W. A critical literature review of focused electron beam induced deposition. J. Appl. Phys. 2008, 104, 081301. [Google Scholar] [CrossRef]
- Tennyson, J. Electron-molecule collision calculations using the R-matrix method. Phys. Rep. 2010, 491, 29–76. [Google Scholar] [CrossRef]
- Song, M.-Y.; Yoon, J.-S.; Cho, H.; Itikawa, Y.; Karwasz, G.P.; Kokoouline, V.; Nakamura, Y.; Tennyson, J. Cross Sections for Electron Collisions with Methane. J. Phys. Chem. Ref. Data 2015, 44, 2. [Google Scholar] [CrossRef]
- Brigg, W.J.; Tennyson, J.; Plummer, M. R-matrix calculations of low-energy electron collisions with methane. J. Phys. B At. Mol. Opt. Phys. 2014, 47, 185203. [Google Scholar] [CrossRef]
- Carr, J.M.; Galiatsatos, P.G.; Gorfinkiel, J.D.; Harvey, A.G.; Lysaght, M.A.; Madden, D.; Mašín, Z.; Plummer, M.; Tennyson, J.; Varambhia, H.N. UKRmol: A low-energy electron- and positron-molecule scattering suite. Eur. Phys. J. D 2012, 66, 58. [Google Scholar] [CrossRef]
- Munro, J.J.; Harrison, S.; Fujimoto, M.M.; Tennyson, J. A dissociative electron attachment cross-sections estimator. J. Phys. Conf. Ser. 2012, 388, 012013. [Google Scholar] [CrossRef]
- Tennyson, J.; Noble, C.J. RESON: A program for the detection and fitting of Breit–Wigner resonances. Comput. Phys. Commun. 1984, 33, 421–424. [Google Scholar] [CrossRef]
- Engmann, S.; Stano, M.; Papp, P.; Brunger, M.J.; Matejčik, S.; Ingólfsson, O. Absolute cross sections for dissociative electron attachment and dissociative ionization of cobalt tricarbonyl nitrosyl in the energy range from 0eV to 140eV. J. Chem. Phys. 2013, 138, 044305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postler, J.; Renzler, M.; Kaiser, A.; Huber, S.E.; Probst, M.; Scheier, P.; Ellis, A.M. Electron-Induced Chemistry of Cobalt Tricarbonyl Nitrosyl (Co(CO)3NO) in Liquid Helium Nanodroplets. J. Phys. Chem. C 2015, 119, 20917–20922. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.A.; Friday, T.O.; Goodman, B.R.; Kooi, S.E.; Blair, R.G.; Polik, W.F. Energy Disposal in the Photodissociation of Co(CO)3NO near 225 nm. J. Phys. Chem. A 1998, 102, 10697–10702. [Google Scholar] [CrossRef]
- Bamford, C.H.; Compton, R.G.; Tipper, C.F.H. Decomposition of Inorganic and Organometallic Compounds; Elsevier: Amsterdam, The Netherlands, 1972; Volume 4, p. 200. [Google Scholar]
- Ehlers, A.W.; Dapprich, S.; Vyboishchikov, S.F.; Frenking, G. Structure and Bonding of the Transition-Metal Carbonyl Complexes M(CO)5L (M)Cr, Mo, W) and M(CO)3L (M) Ni, Pd, Pt; L) CO, SiO, CS, N2, NO+, CN−, NC−, HCCH, CCH2, CH2, CF2, H2). Organometallics 1996, 15, 105–117. [Google Scholar] [CrossRef]
- Sherwood, D.E.; Hall, M.B. Theoretical Study of the Dissociation of a Single Carbonyl from Chromium Hexacarbonyl. Inorg. Chem. 1983, 22, 93–100. [Google Scholar] [CrossRef]
- Barnes, L.A.; Liu, B.; Lindh, R. The Structure and Energetics of Cr(CO)6 and Cr(CO)5. J. Chem. Phys. 1993, 98, 3978. [Google Scholar] [CrossRef]
- Mcdowell, R.S.; Horrocks, W.D.; Yates, J.T., Jr. Infrared Spectrum of Co(CO)3NO. J. Chem. Phys. 1961, 34, 530–534. [Google Scholar] [CrossRef]
