1. Introduction
Glycerol is a highly precious and industrially important biomass-derived molecule since it has numerous applications in the pharmaceuticals, cosmetics, and food industries [
1]. Besides that, it is also serving a vital role in the production of various low molecular weight commodity chemicals, e.g., ethylene glycol, 1,2- and 1,3-propanediol, acrylic acid, glycerol carbonate, glyceraldehyde, dihydroxyacetone, etc. [
2,
3,
4]. Glycerol is a co-product of the biodiesel synthesis process and since the production of biodiesel increased tremendously, the glycerol is also produced in huge amounts simultaneously [
3]. Albeit, excess glycerol is often disposed of as a waste, it is not economically beneficial for the biodiesel industries considering the overall cost of the process as well as the negligible value addition to such a vital and renewable chemical. Hence, it is necessary to establish more efficient and alternative pathways to utilize glycerol, for example in the synthesis of value-added chemicals, fuel additives, etc. Considering the excellent source of C
3 carbon backbone, glycerol is used to produce lactic acid, carbonates (linear and cyclic), diols, esters, and epichlorohydrin (ECH), which further reduces the dependency on fossil-derived routes upon their production [
5,
6].
The chlorination of the liquid glycerol to di-chlorinated analogies such as 2,3-dichloro-1-propanol and 1,3-dichloro-2-propanol is a well industrially applied process [
5,
6,
7] (
Scheme 1). This process is a part of Solvay’s Epicerol process, which is applied for the synthesis of industrially important ECH (epoxy resin monomer) where the annual production has reached more than 100 kt [
8,
9]. This process not only replaced the traditional method for the synthesis of ECH, such as the chlorination of propene at high temperatures, but also increased the renewable nature of ECH since the processes use glycerol as one of the reagents in the synthesis.
As shown in
Scheme 1, the chlorination of glycerol is carried out with two moles of hydrochloric acid using Lewis acid catalysts such as carboxylic acid (e.g., acetic acid) to form a mixture of 2, 3-dichloro-1-propanol and 1,3-dichloro-2-propanol. Further, out of these chlorinated derivatives of glycerol, 1,3-dichloro-2-propanol is converted to ECH following the alkaline hydrolysis process [
7]. The synthesis of these chlorinated analogues of the glycerol and their further application is only limited to the ECH synthesis, whereas these analogues are not explored for other fruitful applications. 2-chloro-1,3-propanol, one of the mono-chlorinated analogues of glycerol, is considered as a waste in the Epicerol process. In this case, 2-chloro-1,3-propanol cannot be converted to the di-chlorinated species because the chlorine at the beta position (β form) inhibits further chlorination. Proto et al. proposed that 2-chloro-1,3-propanol can be converted to glycidol, which is also considered a highly active and vital chemical entity in polymer, rubber, as well as in dye industries [
7]. In other words, identical to the synthesis of ECH from glycerol, the processing of 2-chloro-1,3-propanol for glycidol synthesis can emerge as a new alternative for the existing allyl alcohol epoxidation using an H
2O
2 precursor and titanium silicate catalyst, TS-1 [
10]. Hence, this integrated approach for the synthesis of ECH and glycidol can increase the atom economy as well as the overall sustainability of the Epicerol process. However, besides synthesis of the ECH and glycidol, it is necessary to implement more available applications of the chlorinated analogs of the glycerol to enhance the applicability of a surplus amount of glycerol from the biodiesel industries.
