
Optoelectronics and Transport Phenomena in Rb2InBiX6 (X = Cl, Br) Compounds for Renewable Energy Applications: A DFT Insight

Abstract
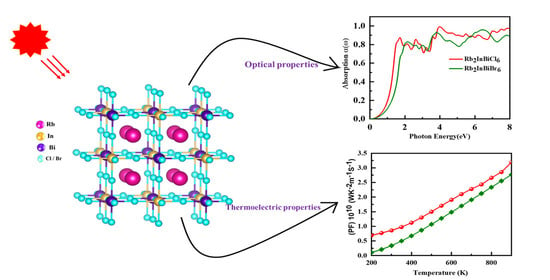
:
1. Introduction
2. Computational Method
3. Result and Discussion
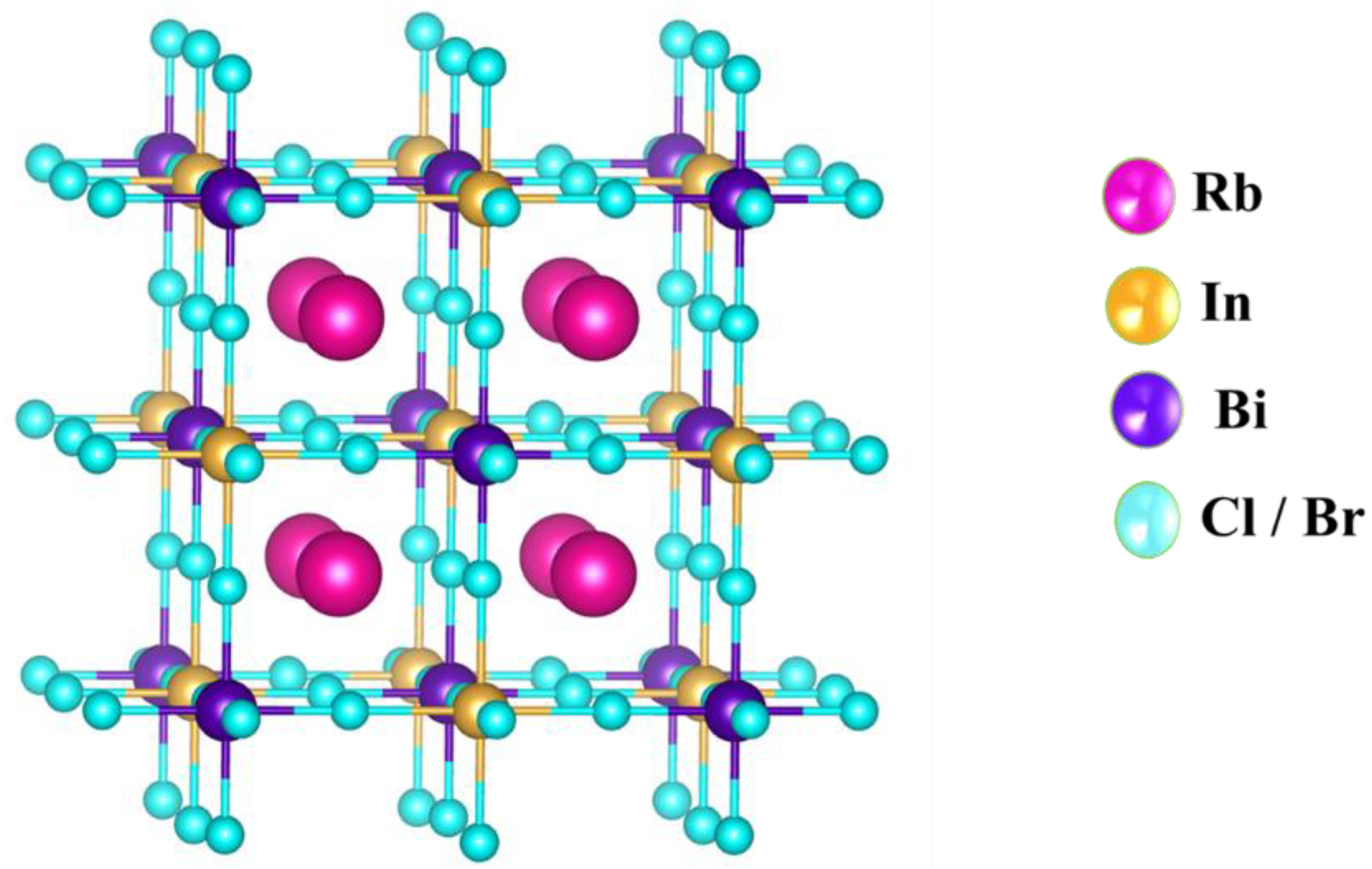
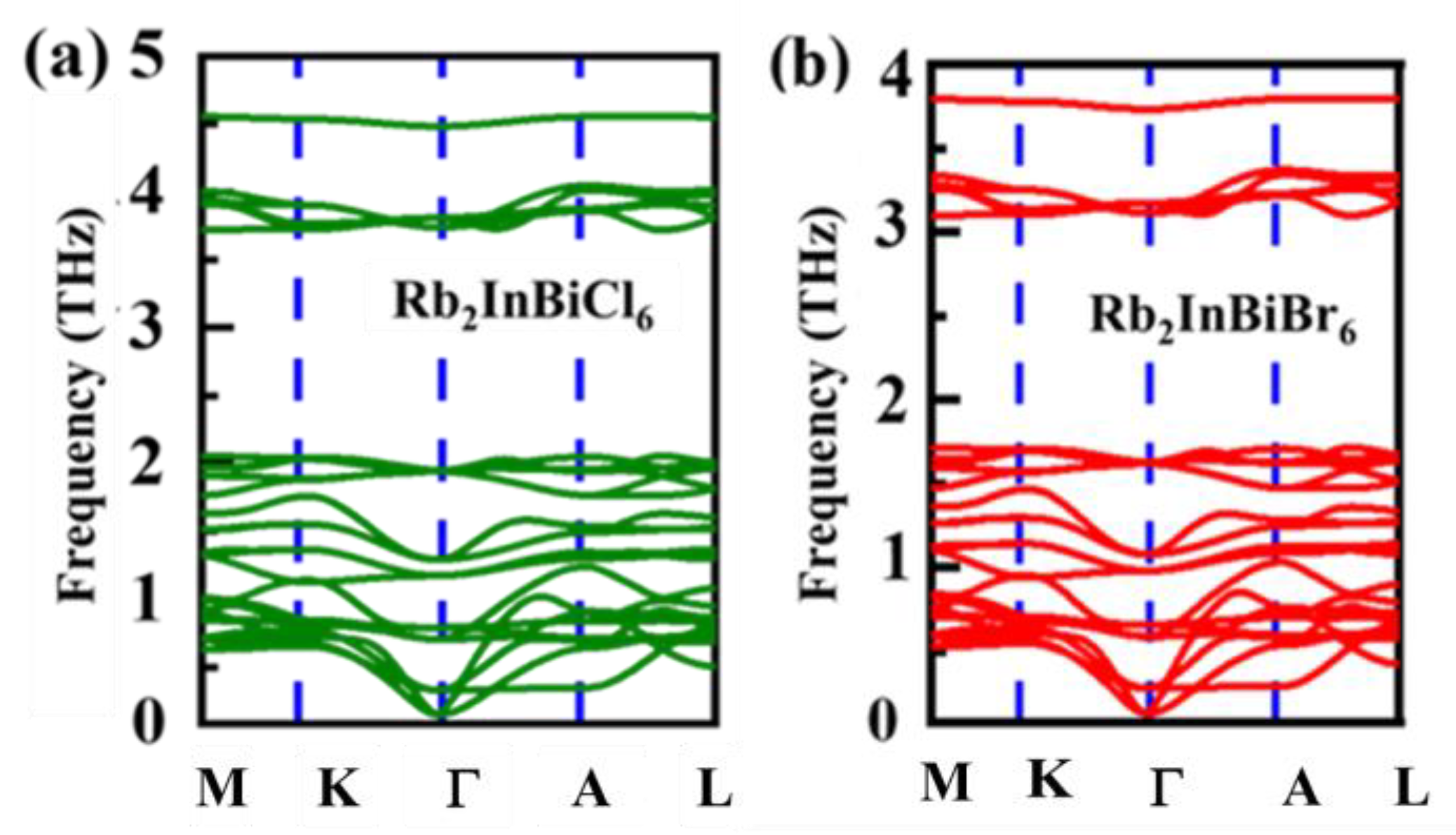
3.1. Structural Properties
3.2. Mechanical Property

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Rb2InBiCl6 | Rb2InBiBr6 | Cs2LiYBr6 [65] |
|---|---|---|---|
| C11 | 44.32 | 61.81 | 45.73 |
| C12 | 4.97 | 20.05 | 11.88 |
| C44 | 1.02 | 1.58 | 10.54 |
| B | 18.08 | 33.97 | 23.16 |
| G | 8.48 | 9.30 | 12.75 |
| Y | 22.01 | 25.56 | 32.33 |
| ν | 0.29 | 0.37 | 0.26 |
| B/G | 2.13 | 3.65 | 1.81 |
| C12 − C44 | 3.95 | 18.47 | 1.33 |
| A | 0.05 | 0.07 | |
| ΘD | 126.6 | 119.1 | |
| Vtran | 1245 | 1203 | |
| Vlong | 2759 | 3202 | |
| Vavg. | 1404 | 1365 | |
| Tm | 494.8 | 643.2 |
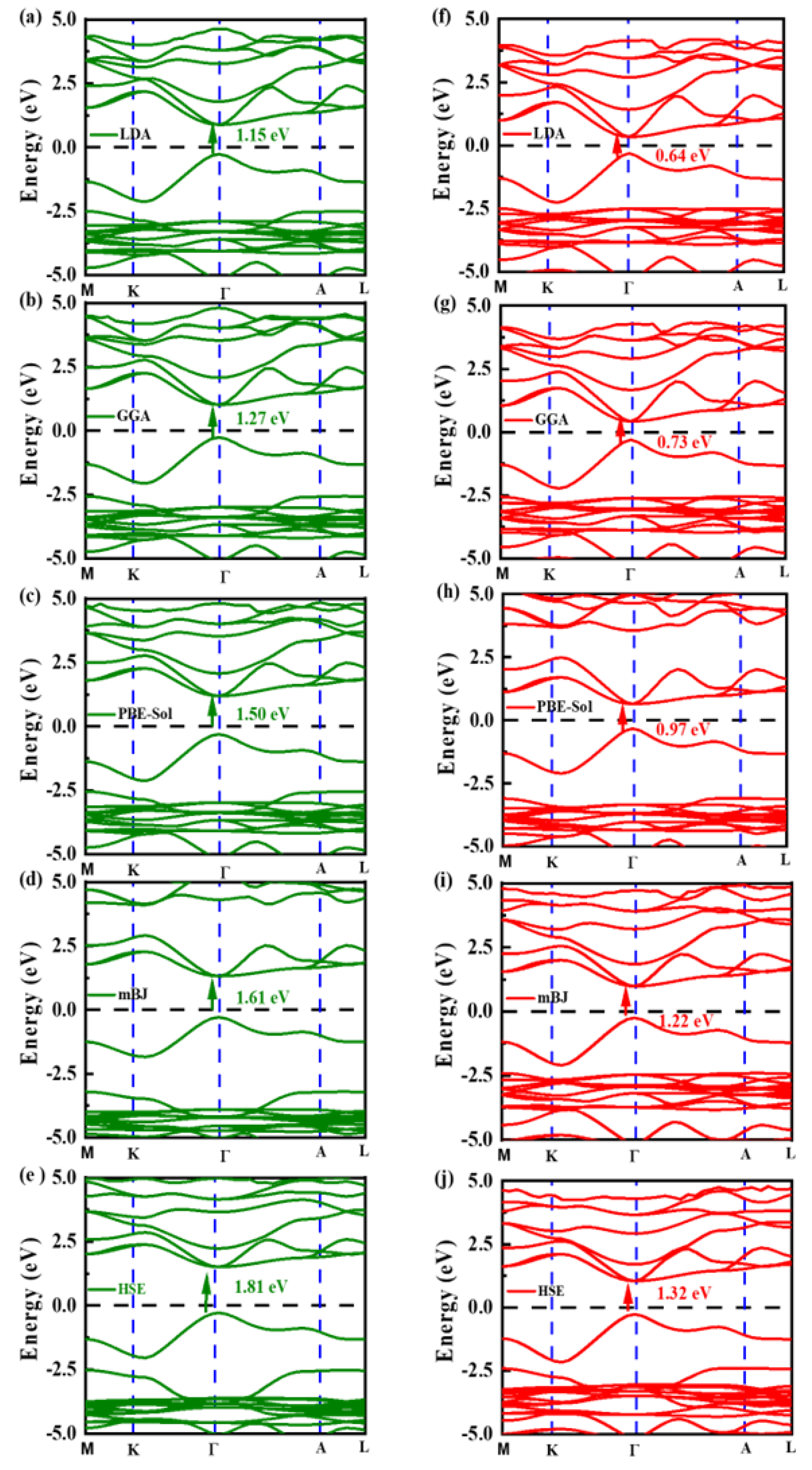
3.3. Electronic Properties
| Compound | LDA | GGA | PBE-Sol | mBJ | HSE | Effective Mass | Bader Charge |
|---|---|---|---|---|---|---|---|
| Rb2InBiCl6 | 1.15 | 1.27 | 1.50 | 1.61 | 1.81 | m*e = 0.76 m*h = 0.91 | Rb = 0.91 In = 0.89 Bi = 1.65 Cl = −0.72 |
| Cs2InAgCl6 [68] | 2.49 | ||||||
| Rb2InBiBr6 | 0.64 | 0.73 | 0.97 | 1.22 | 1.32 | m*e = 0.72 m*h = 0.80 | Rb = 0.88 In = 0.67 Bi = 1.34 Br = −0.63 |
| Cs2InAgBr6 [68] | 1.38 |
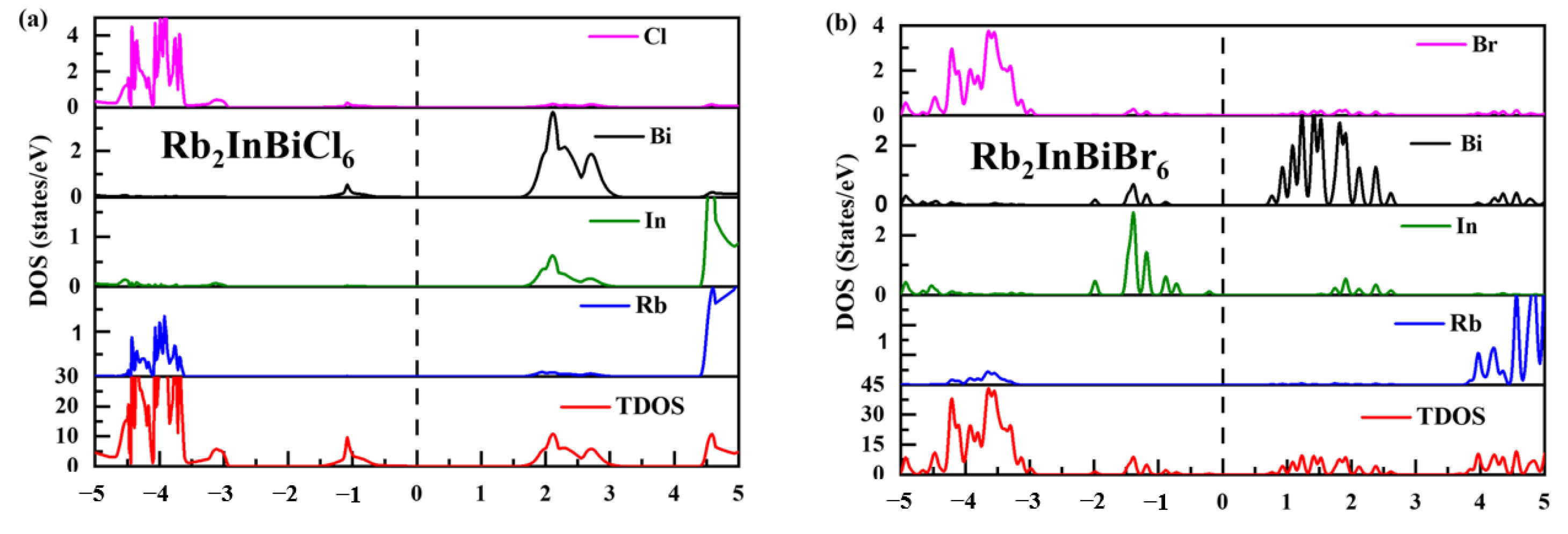
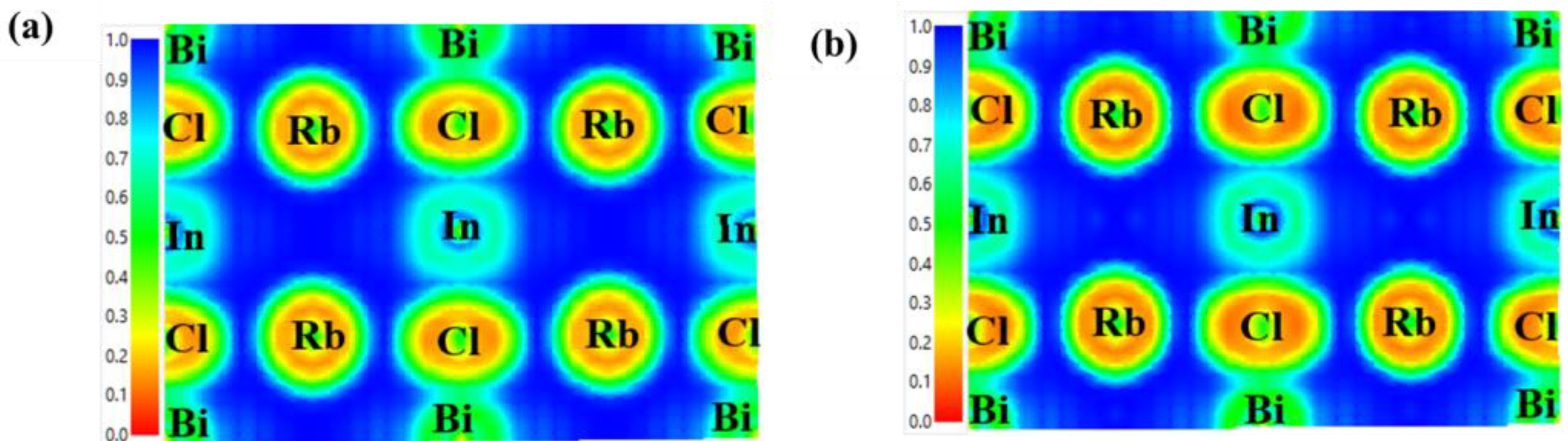
3.4. ELF and Bader Charge
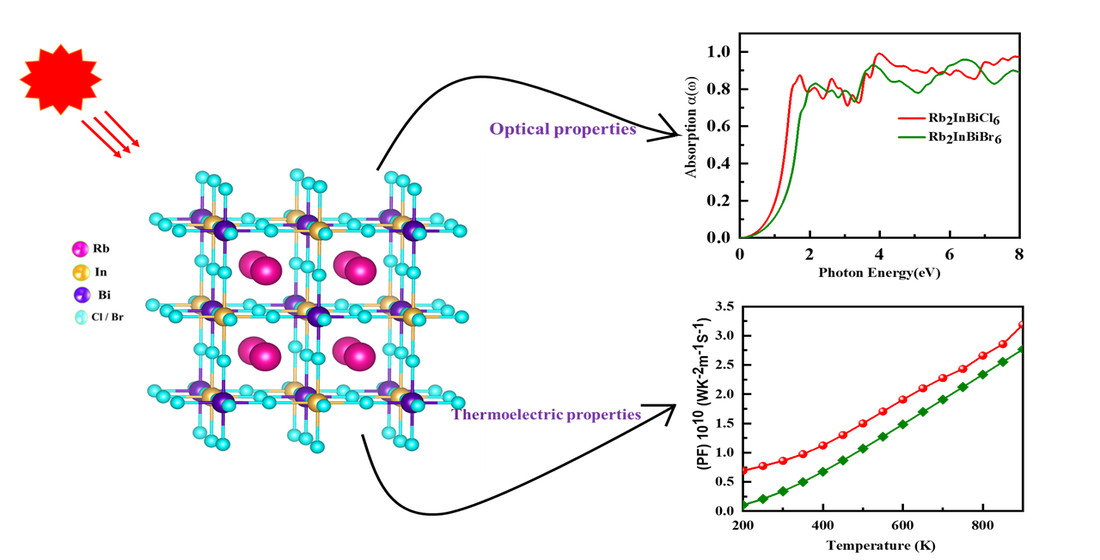
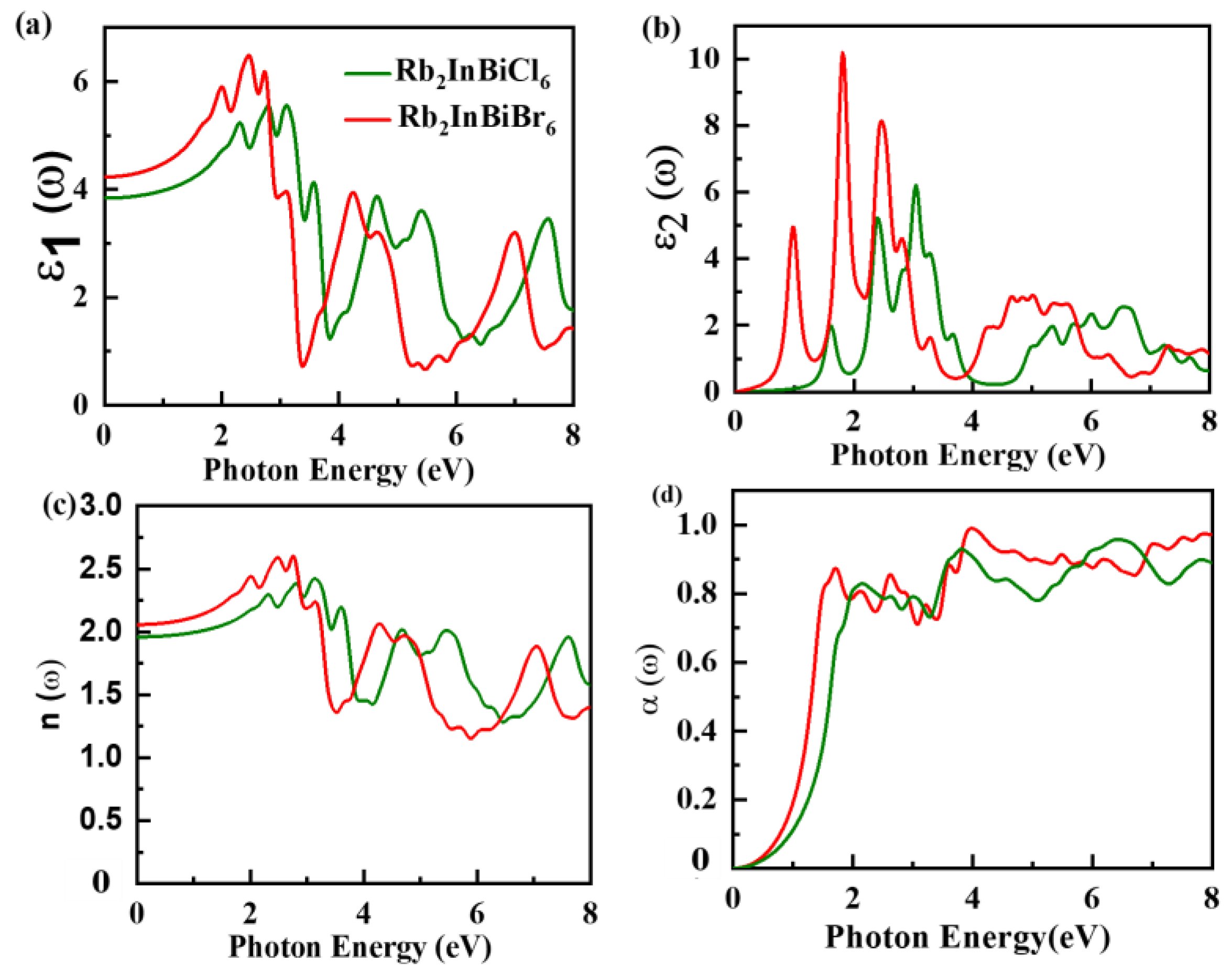
3.5. Optical Properties
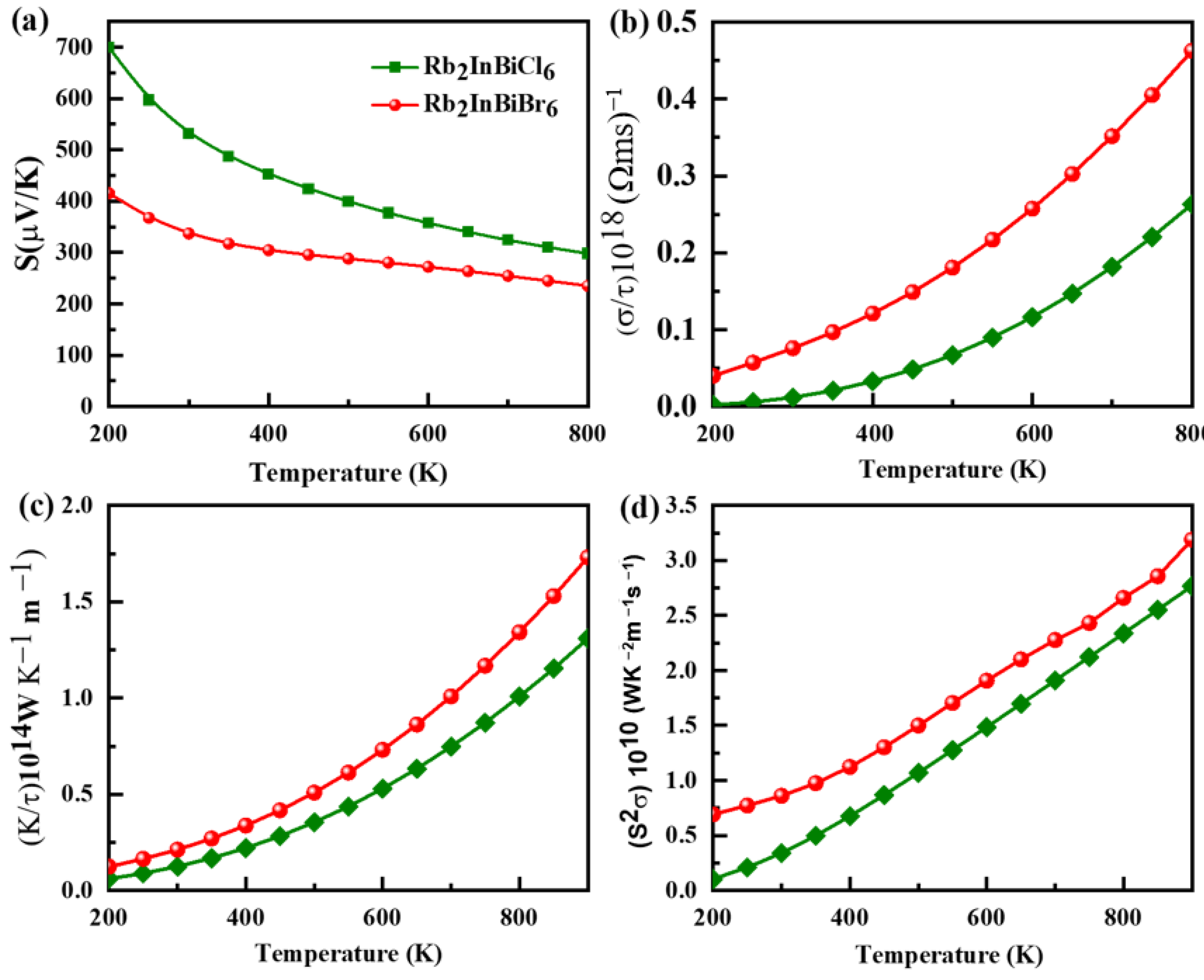
3.6. Thermoelectric Property
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elsheikh, M.H.; Shnawah, D.A.; Sabri, M.F.M.; Said, S.B.M.; Hassan, M.H.; Bashir, M.B.A.; Mohamad, M. A review on thermoelectric renewable energy: Principle parameters that affect their performance. Renew. Sustain. Energy Rev. 2014, 30, 337–355. [Google Scholar] [CrossRef]
- Zheng, X.F.; Liu, C.X.; Yan, Y.Y.; Wang, Q. A review of thermoelectrics research–Recent developments and potentials for sustainable and renewable energy applications. Renew. Sustain. Energy Rev. 2014, 32, 486–503. [Google Scholar] [CrossRef]
- Goldsmid, H.J.; Goldsmid, H.J. The Seebeck and Peltier Effects; Morgan & Claypool Publishers: New York, NY, USA, 2017. [Google Scholar]
- Steele, W.H. A new thermoelectric phenomenon. Science 1893, 562, 256. [Google Scholar] [CrossRef] [PubMed]
- Champier, D. Thermoelectric generators: A review of applications. Energy Convers. Manag. 2017, 140, 167–181. [Google Scholar] [CrossRef]