- Horrocks, W.D.; Taylor, R.C. Infrared Spectroscopic Study of Derivatives of Cobalt Tricarbonyl Nitrosyl. Inorg. Chem. 1963, 2, 723–727. [Google Scholar] [CrossRef]
- Sawyer, K.R.; Steele, R.P.; Glascoe, E.A.; Cahoon, J.F.; Schlegel, J.P.; Head-Gordon, M.; Harris, C.B. Direct Observation of Photoinduced Bent Nitrosyl Excited–State Complexes. J. Phys. Chem. A 2008, 112, 8505–8514. [Google Scholar] [CrossRef]
- Papp, P.; Engmann, S.; Kučera, M.; Stano, M.; Matejčik, Š.; Ingólfsson, O. An experimental and theoretical study on structural parameters and energetics in ionization and dissociation of cobalt tricarbonyl nitrosyl. Int. J. Mass Spectrom. 2013, 356, 24–32. [Google Scholar] [CrossRef]
- Compton, R.N.; Stockdale, J.A.D. Formation of gas–phase negative ions in Fe(CO)5 and Ni(CO)4. Intl. J. Mass Spectrom. Ion Phys. 1976, 22, 47–55. [Google Scholar] [CrossRef]
- Khreis, J.M.; Ameixa, J.; Ferreira da Silva, F.; Denifl, S. Interactions of low-energy electrons with the FEBID precursor chromium hexacarbonyl (Cr(CO)6). Beilstein J. Nanotechnol. 2017, 8, 2583–2590. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.J.; Martel, A.A.; Waller, I.M. Electron Affinity of Co(CO)2NO Measured by Negative-Ion Photoelectron Spectroscopy. J. Phys. Chem. 1994, 98, 474–477. [Google Scholar] [CrossRef]
- Nakata, H.; Nagamori, K.; Yamasaki, K.; Kohguchi, H. Detection of direct NO ligand loss in the ultraviolet photodissociation of Co(CO)3NO. Chem. Phys. Lett. 2018, 707, 150–153. [Google Scholar] [CrossRef]
- Crespo-Otero, R.; Barbatti, M. Cr(CO)6 photochemistry: Semi-classical study of UV absorption spectral intensities and dynamics of photodissociation. J. Chem. Phys. 2011, 134, 164305. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.J.V.; Henning, N.; Krüger, B.C.; Barratt Park, G.; Altschäffel, J.; Kandratsenka, A.; Wodtke, A.M.; Schäfer, T. Vibrational Relaxation of Highly Vibrationally Excited CO Scattered from Au(111): Evidence for CO- Formation. J. Phys. Chem. Lett. 2017, 8, 4887–4892. [Google Scholar] [CrossRef]
- McKinlay, R.G.; Almeida, N.M.S.; Coe, J.P.; Paterson, M.J. Excited States of the Nickel Carbonyls Ni(CO) and Ni(CO)4: Challenging Molecules for Electronic Structure Theory. J. Phys. Chem. A 2015, 119, 10076–10083. [Google Scholar] [CrossRef]
- Whitaker, A.; Jefferey, J.W. The Crystal Structure of Chromium Hexacarbonyl. Acta Cryst. 1967, 23, 977. [Google Scholar] [CrossRef]
- Pierloot, K.; Tsokos, E.; Vanquickenborne, L.G. Optical Spectra of Ni(CO)4 and Cr(CO)6 Revisited. J. Phys. Chem. 1996, 100, 16545–16550. [Google Scholar] [CrossRef]
- Beach, N.A.; Gray, H.B. Electronic structures of metal hexacarbonyls. J. Am. Chem. Soc. 1968, 90, 5713–5721. [Google Scholar] [CrossRef]
- Vilaume, S.; Strich, A.; Daniel, C.; Perera, S.A.; Bartlett, R.J. A coupled cluster study of the electronic spectroscopy and photochemistry of Cr(CO)6. Phys. Chem. Chem. Phys. 2007, 9, 6115–6122. [Google Scholar] [CrossRef]