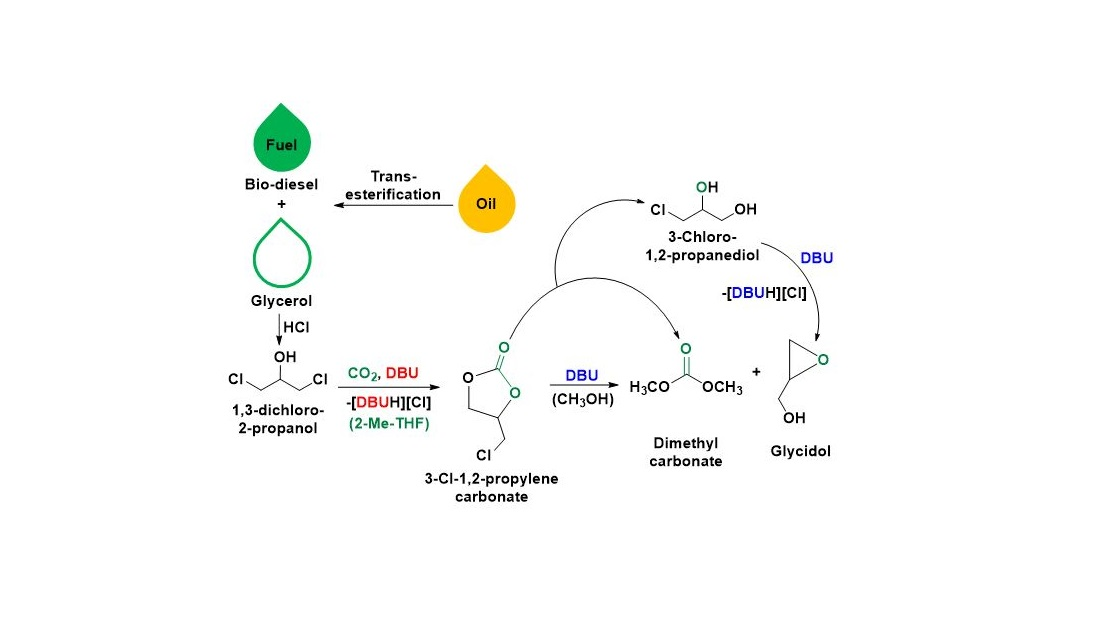
In this work, we report the integrated method for the valorization of 1,3-dichloro-2-propanol to dimethyl carbonate (DMC) and glycidol through an organic superbase involving CO
2 capture and a base-catalyzed process. Being less toxic as well as having versatile reactivity, the use of DMC as a reagent as well as a solvent in various organic transformations is considered a green, sustainable, and environmentally friendly approach. Besides having combined the functionality of CO
2 and the methyl group, DMC is successfully utilized for the valorization of bio-based building blocks to value-added chemicals and fuels as well as for the derivatization of cellulose-to-cellulose methyl carbonate [
11,
12]. Considering the vital role of DMC in synthetic chemistry, several catalytic processes with and without the use of CO
2 have been developed for the DMC synthesis where some of the methods have been commercialized [
13,
14].
In this reaction approach, the CO
2 molecule was initially activated through the equivalent interaction between 1,3-dichloro-2-propanol and organic superbase diazabicyclo [5.4.0] undec-7-ene (DBU), where the resultant 3-chloro-1,2-propylenecarbonate was further transesterified with methanol to form DMC and glycidol (
Scheme 2).
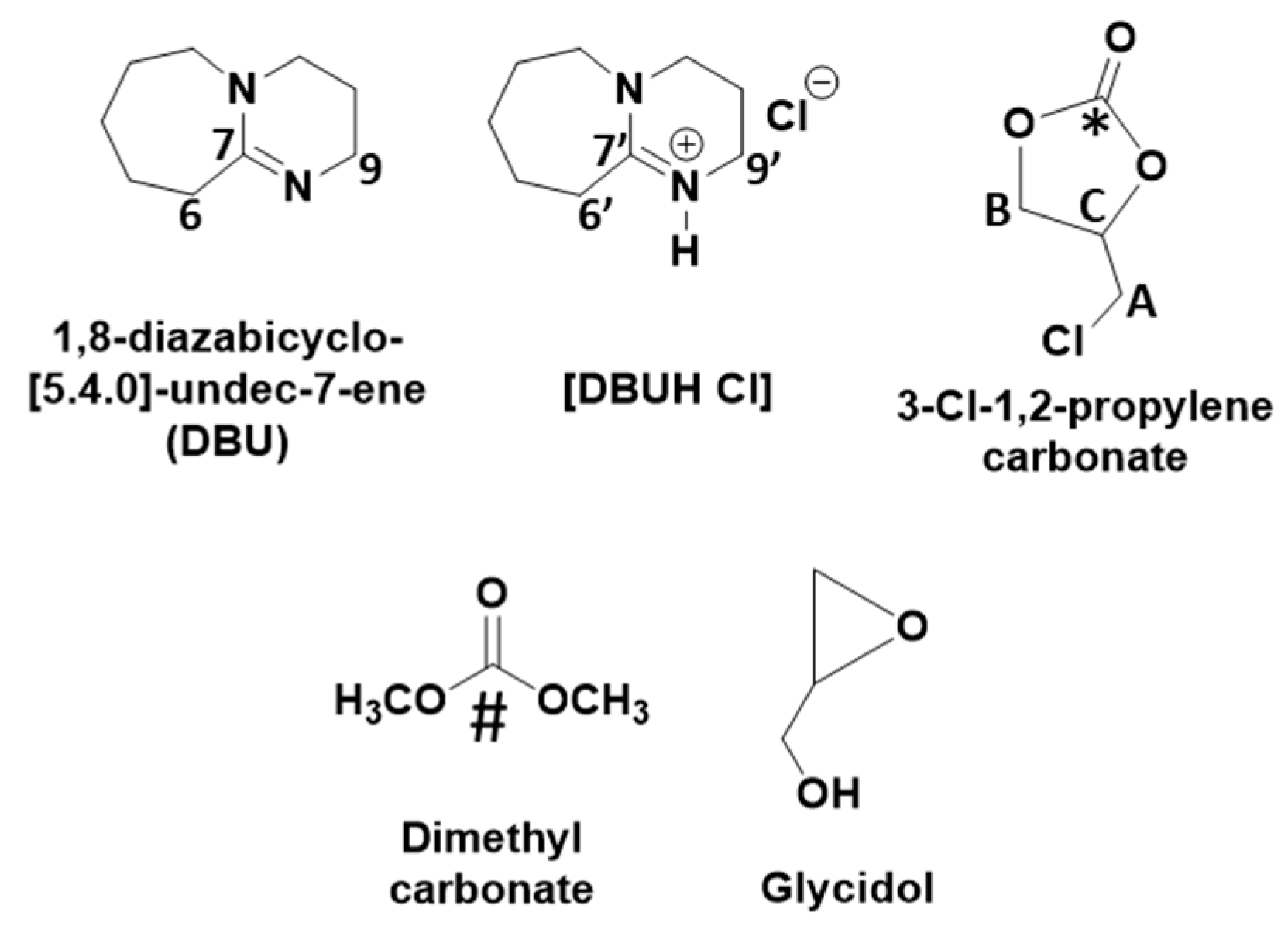
The DBU superbase (
pka = 23)-mediated activation of CO
2 is a well-studied process where it not only emerged as a new and greener pathway for the up-gradation of CO
