Potential Dependent Structure and Stability of Cu(111) in Neutral Phosphate Electrolyte

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
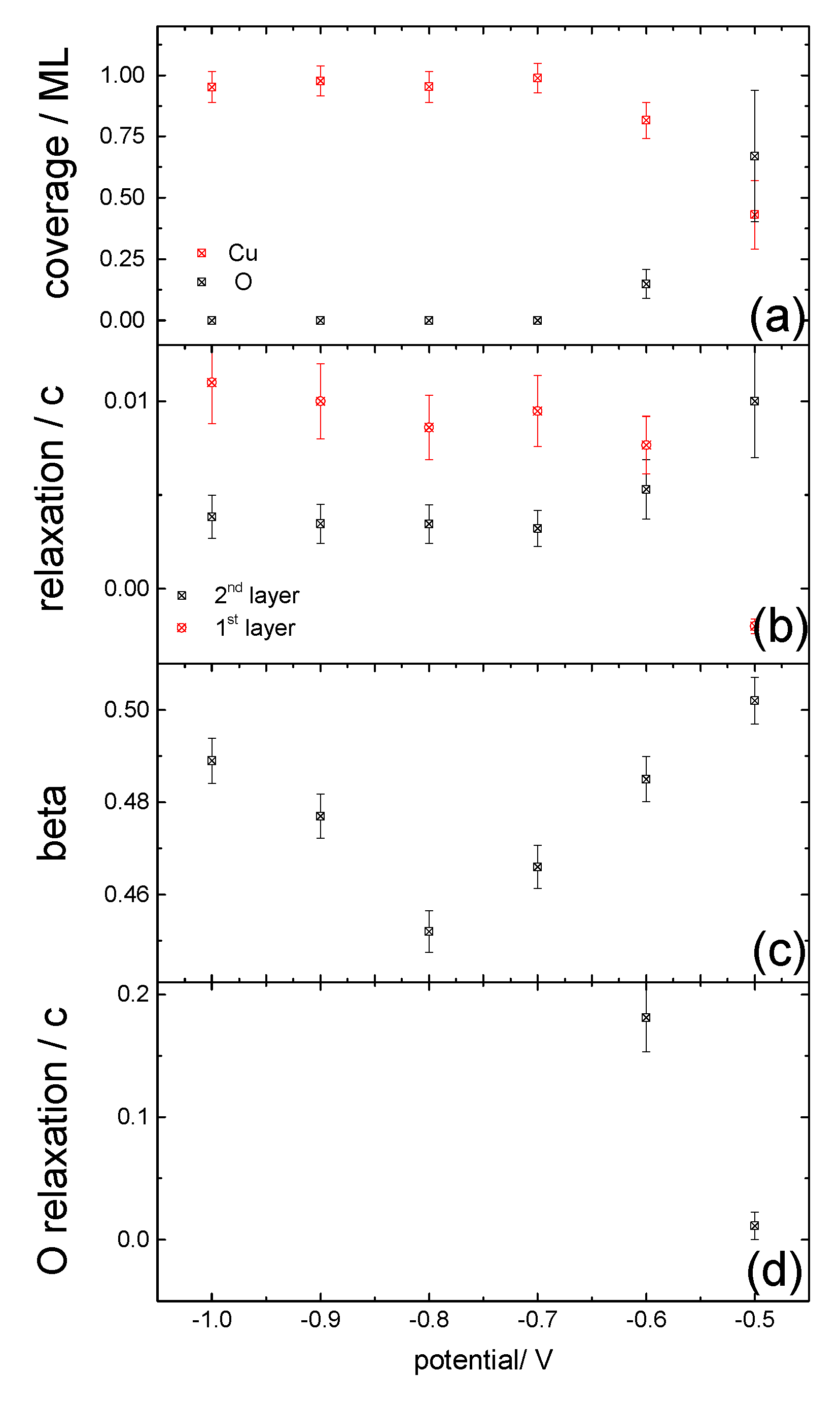{kind=link}
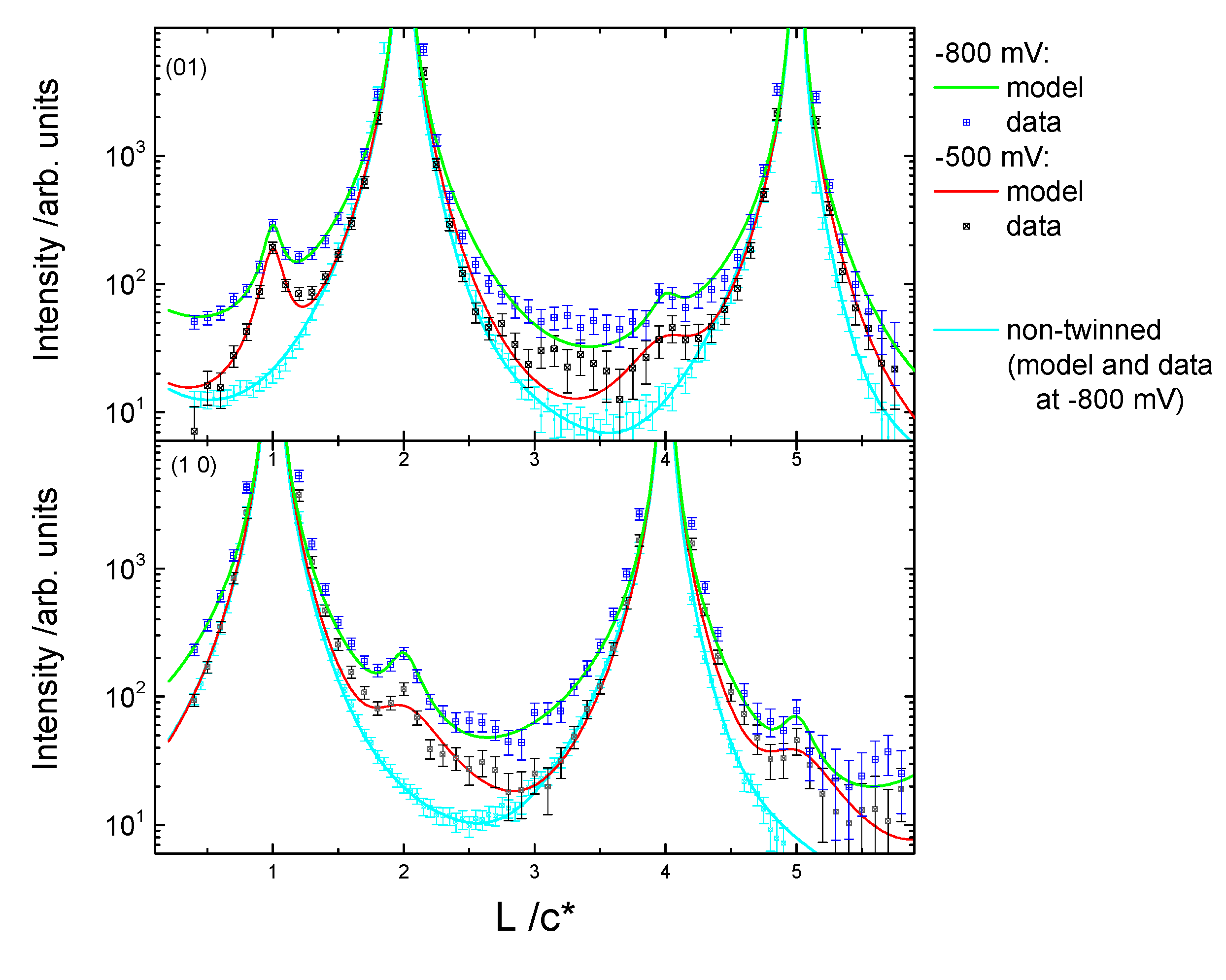{kind=link}
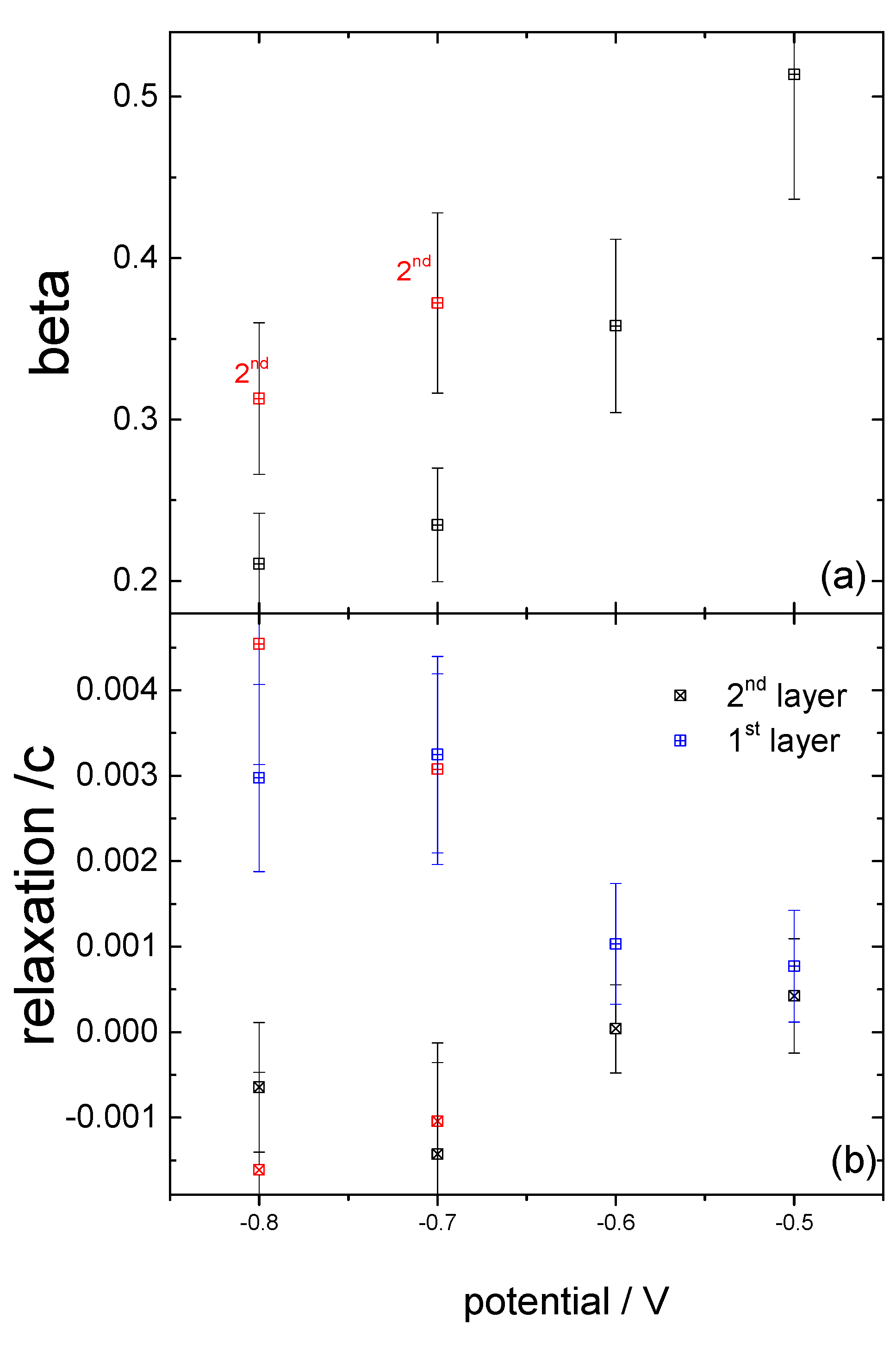{kind=link}
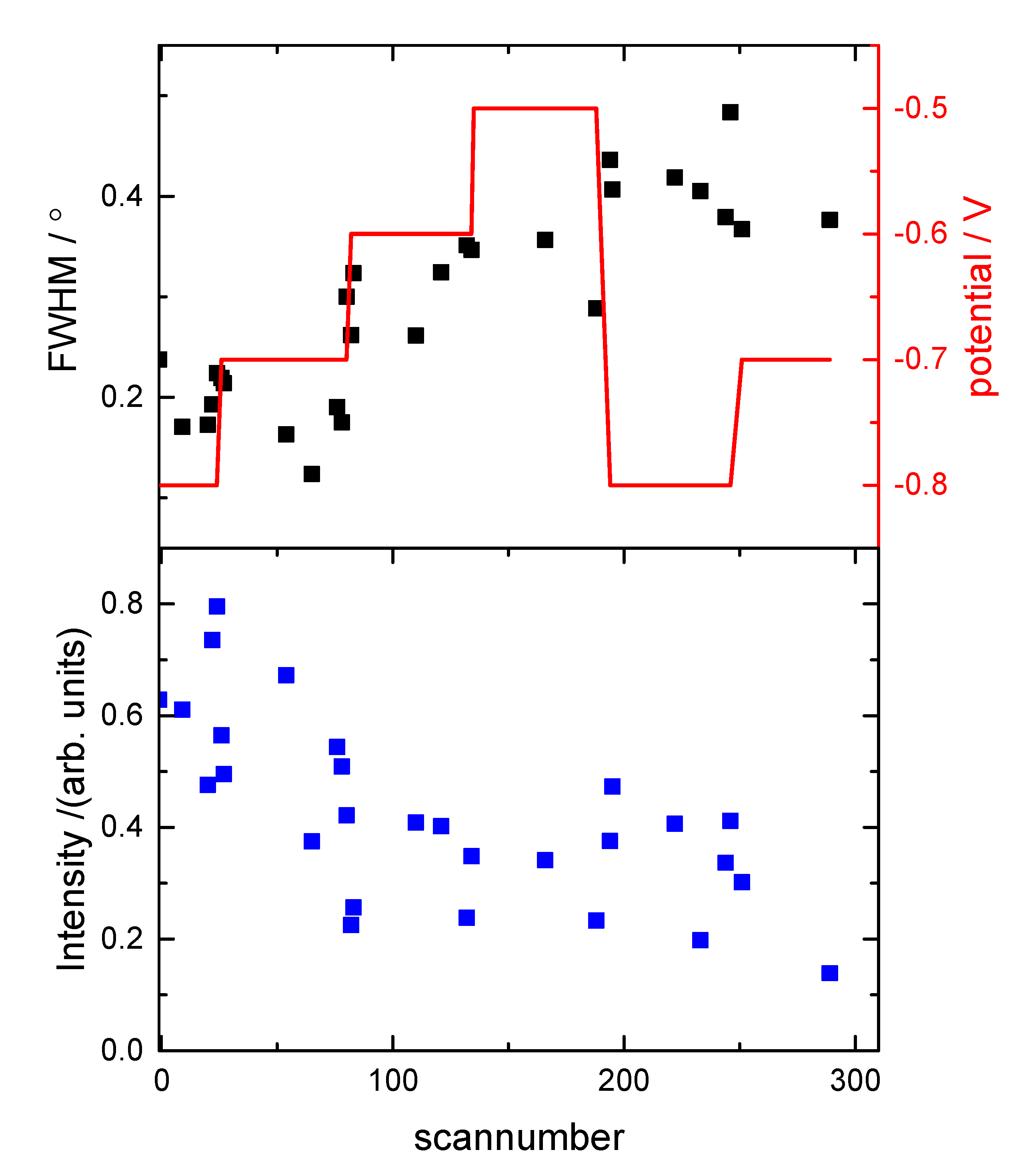{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
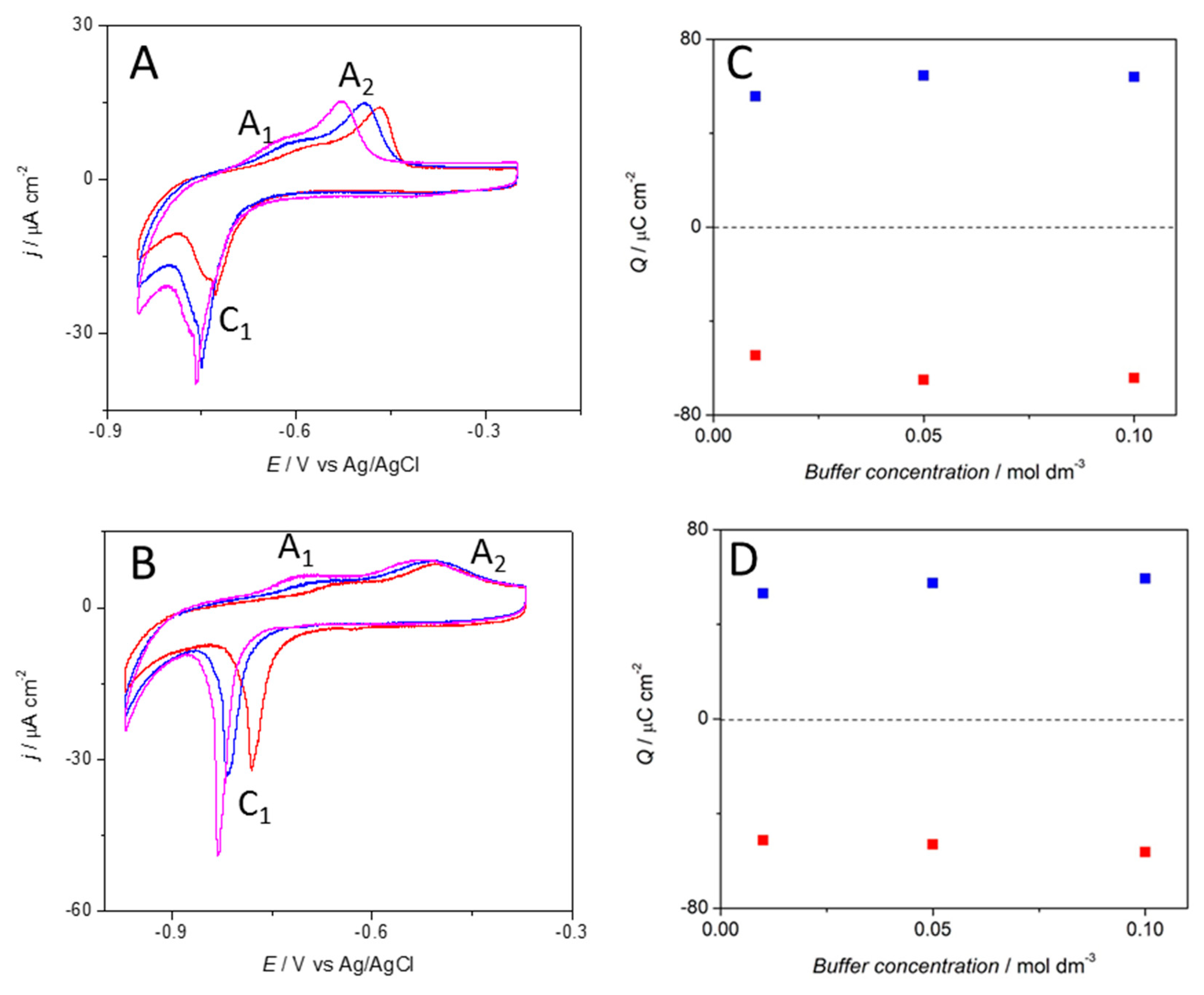
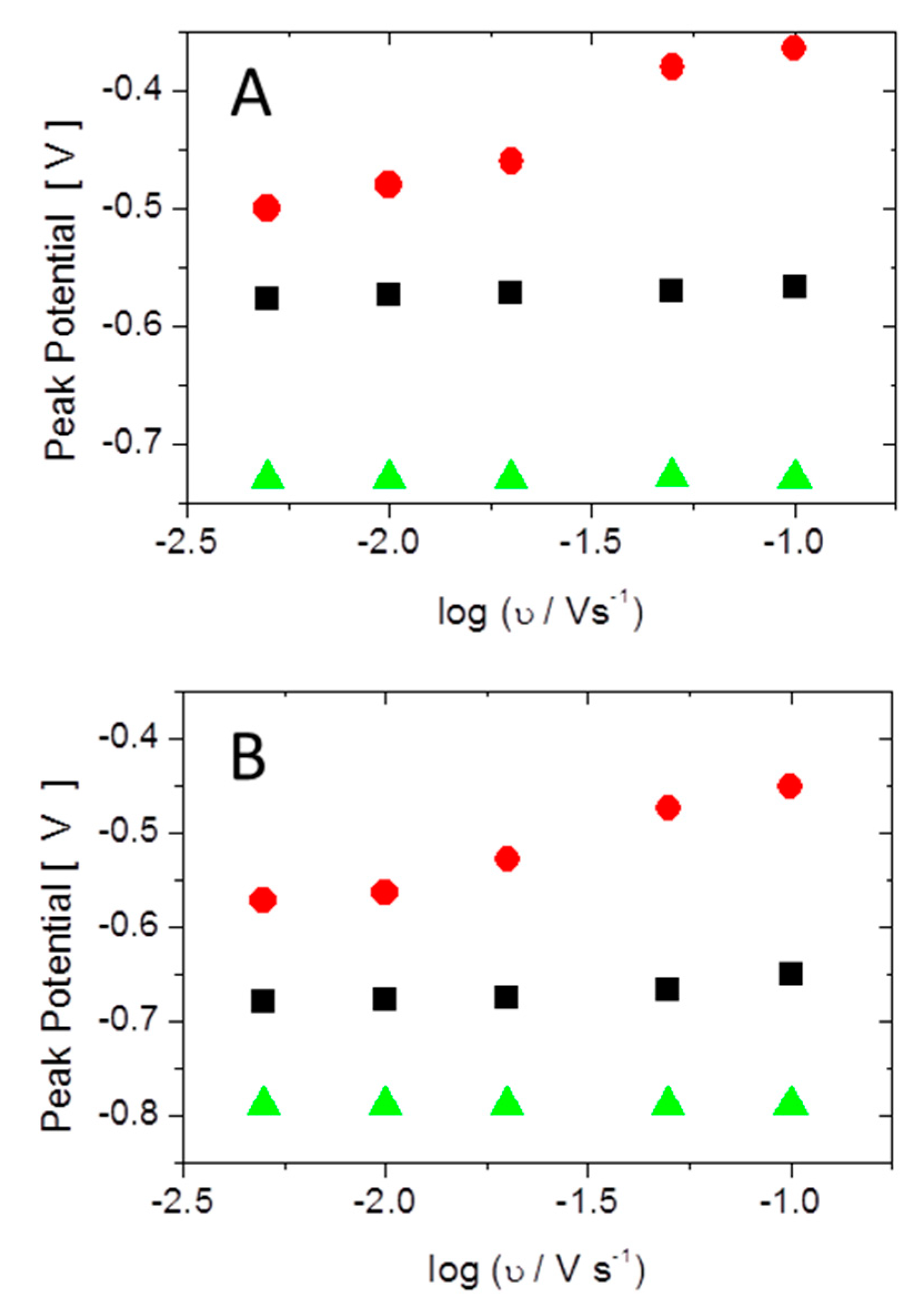
3.1. Electrochemical Characterization
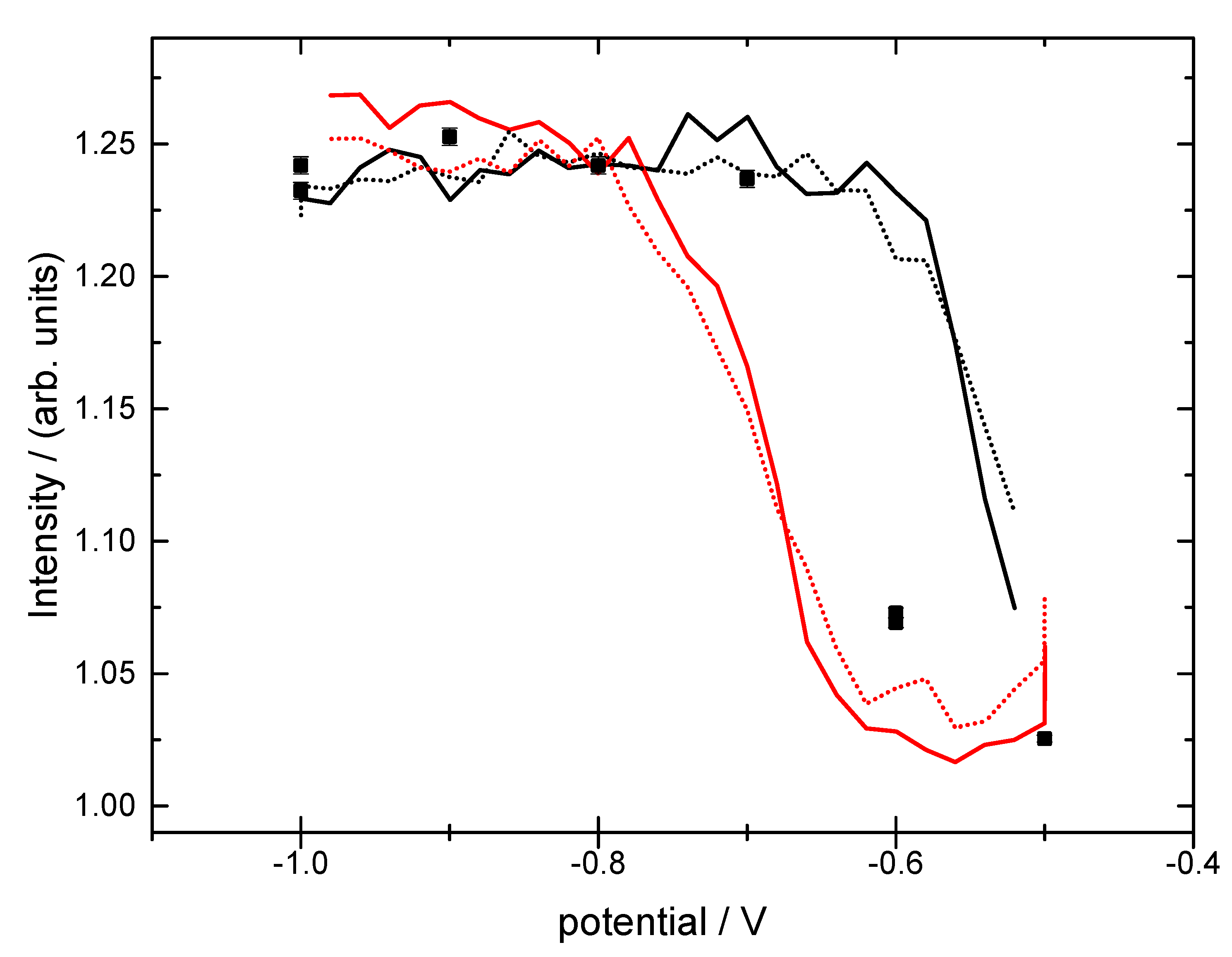
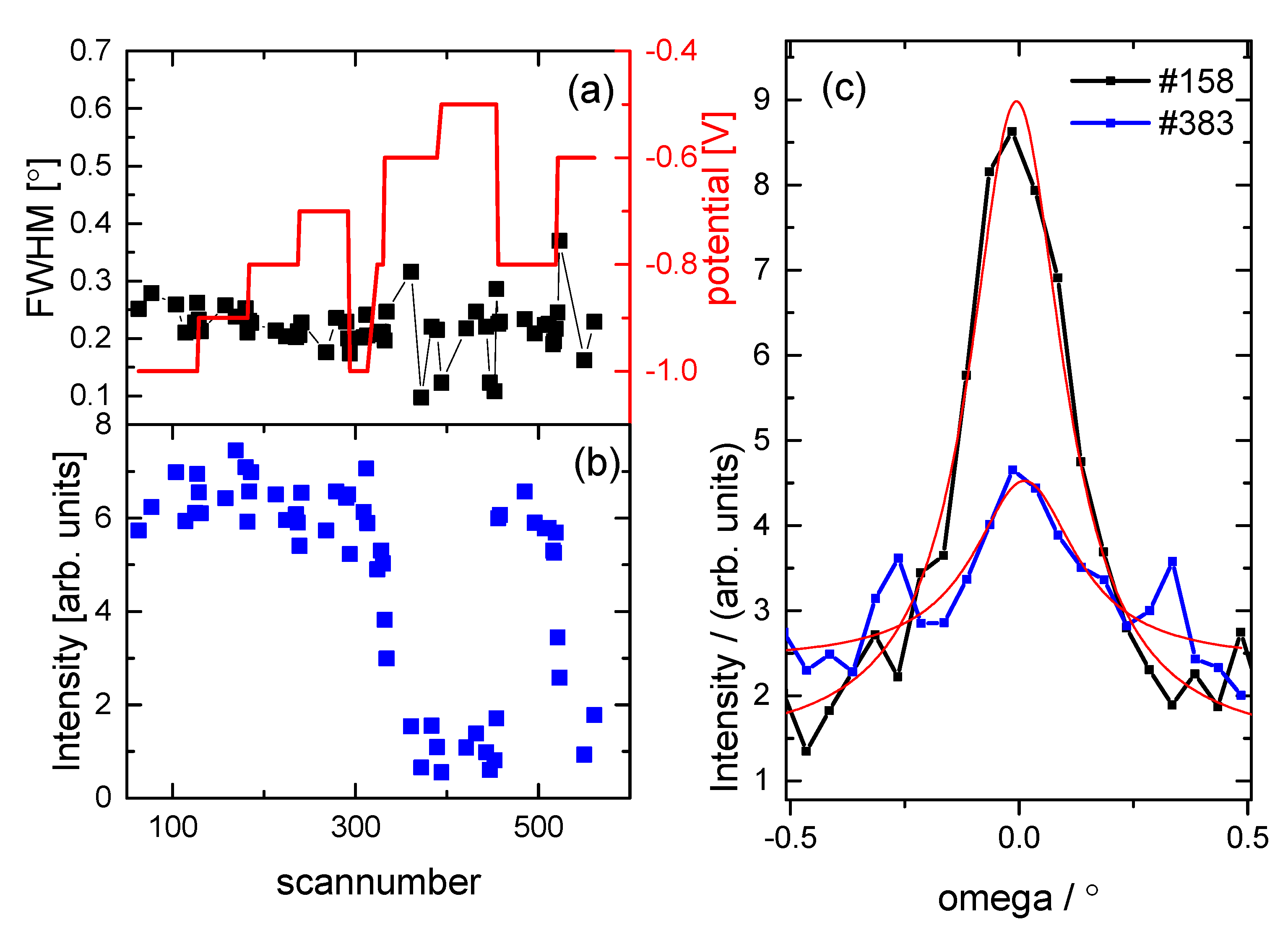
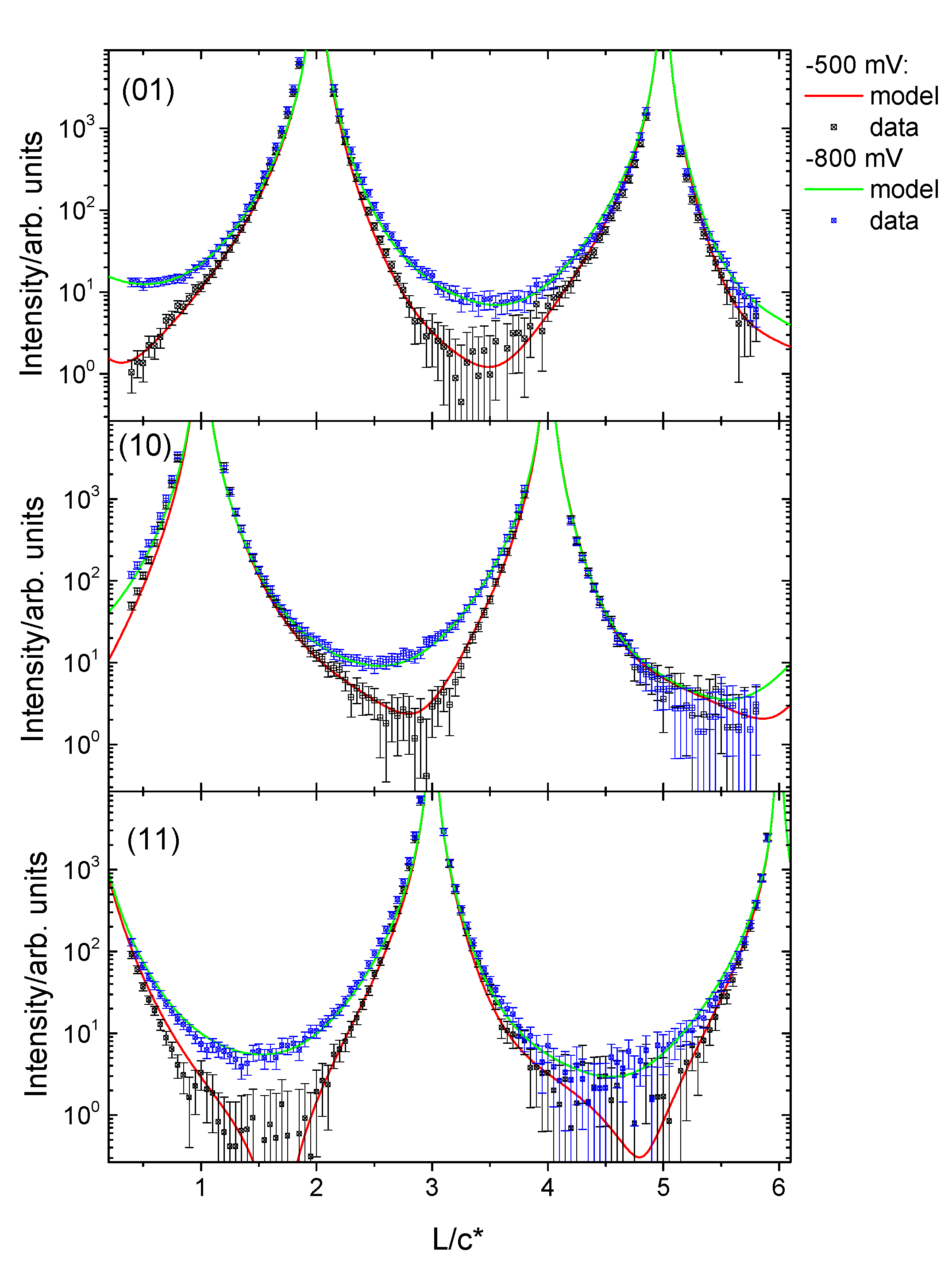
3.2. Structural Characterization
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Larrazábal, G.O.; Martín, A.J.; Pérez-Ramírez, J. Building blocks for high performance in electrocatalytic CO2 reduction: materials, optimization strategies, and device engineering. J. Phys. Chem. Lett. 2017, 8, 3933–3944. [Google Scholar]
- Arán-Ais, R.M.; Gao, D.; Roldan cuenya, B. structure- and electrolyte-sensitivity in CO2 electroreduction. Acc. Chem. Res. 2018, 51, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Engstfeld, A.K.; Maagaard, T.; Horch, S.; Chorkendorff, I.; Stephens, I.E.L. Polycrystalline