- Mingo, N. Thermoelectric figure of merit and maximum power factor in III–V semiconductor nanowires. Appl. Phys. Lett. 2004, 84, 2652–2654. [Google Scholar] [CrossRef]
- Nemir, D.C.; Beck, J. On the Significance of the Thermoelectric Figure of Merit Z. J. Electron. Mater. 2010, 39, 1897–1901. [Google Scholar] [CrossRef]
- Zhao, L.-D.; Lo, S.-H.; Zhang, Y.; Sun, H.; Tan, G.; Uher, C.; Wolverton, C.; Dravid, V.P.; Kanatzidis, M.G. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 2014, 508, 373–377. [Google Scholar] [CrossRef]
- Aswal, D.K.; Basu, R.; Singh, A. Key issues in development of thermoelectric power generators: High figure-of-merit materials and their highly conducting interfaces with metallic interconnects. Energy Convers. Manag. 2016, 114, 50–67. [Google Scholar] [CrossRef]
- Dahbi, S.; Tahiri, N.; El Bounagui, O.; Ez-Zahraouy, H. Electronic, optical, and thermoelectric properties of perovskite BaTiO3 compound under the effect of compressive strain. Chem. Phys. 2021, 544, 111105. [Google Scholar] [CrossRef]
- Maiti, T.; Saxena, M.; Roy, P. Double perovskite (Sr2B′ B ″O6) oxides for high-temperature thermoelectric power generation—A review. J. Mater. Res. 2019, 34, 107–125. [Google Scholar] [CrossRef] [Green Version]
- Arribi, P.V.; García-Fernández, P.; Junquera, J.; Pardo, V. Efficient thermoelectric materials using nonmagnetic double perovskites with d0/d6 band filling. Phys. Rev. 2016, 94, 35124. [Google Scholar] [CrossRef]
- Ricciarelli, D.; Kaiser, W.; Mosconi, E.; Wiktor, J.; Ashraf, M.W.; Malavasi, L.; Ambrosio, F.; De Angelis, F. Reaction Mechanism of Photocatalytic Hydrogen Production at Water/Tin Halide Perovskite Interfaces. ACS Energy Lett. 2022, 7, 1308–1315. [Google Scholar] [CrossRef]
- Irfan, R.M.; Tahir, M.H.; Iqbal, S.; Nadeem, M.; Bashir, T.; Maqsood, M.; Zhao, J.; Gao, L. Co3C as a promising cocatalyst for superior photocatalytic H2 production based on swift electron transfer processes. J. Mater. Chem. 2021, 9, 3145–3154. [Google Scholar] [CrossRef]
- Bhamu, K.C.; Soni, A.; Sahariya, J. Revealing optoelectronic and transport properties of potential perovskites Cs2PdX6 (X = Cl, Br): A probe from density functional theory (DFT). Sol. Energy 2018, 162, 336–343. [Google Scholar] [CrossRef]
- Usman, M.; Yan, Q. Recent Advancements in Crystalline Pb-Free Halide Double Perovskites. Crystals 2020, 10, 62. [Google Scholar] [CrossRef]
- Yan, L.; Zhao, L.; Zhao, C.; Lin, S. Theoretical Understanding of Thermoelectric Energy Conversion Efficiency in Lead-Free Halide Double Perovskites Showing Intrinsic Defect Tolerance. Appl. Therm. Eng. 2022, 215, 119024. [Google Scholar] [CrossRef]