2, but it also introduced new reversible solvent media called switchable ionic liquid (SIL), which was efficiently used for the processing of lignocellulosic biomass such as wood and cellulose esters synthesis [
15,
16]. In this actual case, DBU initially deprotonates alcohols (R-OH) where the resultant alkoxide anion equivalently reacts with CO
2 to form [DBUH][ROCO
2] salt in the form of an SIL [
17,
18]. Besides the CO
2 capture, the synthesis of SIL has also been further explored upon the synthesis of linear as well as cyclic carbonates, methyl formate, as well as acrylic plastic precursors synthesis [
19,
20,
21,
22]. In this regard, the synthesis of cyclic carbonates using 1,2 chlorohydrins has been also previously reported for the synthesis of various cyclic carbonates [
20]. Similar work of the synthesis of cyclic carbonates now mimicked in this report in the case of the synthesis of the 3-chloro-1,2-propylenecarbonate where 1,3-dichloro-2-propanol is assumed as the 1,2 chlorohydrin. After the complete synthesis, the recovery method has also been further set up for the separation of DMC and glycidol following solvent extraction techniques. In addition, as shown in
Scheme 2, the overall process of the synthesis of DMC and glycidol was also accompanied by the formation of [DBUH][Cl] salt, which was further separated from the reaction mixture and further used for the recovery of DBU. The progress of the reaction as well as the purity of the recovered chemical species was confirmed by means of NMR analysis techniques.