and Single-Crystal Cu Electrodes: Influence of Experimental Conditions on the Electrochemical Properties in Alkaline Media. Chem. A Eur. J. 2018, 24, 17743–17755. [Google Scholar] [CrossRef] [PubMed]
- Grosse, P.; Gao, D.; Scholten, F.; Sinev, I.; Mistry, H.; Roldan Cuenya, B. Dynamic Changes in the Structure, Chemical State and Catalytic Selectivity of Cu Nanocubes during CO2 Electroreduction: Size and Support Effects. Angew. Chem. 2018, 130, 6300–6305. [Google Scholar] [CrossRef]
- Mistry, H.; Varela, A.S.; Bonifacio, C.S.; Zegkinoglou, I.; Sinev, I.; Choi, Y.-W.; Kisslinger, K.; Stach, E.A.; Yang, J.C.; Strasser, P.; et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 2016, 7, 12123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resasco, J.; Chen, L.D.; Clark, E.; Tsai, C.; Hahn, C.; Jaramillo, T.F.; Chan, K.; Bell, A.T. Promoter Effects of Alkali Metal Cations on the Electrochemical Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2017, 139, 11277–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Duff, C.S.; Lawrence, M.J.; Rodriguez, P. Role of the Adsorbed Oxygen Species in the Selective Electrochemical Reduction of CO2to Alcohols and Carbonyls on Copper Electrodes. Angew. Chem. Int. Ed. 2017, 56, 12919–12924. [Google Scholar] [CrossRef] [PubMed]
- Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M.T.M.; Mul, G.; Baltrusaitis, J. Electrochemical CO2 reduction on Cu2 O-derived copper nanoparticles: controlling the catalytic selectivity of hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. [Google Scholar] [CrossRef] [PubMed]
- Mandal, L.; Yang, K.R.; Motapothula, M.R.; Ren, D.; Lobaccaro, P.; Patra, A.; Sherburne, M.; Batista, V.S.; Yeo, B.S.; Ager, J.W.; et al. Investigating the Role of Copper Oxide in Electrochemical CO2 Reduction in Real Time. ACS Appl. Mater. Interfaces 2018, 10, 8574–8584. [Google Scholar] [CrossRef] [PubMed]
- Verdaguer-Casadevall, A.; Li, C.W.; Johansson, T.P.; Scott, S.B.; McKeown, J.T.; Kumar, M.; Stephens, I.E.L.; Kanan, M.W.; Chorkendorff, I. Probing the Active Surface Sites for CO Reduction on Oxide-Derived Copper Electrocatalysts. J. Am. Chem. Soc. 2015, 137, 9808–9811. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, D.; Lee, J. Electrocatalytic Production of C3-C4 Compounds by Conversion of CO2 on a Chloride-Induced Bi-Phasic Cu2 O-Cu Catalyst. Angew. Chem. Int. Ed. 2015, 54, 14701–14705. [Google Scholar] [CrossRef] [PubMed]