- Zhao, X.-G.; Yang, J.-H.; Fu, Y.; Yang, D.; Xu, Q.; Yu, L.; Wei, S.-H.; Zhang, L. Design of Lead-Free Inorganic Halide Perovskites for Solar Cells via Cation-Transmutation. J. Am. Chem. Soc. 2017, 139, 2630–2638. [Google Scholar] [CrossRef]
- Ghosh, S.; Shankar, H.; Kar, P. Recent developments of lead-free halide double perovskites: A new superstar in the optoelectronic field. Mater. Adv. 2022, 3, 3742–3765. [Google Scholar] [CrossRef]
- Ravi, V.K.; Singhal, N.; Nag, A. Initiation and future prospects of colloidal metal halide double-perovskite nanocrystals: Cs2 AgBiX6 (X = Cl, Br, I). J. Mater. Chem. 2018, 6, 21666–21675. [Google Scholar] [CrossRef]
- Haque, E.; Hossain, M.A. Origin of ultra-low lattice thermal conductivity in Cs2BiAgX6 (X = Cl, Br) and its impact on thermoelectric performance. J. Alloys Compd. 2018, 748, 63–72. [Google Scholar] [CrossRef]
- Murtaza, G.; Alshahrani, T.; Khalil, R.A.; Mahmood, Q.; Flemban, T.H.; Althib, H.; Laref, A. Lead Free Double Perovsites Halides X2AgTlCl6 (X = Rb, Cs) for solar cells and renewable energy applications. J. Solid State Chem. 2021, 297, 121988. [Google Scholar] [CrossRef]
- Noor, N.; Iqbal, M.W.; Zelai, T.; Mahmood, A.; Shaikh, H.; Ramay, S.M.; Al-Masry, W. Analysis of Direct Band Gap A2ScInI6 (A = Rb, Cs) Double Perovskite Halides Using DFT Approach for Renewable Energy Devices. J. Mater. Res. Technol. 2021, 13, 2491–2500. [Google Scholar] [CrossRef]
- Aslam, F.; Ullah, H.; Hassan, M. Theoretical investigation of Cs2InBiX6 (X = Cl, Br, I) double perovskite halides using first-principle calculations. Mater. Sci. Eng. 2021, 274, 115456. [Google Scholar] [CrossRef]
- Zhou, X.; Jankowska, J.; Dong, H.; Prezhdo, O.V. Recent theoretical progress in the development of perovskite photovoltaic materials. J. Energy Chem. 2018, 27, 637–649. [Google Scholar] [CrossRef]
- Shi, W.; Cai, T.; Wang, Z.; Chen, O. The effects of monovalent metal cations on the crystal and electronic structures of Cs2MBiCl6 (M = Ag, Cu, Na, K, Rb, and Cs) perovskites. J. Chem. Phys. 2020, 153, 141101. [Google Scholar] [CrossRef]
- Romani, L.; Speltini, A.; Dibenedetto, C.N.; Listorti, A.; Ambrosio, F.; Mosconi, E.; Simbula, A.; Saba, M.; Profumo, A.; Quadrelli, P. Experimental Strategy and Mechanistic View to Boost the Photocatalytic Activity of Cs3Bi2Br9 Lead-Free Perovskite Derivative by g-C3N4 Composite Engineering. Adv. Funct. Mater. 2021, 31, 2104428. [Google Scholar] [CrossRef]