3. Results
DMC and glycidol synthesis proceeded via the integrated two-step process approach. In this case, initially, the synthesis of 3-chloro-1,2-propylenecarbonate was prepared through the equivalent interaction of 1,3-dichloro-2-propanol, DBU, and CO
2, at room temperature. Further, the DMC along with glycidol were synthesized via a base-catalyzed transesterification of 3-chloro-1,2-propylenecarbonate in methanol. The synthesis of 3-chloro-1,2-propylenecarbonate was initially carried out in DMSO as a solvent and a similar synthesis was further studied in other solvents such as 2-Me-THF. After the complete addition of DBU in the reaction mixture containing DMSO and 1,3-dichloro-2-propanol under CO
2 bubbling, the composition of the resultant reaction mixture was confirmed by one- as well as two-dimensional NMR analysis. The
1H and
13C NMR spectra of the reaction mixture are shown in
Figure 2 and supporting information
Figure S2, respectively. As shown in
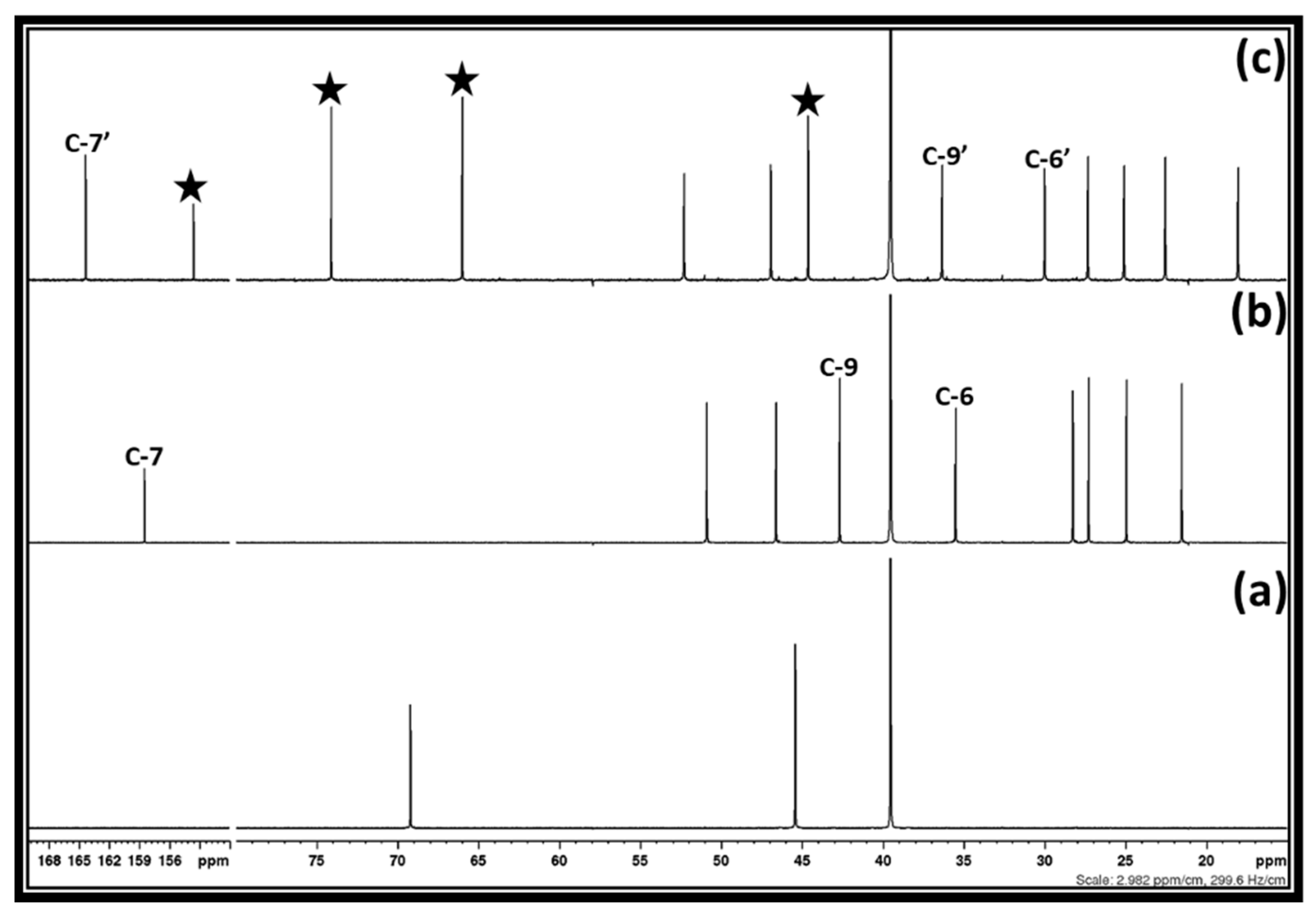
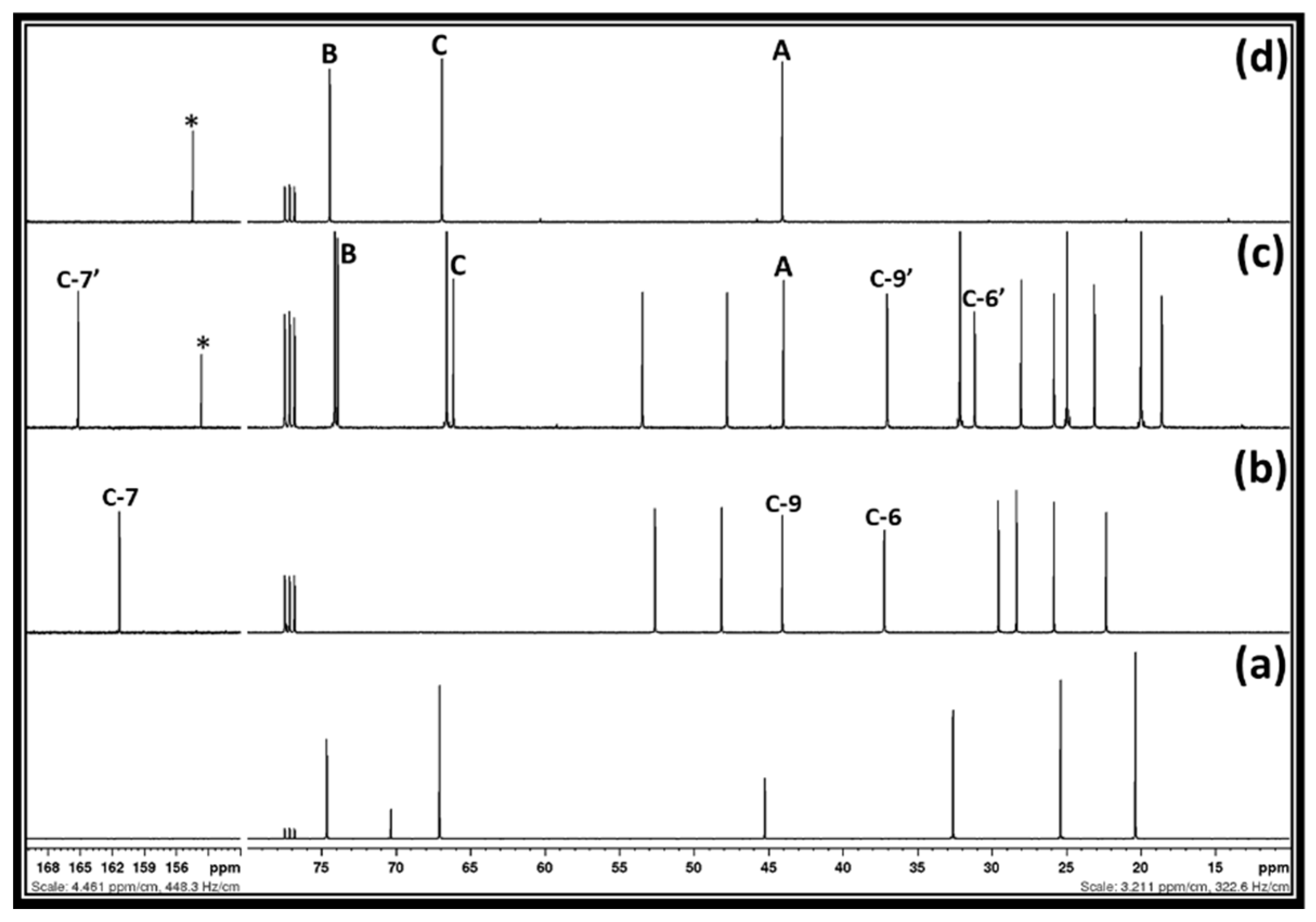
Figure 2, after the interaction between 1,3-dichloro-2-propanol, DBU, and CO
2, DBU as well as 1,3-dichloro-2-propanol were completely consumed in the reaction mixture as their corresponding signals for the carbon atoms disappeared. In this case, the characteristics signals for the carbon atoms at positions six, seven, and nine, respectively, in the molecular DBU disappeared and new shielded signals for the carbon atoms at position six and seven (C-6′ and C-7′) as well as a de-shielded signal for carbon atom at position nine (C-9′), respectively, were observed.
This observation represents that the sp
2-N atom in the DBU molecule became protonated, which is in agreement with the previous reports [
20]. Besides the signals for the protonated DBU, the signals for the unknown chemical species were also observed in the
13C NMR analysis (shown by a filled star).