- Eilert, A.; Cavalca, F.; Roberts, F.S.; Osterwalder, J.; Liu, C.; Favaro, M.; Crumlin, E.J.; Ogasawara, H.; Friebel, D.; Pettersson, L.G.M.; et al. Subsurface Oxygen in Oxide-Derived Copper Electrocatalysts for Carbon Dioxide Reduction. J. Phys. Chem. Lett. 2017, 8, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Friebel, D.; Broekmann, P.; Wandelt, K. Electrochemical in situ STM study of a Cu(111) electrode in neutral sulfate containing electrolyte. Phys. Status Solidi 2004, 201, 861–869. [Google Scholar] [CrossRef]
- Lucas, C.A.; Markovic, N.M. Structure Relationships in Electrochemical Reactions. In Encyclopedia of Electrochemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; ISBN 9783527610426. [Google Scholar]
- Gründer, Y.; Lucas, C.A. Surface X-ray diffraction studies of single crystal electrocatalysts. Nano Energy 2016, 29, 378–393. [Google Scholar] [CrossRef]
- Lucas, C.A.; Thompson, P.; Gründer, Y.; Markovic, N.M. The structure of the electrochemical double layer: Ag(111) in alkaline electrolyte. Electrochem. Commun. 2011, 13, 1205–1208. [Google Scholar] [CrossRef]