- Shafi, Z.; Ahmad, Z.; Joya, K.S.; Iqbal, S.; Khan, M.N. Designing of noble metal free high performance mesoporous electrocatalysts for water splitting. Int. J. Hydrogen Energy 2021, 46, 39799–39809. [Google Scholar] [CrossRef]
- Bibi, A.; Lee, I.; Nah, Y.; Allam, O.; Kim, H.; Quan, N.L.; Tang, J.; Walsh, A.; Jang, S.S.; Sargent, E.H. Lead-free halide double perovskites: Toward stable and sustainable optoelectronic devices. Mater. Today 2021, 49, 123–144. [Google Scholar] [CrossRef]
- Savory, C.N.; Walsh, A.; Scanlon, D.O. Can Pb-Free Halide Double Perovskites Support High-Efficiency Solar Cells? ACS Energy Lett. 2016, 1, 949. [Google Scholar] [CrossRef]
- Xiao, Z.; Meng, W.; Wang, J.; Yan, Y. Thermodynamic stability and defect chemistry of bismuth-based lead-free double perovskites. ChemSusChem 2016, 9, 2628–2633. [Google Scholar] [CrossRef]
- Filip, M.R.; Hillman, S.; Haghighirad, A.A.; Snaith, H.J.; Giustino, F. Band gaps of the lead-free halide double perovskites Cs2BiAgCl6 and Cs2BiAgBr6 from theory and experiment. J. Phys. Chem. Lett. 2016, 7, 2579–2585. [Google Scholar] [CrossRef]
- Wei, F.; Deng, Z.; Sun, S.; Hartono, N.T.P.; Seng, H.L.; Buonassisi, T.; Bristowe, P.D.; Cheetham, A.K. Enhanced visible light absorption for lead-free double perovskite Cs2AgSbBr6. Chem. Commun. 2019, 55, 3721–3724. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, S.; Wu, H.; Zhou, Y.; Li, Y.; Liu, J.; Li, J.; Li, K.; Yi, F.; Niu, G. Cs2AgInCl6 double perovskite single crystals: Parity forbidden transitions and their application for sensitive and fast UV photodetectors. ACS Photon. 2018, 5, 398–405. [Google Scholar] [CrossRef]
- Haque, E.; Hossain, M.A. High Seebeck coefficient and ultra-low lattice thermal conductivity in Cs2InAgCl6. arXiv 2018, arXiv:1802.08136. [Google Scholar]
- Mukhtar, M.W.; Ramzan, M.; Rashid, M.; Hussain, A.; Naz, G.; Ciftci, Y.O.; Dahshan, A.; Znaidia, S. Systematic study of optoelectronic and thermoelectric properties of new lead-free halide double perovskites A2KGaI6 (A = Cs, Rb) for solar cell applications via ab-initio calculations. Mater. Sci. Eng. 2022, 285, 115957. [Google Scholar] [CrossRef]
- Saeed, M.; Haq, I.U.; Saleemi, A.S.; Rehman, S.U.; Haq, B.U.; Chaudhry, A.R.; Khan, I. First-principles prediction of the ground-state crystal structure of double-perovskite halides Cs2AgCrX6 (X = Cl, Br, and I). J. Phys. Chem. Solids 2022, 160, 110302. [Google Scholar] [CrossRef]
- Iqbal, S.; Mustafa, G.M.; Asghar, M.; Noor, N.A.; Iqbal, M.W.; Mahmood, A.; Shin, Y.-H. Tuning the optoelectronic and thermoelectric characteristics of narrow bandgap Rb2AlInX6 (X = Cl, Br, I) double perovskites: A DFT study. Mater. Sci. Semicond. Process. 2022, 143, 106551. [Google Scholar] [CrossRef]