- Winters, R.E.; Kiser, R.W. Ions Produced by Electron Impact with the Dimetallic Carbonyls of Cobalt and Manganese. J. Phys. Chem. 1965, 69, 1618–1622. [Google Scholar] [CrossRef]
- Fukuda, R.; Hayaki, S.; Nakatsuji, H. Valence ionization spectra of group six metal hexacarbonyls studied by the symmetry-adapted cluster-configuration interaction method. J. Chem. Phys. 2009, 131, 174303. [Google Scholar] [CrossRef]
- Foffani, A.; Pignataro, S.; Cantone, B.; Grasso, F. Calculation of Ionization Potentials for Aromatic Compounds. Z. Physik. Chem. Neue Folge 1964, 42, 221. [Google Scholar] [CrossRef]
- Junk, G.A.; Svec, H.J. Energetics of the Ionization and Dissociation of Ni(CO)4, Fe(CO)5, Cr(CO)6, Mo(CO)6 and W(CO)6. Z. Für Nat. 1968, 23b, 1–9. [Google Scholar] [CrossRef]
- Pollak, C.; Rosa, A.; Baerends, E.J. Cr-CO Photodissociation in Cr(CO)6: Reassessment of the Role of Ligand Field Excited States in the Photochemical Dissociation of Metal–Ligand Bonds. J. Am. Chem. Soc. 1997, 119, 7324–7329. [Google Scholar] [CrossRef]
- Büker, H.H.; Maître, P.; Ohanessian, G. Theoretical Study of Tungsten Carbonyl Complexes W(CO)+n(n = 1–6): Structures, Binding Energies, and Implications for Gas Phase Reactivities. J. Phys. Chem. A 1997, 101, 3966–3976. [Google Scholar] [CrossRef]
- Kopyra, J.; Maciejewska, P.; Maljković, J. Dissociative electron attachment to coordination complexes of chromium: Chromium(0) hexacarbonyl and benzene-chromium(0) tricarbonyl. Beilstein J. Nanotechnol. 2017, 8, 2257–2263. [Google Scholar] [CrossRef]
- Reutt, J.E.; Wang, L.S.; Lee, Y.T.; Shirley, D.A. Molecular beam photoelectron spectroscopy of Ni(CO)4. Chem. Phys. Lett. 1986, 126, 5. [Google Scholar] [CrossRef]
- Distefano, G. Photoionization Study of Fe(CO)5 and Ni(CO)4. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1970, 74A, 233–239. [Google Scholar] [CrossRef]
- Mihaylov, M.; Hadjiivanov, K.; Knözinger, H. Formation of Ni(CO)4 during the interaction between CO and silica-supported nickel catalyst: An FTIR spectroscopic study. Catal. Lett. 2001, 76, 59–63. [Google Scholar] [CrossRef]
- Fuss, W.; Schmid, W.E.; Trushin, S.A. Ultrafast Photodissociation Dynamics of Ni(CO)4. J. Phys. Chem. A 2001, 105, 333–339. [Google Scholar] [CrossRef]
- Kotzian, M.; Rösch, N.; Schröder, H.; Zerner, M.C. Optical Spectra of Transition-Metal Carbonyls: Cr(CO)6, Fe(CO)5 and Ni(CO)4. J. Am. Chem. Soc. 1989, 111, 7687–7696. [Google Scholar] [CrossRef]
- Yamazaki, E.; Okabayashi, T.; Tanimoto, M. Detection of free nickel monocarbonyl, NiCO: Rotational spectrum and structure. J. Am. Chem. Soc. 2004, 126, 1028–1029. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, L.; Iijima, T.; Hedberg, K. Nickel tetracarbonyl, Ni(CO)4. I. Molecular Structure by gaseous electron diffraction. Ii. Refinement of quadratic force field. J. Chem. Phys. 1979, 70, 3224–3229. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Orpen, G. Ni(C0)4 and Fe(CO)s: Molecular Structures in the Solid State. Organometallics 1993, 12, 1481–1483. [Google Scholar] [CrossRef]