- Sisson, N.; Gründer, Y.; Lucas, C.A. Structure and Stability of Underpotentially Deposited Ag on Au(111) in Alkaline Electrolyte. J. Phys. Chem. C 2016, 120, 16100–16109. [Google Scholar] [CrossRef]
- Nicklin, C.; Arnold, T.; Rawle, J.; Warne, A. Diamond beamline I07: A beamline for surface and interface diffraction. J. Synchrotron Radiat. 2016, 23, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Vlieg, E. A (2 + 3)-Type Surface Diffractometer: Mergence of the z-Axis and (2 + 2)-Type Geometries. J. Appl. Crystallogr. 1998, 31, 198–203. [Google Scholar] [CrossRef]
- Schlepütz, C.M.; Herger, R.; Willmott, P.R.; Patterson, B.D.; Bunk, O.; Brönnimann, C.; Henrich, B.; Hülsen, G.; Eikenberry, E.F. Improved data acquisition in grazing-incidence X-ray scattering experiments using a pixel detector. Acta Crystallogr. Sect. A Found. Crystallogr. 2005, 61, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Schlepütz, C.M.; Mariager, S.O.; Pauli, S.A.; Feidenhansl, R.; Willmott, P.R. Angle calculations for a (2+3)-type diffractometer: Focus on area detectors. J. Appl. Crystallogr. 2011, 44, 73–83. [Google Scholar] [CrossRef]