- Harikesh, P.C.; Mulmudi, H.K.; Ghosh, B.; Goh, T.W.; Teng, Y.; Thirumal, K.; Lockrey, M.; Weber, K.; Koh, T.M.; Li, S. Rb as an Alternative Cation for Templating Inorganic Lead-Free Perovskites for Solution Processed Photovoltaics. Chem. Mater. 2016, 28, 7496–7504. [Google Scholar] [CrossRef]
- Saliba, M.; Matsui, T.; Domanski, K.; Seo, J.-Y.; Ummadisingu, A.; Zakeeruddin, S.M.; Correa-Baena, J.-P.; Tress, W.R.; Abate, A.; Hagfeldt, A. Incorporation of rubidium cations into perovskite solar cells improves photovoltaic performance. Science 2016, 354, 206–209. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.K.H.; Kvasnicka, D.; Luitz, J.; Laskowski, R.; Tran, F.; Marks, L.D. Wien2k. In An Augmented Plane Wave+ Local Orbitals Program for Calculating Crystal Properties; Schwarz, S., Ed.; Vienna University of Technology: Vienna, Austria, 2001. [Google Scholar]
- Hohenberg, P.; Kohn, W. Density functional theory (DFT). Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Koller, D.; Tran, F.; Blaha, P. Improving the modified Becke-Johnson exchange potential. Phys. Rev. 2012, 85, 155109. [Google Scholar] [CrossRef] [Green Version]
- Blaha, P.; Schwarz, K.; Sorantin, P.; Trickey, S. Full-potential, linearized augmented plane wave programs for crystalline systems. Comput. Phys. Commun. 1990, 59, 399–415. [Google Scholar] [CrossRef]
- Jamal, M.; Bilal, M.; Ahmad, I.; Jalali-Asadabadi, S. IRelast package. J. Alloys Compd. 2018, 735, 569–579. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initiosimulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Madsen, G.K.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]
- Anderson, M.; Greenwood, K.; Taylor, G.; Poeppelmeier, K. B-cation arrangements in double perovskites. Prog. Solid State Chem. 1993, 22, 197–233. [Google Scholar] [CrossRef]
- Haque, E.; Hossain, M.A. Electronic, phonon transport and thermoelectric properties of Cs2InAgCl6 from first-principles study. Comput. Condens. Matter 2019, 19, e00374. [Google Scholar] [CrossRef]
- Albalawi, H.; Mustafa, G.M.; Saba, S.; Kattan, N.A.; Mahmood, Q.; Somaily, H.H.; Morsi, M.; Alharthi, S.; Amin, M.A. Study of Optical and Thermoelectric Properties of Double Perovskites Cs2KTlX6 (X = Cl, Br, I) for Solar Cell and Energy Harvesting. Mater. Today Commun. 2022, 32, 104083. [Google Scholar] [CrossRef]
- Mahmood, Q.; Ghrib, T.; Rached, A.; Laref, A.; Kamran, M. Probing of mechanical, optical and thermoelectric characteristics of double perovskites Cs2GeCl/Br6 by DFT method. Mater. Sci. Semicond. Process. 2020, 112, 105009. [Google Scholar] [CrossRef]