1H NMR spectra also depict that the characteristics signals for the protons in both 1,3-dichloro-2-propanol and DBU molecules, respectively, disappeared, while signals for the unknown chemical species as well as protonated DBU appeared. As described previously, the DBU molecule is popularly known as a superbase to activate the CO
2 molecule through the formation of SIL in the presence of proton sources such as water or alcohol. Besides that, it was also previously confirmed that the equivalent interaction between 1, 2-halohydrin and DBU molecule in the presence of CO
2 results in the formation of cyclic carbonate [
20]. Since 1,3-dichloro-2-propanol molecule has a similar structure to the 1,2-halohydrin, i.e., –OH and halide groups are attached to the adjacent carbon atoms, its interaction with DBU and CO
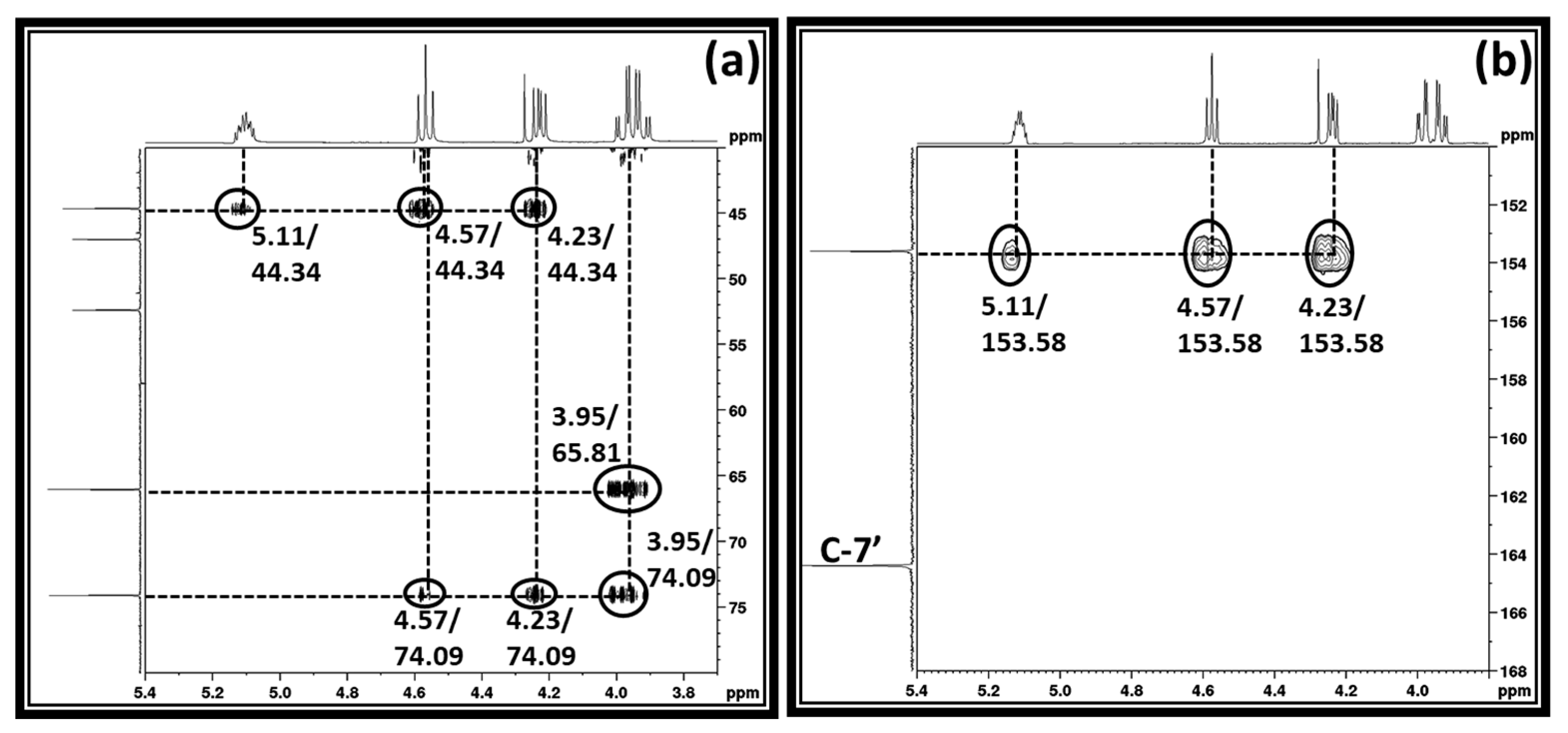
2 molecules could result in the formation of cyclic carbonate such as 3-chloro-1,2-propylenecarbonate. To confirm the formation of 3-chloro-1,2-propylenecarbonate, the