- Robinson, I.K. Crystal truncation rods and surface roughness. Phys. Rev. B 1986, 33, 3830–3836. [Google Scholar] [CrossRef]
- Platzman, I.; Brener, R.; Haick, H.; Tannenbaum, R. Oxidation of Polycrystalline Copper Thin Films at Ambient Conditions. J. Phys. Chem. C 2008, 112, 1101–1108. [Google Scholar] [CrossRef]
- Pérez León, C.; Sürgers, C.; Löhneysen, H. Formation of copper oxide surface structures via pulse injection of air onto Cu(111) surfaces. Phys. Rev. B 2012, 85, 035434. [Google Scholar]
- Matsumoto, T.; Bennett, R.A.; Stone, P.; Yamada, T.; Domen, K.; Bowker, M. Scanning tunneling microscopy studies of oxygen adsorption on Cu(1 1 1). Surf. Sci. 2001, 471, 225–245. [Google Scholar] [CrossRef]
- Gamoke, B.; Neff, D.; Simons, J. Nature of PO Bonds in Phosphates. J. Phys. Chem. A 2009, 113, 5677–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pye, C.C.; Rudolph, W.W. An ab Initio, Infrared, and Raman Investigation of Phosphate Ion Hydration. J. Phys. Chem. A 2003, 107, 8746–8755. [Google Scholar] [CrossRef]
- Rose, J.; Flank, A.M.; Masion, A.; Bottero, J.Y.; Elmerich, P. Nucleation and Growth Mechanisms of Fe Oxyhydroxide in the Presence of PO4 Ions. 2. P K-Edge EXAFS Study. Langmuir 1997, 13, 1827–1834. [Google Scholar] [CrossRef]