- Stevens, A.E.; Feigerle, C.S.; Lineberger, W.C. Laser Photoelectron Spectrometry of Ni(CO)n, n = 1–3. J. Am. Chem. Soc. 1982, 104, 5026–5031. [Google Scholar] [CrossRef]
- Ramsier, R.D.; Henderson, M.A.; Yates, J.T., Jr. Electron induced decomposition of Ni(CO)4 adsorbed on Ag(111). Surf. Sci. 1991, 257, 9–21. [Google Scholar] [CrossRef]
- Utke, I.; Melngailis, J.; Hoffmann, P. Gas-assisted focused electron beam and ion beam processing and fabrication. J. Vac. Sci. Technol. B 2008, 26, 1197. [Google Scholar] [CrossRef] [Green Version]











| Compound | Dissociation | BDE (kcal/mol) |
|---|---|---|
| Co(CO)3NO | Co + 3CO + NO | 144.8–154.4 [12] |
| Ni(CO)4 | Ni(CO)3 + CO | 35 [13], 22.3 [14] |
| Co(CO)3NO | Co(CO) + 2CO + NO | 115 [12] |
| Cr(CO)6 | Cr(CO)5 + CO | 49.8 [15], 38 [16] |
| Compound | Fukuda et al. (2009) [33] | Winters and Kiser (1965) [32] | Foffani et al. (1964) [34] | Electron Ionization [35] | Photon Impact [35] | Metal Atom [35] | Junk and Svec (1968) [35] |
|---|---|---|---|---|---|---|---|
| Cr(CO)6 | 8.5 | 8.15 ± 0.17 | 8.18 ± 0.07 | 8.23 | 8.03 | 6.76 | 8.44 ± 0.05 |
| Negative Ion | Dissociation Mechanism (eV) from Cr(CO)6 | Experimental Incident Electron Energy (eV) [22] | Electron Affinity (eV) | Vibrational Frequency (cm−1) |
|---|---|---|---|---|
| Cr(CO)5− | Cr(CO)−5 + CO | 0.1 | >1.6 eV [34] | 2000 |
| Cr(CO)4− | Cr(CO)−4 + 2(CO) | 1.5 | ||
| Cr(CO)3− | Cr(CO)−3 + 3(CO) | 4.7 | ||
| Cr(CO)2− | Cr(CO)−2 + 4(CO) | 5.9 | ||
| CrCO− | Cr(CO)− + 5(CO) | 8.5 | ||
| Cr- | Cr− + 6(CO) | 8.8 |
| State | TDDFT/B3LYP–ΔE (eV) [25] | Experimental [25] ΔE (eV) | Experimental [22]/Our Q-N* Sim. ΔE (eV) |
|---|---|---|---|
| 11Eu | 4.14 | ||
| 11T2u | 4.2 | ||
| 11A2u | 4.25 | ||
| 11T1u | 4.5 | 4.44 | 4.7 |
| 11T1g | 4.65 | ||
| 11A1u | 4.7 | ||
| 21T2u | 4.82 | ||
| 21Eu | 4.71 | ||
| 11Eg | 4.99 | ||
| 11T2g | 4.74 | ||
| 21T1g | 4.91 | ||
| 21T2g | 5.59 | ||
| 21T1u | 6.02 | 5.48 | 5.9 |
| Negative Ion | Incident Electron Energy (eV) [21] | Appearance Potentials (eV) [47] | Electron Affinity (eV) [47] | Vibrational Frequency (cm−1) [47] |
|---|---|---|---|---|
| Ni(CO)3− | 0.8 | 0 | 0.804 ± 0.012 | 2100 ± 80 |
| Ni(CO)2− | 1.7 | 1.0 ± 0.4 | 0.643 ± 0.014 | 2100 ± 80 |
| Ni(CO)− | 4.6 | 3.2 ± 0.5 | 1.077 ± 0.013 | 1940 ± 80 |
| Ni− | 5.4 | 4.1 ± 0.3 | 1.157 ± 0.010 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pintea, M.; Mason, N.; Tudorovskaya, M. Dissociative Electron Attachment Cross Sections for Ni(CO)4, Co(CO)3NO, Cr(CO)6. Chemistry 2022, 4, 1060-1075. https://doi.org/10.3390/chemistry4030072
Pintea M, Mason N, Tudorovskaya M. Dissociative Electron Attachment Cross Sections for Ni(CO)4, Co(CO)3NO, Cr(CO)6. Chemistry. 2022; 4(3):1060-1075. https://doi.org/10.3390/chemistry4030072
Chicago/Turabian StylePintea, Maria, Nigel Mason, and Maria Tudorovskaya. 2022. "Dissociative Electron Attachment Cross Sections for Ni(CO)4, Co(CO)3NO, Cr(CO)6" Chemistry 4, no. 3: 1060-1075. https://doi.org/10.3390/chemistry4030072