- Fedorovskiy, A.E.; Drigo, N.A.; Nazeeruddin, M.K. The role of Goldschmidt’s tolerance factor in the formation of A2BX6 double halide perovskites and its optimal range. Small Methods 2020, 4, 1900426. [Google Scholar] [CrossRef]
- Varadwaj, P.R. A2AgCrCl6 (A = Li, Na, K, Rb, Cs) halide double perovskites: A transition metal-based semiconducting material series with appreciable optical characteristics. Phys. Chem. Chem. Phys. 2020, 22, 24337–24350. [Google Scholar] [CrossRef]
- Behera, D.; Sharma, R.; Ullah, H.; Waheed, H.S.; Mukherjee, S.K. Electronic, optical, and thermoelectric investigations of Zintl phase AAg2Se2 (A = Sr, Ba) compounds: A first first-principles approach. J. Solid State Chem. 2022, 312, 123259. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. 2014, 90, 224104. [Google Scholar] [CrossRef]
- Waller, I. Dynamical theory of crystal lattices by M. Born and K. Huang. Acta Crystallogr. 1956, 9, 837–838. [Google Scholar] [CrossRef]
- Choudary, M.Z.; Aldaghfag, S.A.; Yaseen, M.; Nazar, M.; Neffati, R. Study of elastic, structural, thermoelectric and optoelectronics characteristics of Na2YCuX6 (X = Br, Cl) halide double perovskites. Phys. Scr. 2022, 97, 105705. [Google Scholar]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Pugh, S.F.; Pugh, S.F.; Philo, M. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science. Taylor Fr. 1954, 45, 823–843. [Google Scholar]
- Eberhart, M.E.; Jones, T.E. Cauchy pressure and the generalized bonding model for nonmagnetic bcc transition metals. Phys. Rev. 2012, 86, 134106. [Google Scholar] [CrossRef]
- Hossain, K.M.; Hasan, M.Z.; Ali, M.L. Narrowing bandgap and enhanced mechanical and optoelectronic properties of perovskite halides: Effects of metal doping. AIP Adv. 2021, 11, 15052. [Google Scholar] [CrossRef]
- Zener, C. Elasticity and Anelasticity of Metals; University of Chicago Press: Chicago, IL, USA, 1948. [Google Scholar]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Schreiber, E.; Anderson, O.L.; Soga, N.; Bell, J.F. Elastic Constants and Their Measurement. J. Appl. Mech. 1975, 42, 747–748. [Google Scholar] [CrossRef]
- Al-Qaisi, S.; Rai, D.P.; Haq, B.U.; Ahmed, R.; Vu, T.V.; Khuili, M.; Tahir, S.A.; Alhashim, H.H. First-principles investigation of structural, elastic, thermodynamic, electronic and optical properties of lead-free double perovskites halides: Cs2LiYX6 (X = Br, I). Mater. Chem. Phys. 2021, 258, 123945. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, J.; Zhong, X.; Wang, Q.; Zhang, H.; Ren, C.; Wang, J. Revealing the structural, electronic and optical properties of lead-free perovskite derivatives of Rb2SnX6 (X = Cl, Br and I): A theory calculation. Sol. Energy 2019, 190, 272–277. [Google Scholar] [CrossRef]