- Maurice, V.; Strehblow, H.H.; Marcus, P. In situ STM study of the initial stages of oxidation of Cu(111) in aqueous solution. Surf. Sci. 2000, 458, 185–194. [Google Scholar] [CrossRef]
- Kunze, J.; Maurice, V.; Klein, L.H.; Strehblow, H.H.; Marcus, P. In situ scanning tunneling microscopy study of the anodic oxidation of Cu(111) in 0.1 M NaOH. J. Phys. Chem. B 2001, 105, 4263–4269. [Google Scholar] [CrossRef]
- Kunze, J.; Maurice, V.; Klein, L.H.; Strehblow, H.-H.; Marcus, P. In situ STM study of the anodic oxidation of Cu(0 0 1) in 0.1 M NaOH. J. Electroanal. Chem. 2003, 554–555, 113–125. [Google Scholar] [CrossRef]
- Yaguchi, M.; Uchida, T.; Motobayashi, K.; Osawa, M. Speciation of Adsorbed Phosphate at Gold Electrodes: A Combined Surface-Enhanced Infrared Absorption Spectroscopy and DFT Study. J. Phys. Chem. Lett. 2016, 7, 3097–3102. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, R.; García, G.; Koper, M.T.M. Adsorption of phosphate species on poly-oriented Pt and Pt(1 1 1) electrodes over a wide range of pH. Electrochim. Acta 2010, 55, 7961–7968. [Google Scholar] [CrossRef]
- Niaura, G.; Gaigalas, A.K.; Vilker, V.L. Surface-Enhanced Raman Spectroscopy of Phosphate Anions: Adsorption on Silver, Gold, and Copper Electrodes. J. Phys. Chem. B 1997, 101, 9250–9262. [Google Scholar] [CrossRef]
- Jensen, F.; Besenbacher, F.; Lægsgaard, E.; Stensgaard, I. Oxidation of Cu(111): two new oxygen induced reconstructions. Surf. Sci. Lett. 1991, 259, L774–L780. [Google Scholar]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grunder, Y.; Beane, J.; Kolodziej, A.; Lucas, C.A.; Rodriguez, P. Potential Dependent Structure and Stability of Cu(111) in Neutral Phosphate Electrolyte. Surfaces 2019, 2, 145-158. https://doi.org/10.3390/surfaces2010012
Grunder Y, Beane J, Kolodziej A, Lucas CA, Rodriguez P. Potential Dependent Structure and Stability of Cu(111) in Neutral Phosphate Electrolyte. Surfaces. 2019; 2(1):145-158. https://doi.org/10.3390/surfaces2010012
Chicago/Turabian StyleGrunder, Yvonne, Jack Beane, Adam Kolodziej, Christopher A. Lucas, and Paramaconi Rodriguez. 2019. "Potential Dependent Structure and Stability of Cu(111) in Neutral Phosphate Electrolyte" Surfaces 2, no. 1: 145-158. https://doi.org/10.3390/surfaces2010012