- Cohen, M.; Ham, F. Electron effective mass in solids—A generalization of Bardeen’s formula. J. Phys. Chem. Solids 1960, 16, 177–183. [Google Scholar] [CrossRef]
- Aslam, F.; Sabir, B.; Hassan, M. Structural, electronic, optical, thermoelectric, and transport properties of indium-based double perovskite halides Cs2InAgX6 (X = Cl, Br, I) for energy applications. Appl. Phys. 2021, 127, 1–12. [Google Scholar] [CrossRef]
- Abraham, J.A.; Behera, D.; Kumari, K.; Srivastava, A.; Sharma, R.; Mukherjee, S.K. A comprehensive DFT analysis on structural, electronic, optical, thermoelectric, SLME properties of new Double Perovskite Oxide Pb2ScBiO6. Chem. Phys. Lett. 2022, 806, 139987. [Google Scholar] [CrossRef]
- Wu, S.; Chen, Z.; Yip, H.-L.; Jen, A.K.-Y. The evolution and future of metal halide perovskite-based optoelectronic devices. Matter 2021, 4, 3814–3834. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. 2006, 73, 045112. [Google Scholar] [CrossRef]
- Kuzmenko, A.B. Kramers–Kronig constrained variational analysis of optical spectra. Rev. Sci. Instrum. 2005, 76, 083108. [Google Scholar] [CrossRef]
- Penn, D.R. Wave-Number-Dependent Dielectric Function of Semiconductors. Phys. Rev. 1962, 128, 2093–2097. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Iqbal, M.W.; Lakra, S.; Mukherjee, S.K. Revealing Excellent Electronic, Optical, and Thermoelectric Behavior of EU Based Euag2y2 (Y = S/Se): For Solar Cell Applications. Comput. Condens. Matter. 2022, 32, e00723. [Google Scholar] [CrossRef]
- Jong, U.-G.; Yu, C.-J.; Kim, Y.-S.; Kye, Y.-H.; Kim, C.-H. First-principles study on the material properties of the inorganic perovskite Rb1–x CsxPbI3 for solar cell applications. Phys. Rev. 2018, 98, 125116. [Google Scholar] [CrossRef]
- Yaseen, M.; Butt, M.K.; Ashfaq, A.; Iqbal, J.; Almoneef, M.M.; Misbah; Iqbal, M.; Murtaza, A.; Larefa, A. Phase transition and thermoelectric properties of cubic KNbO3 under pressure: DFT approach. J. Mater. Res. Technol. 2021, 11, 2106–2113. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, L.-D. Thermoelectric materials: Energy conversion between heat and electricity. J. Mater. 2015, 1, 92–105. [Google Scholar] [CrossRef]
- Wu, G.; Yu, X. Contributions of chemical potential to the diffusive Seebeck coefficient for bulk semiconductor materials. Eur. Phys. J. Plus 2020, 135, 1–15. [Google Scholar] [CrossRef]
- Sajjad, M.; Mahmood, Q.; Singh, N.; Larsson, J.A. Ultralow Lattice Thermal Conductivity in Double Perovskite Cs2PtI6: A Promising Thermoelectric Material. ACS Appl. Energy Mater. 2020, 3, 11293–11299. [Google Scholar] [CrossRef]
- Yaseen, M.S.; Murtaza, G.; Khalil, R.M.A. First principle study of structural, electronic, optical, and transport properties of ternary compounds NaGaX2 (X = S, Se, and Te) in tetragonal chalcopyrite phase. Opt. Quantum Electron. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Stojanovic, N.; Maithripala, D.H.S.; Berg, J.M.; Holtz, M. Thermal conductivity in metallic nanostructures at high temperature: Electrons, phonons, and the Wiedemann-Franz law. Phys. Rev. 2010, 82, 75418. [Google Scholar] [CrossRef]
- Dehkordi, A.M.; Zebarjadi, M.; He, J.; Tritt, T.M. Thermoelectric power factor: Enhancement mechanisms and strategies for higher performance thermoelectric materials. Mater. Sci. Eng. Rep. 2015, 97, 1–22. [Google Scholar] [CrossRef] [Green Version]







| Compound Name | a (Å) | V(a.u3) | Ef (eV/atom) | Tolerance Factor (τ) | Bond Length (Å) |
|---|---|---|---|---|---|
| Rb2InBiCl6 | 11.38 | 1474.04 | −1.57 | 0.95 | In-Cl = 2.97 Bi-Cl = 2.71 |
| Rb2InBiBr6 | 11.87 | 1673.30 | −1.37 | 0.93 | In-Br = 3.05 Bi-Br = 2.88 |
| Cs2KTiCl6 [50] | 11.31 | 0.94 |
| Material Property | Rb2InBiCl6 | Rb2AlInCl6 [38] | Rb2InBiBr6 | Rb2AlInBr6 [38] | |
|---|---|---|---|---|---|
| Optical properties | ε1 (0) | 3.81 | 4.52 | 4.24 | 5.51 |
| n (0) | 1.94 | 2.12 | 2.05 | 2.34 | |
| Transport properties (300 K) | S (µVK) | 335 | 232.13 | 550 | 229.15 |
| σ/τ (Ωms)−1 (1018) | 0.01 | 0.03 | 0.07 | 0.046 | |
| k/τ (W/mKs) (1015) | 0.13 | 9.6 | 0.20 | 10.6 | |
| PF (1010) (W/K2ms) | 0.34 | 0.23 | 0.86 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behera, D.; Mukherjee, S.K. Optoelectronics and Transport Phenomena in Rb2InBiX6 (X = Cl, Br) Compounds for Renewable Energy Applications: A DFT Insight. Chemistry 2022, 4, 1044-1059. https://doi.org/10.3390/chemistry4030071
Behera D, Mukherjee SK. Optoelectronics and Transport Phenomena in Rb2InBiX6 (X = Cl, Br) Compounds for Renewable Energy Applications: A DFT Insight. Chemistry. 2022; 4(3):1044-1059. https://doi.org/10.3390/chemistry4030071
Chicago/Turabian StyleBehera, Debidatta, and Sanat Kumar Mukherjee. 2022. "Optoelectronics and Transport Phenomena in Rb2InBiX6 (X = Cl, Br) Compounds for Renewable Energy Applications: A DFT Insight" Chemistry 4, no. 3: 1044-1059. https://doi.org/10.3390/chemistry4030071