reaction mixture obtained was analyzed using two dimensional (2D) HMBC (Heteronuclear Multiple Bond Correlation), HSQC (Heteronuclear Single Quantum Coherence), and COSY (Correlated Spectroscopy) NMR analysis techniques and corresponding spectra are shown in
Figure 3 and supporting information is shown in
Figure S3a,b.
The HSQC NMR analysis shows that the protons in an unknown chemical species with chemical shifts 3.95, 4.23, 4.57, and 5.11 ppm, respectively, belong to the proton–carbon correlation signals with their corresponding carbon atoms (supporting information S3a). In the case of COSY NMR analysis, the proton with the chemical shift 5.11 ppm proton–proton correlated with all the remaining protons, whereas the protons at 4.23 and 4.57 ppm did not show any correlation with the proton resonating at 3.95 ppm (supporting information S3b). This suggests that the distribution of the protons in this unknown chemical species is identical to the 1,3-dichloro-2-propanol that was used in the synthesis. The HMBC NMR analysis showed that protons with chemical shifts 4.23, 4.57, and 5.11, respectively, are in correlation with the carbon atom resonating at 153.5 ppm. The signal for the carbon atom at 153.5 ppm was a new one and it usually belongs to the carbon atom in a carbonyl group. This observation suggests that the activation of the CO
2 molecule took place through the equivalent interaction between the reagents applied in the synthesis. Since the [DBUH]
+ cation forms in the reaction composition, the formation of this cation proceeds through the activation of CO
2 by the DBU superbase. In this case, as shown in
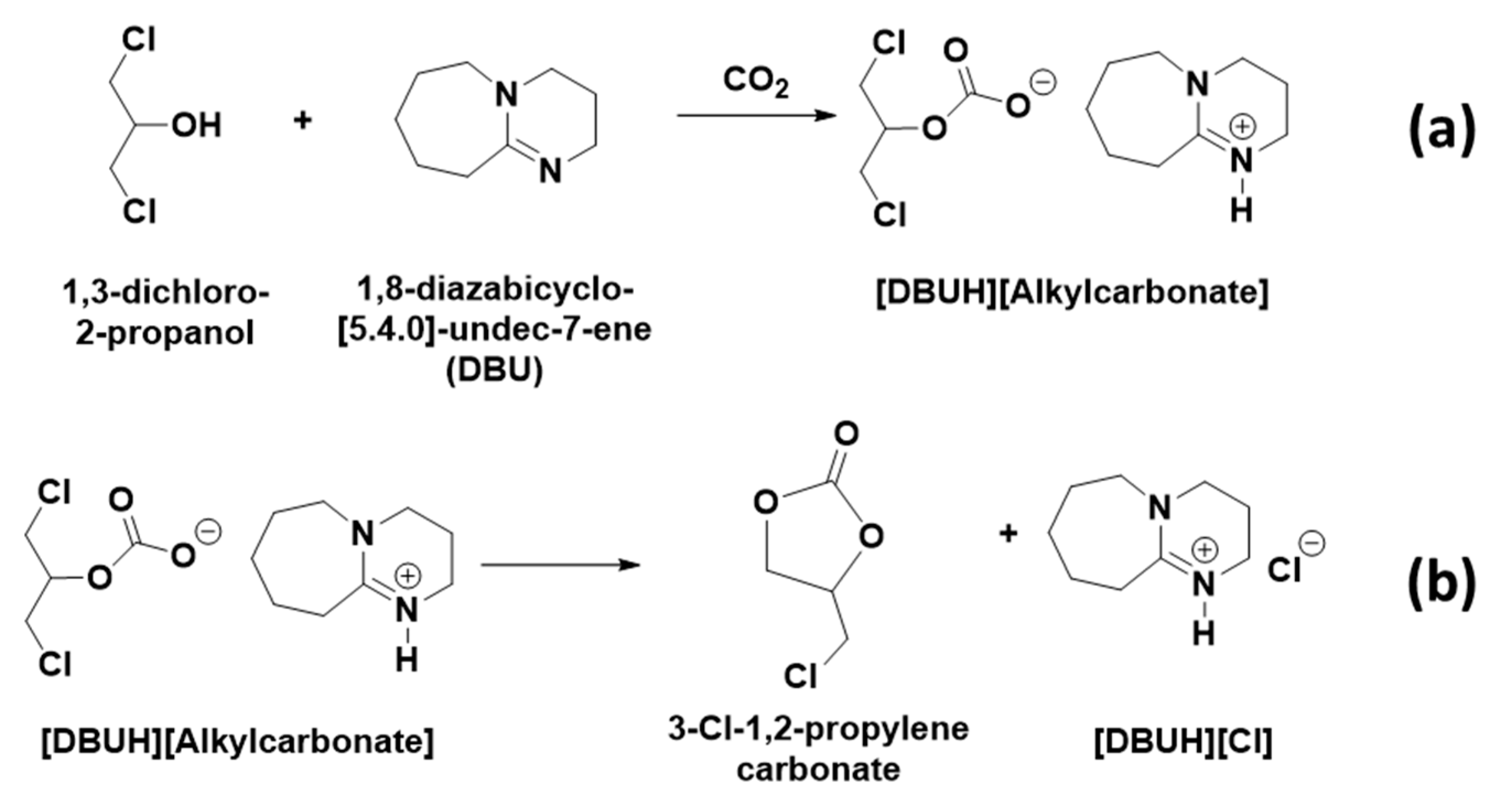
Scheme 3a, DBU removed the proton from the –OH group in 1,3-dichloro-2-propanol and the resultant alkoxide species reacted with CO
2 and alkyl carbonate anion, whereupon the [DBUH]
+ cation formed in the reaction mixture. However, since the COSY and HMBC NMR analysis suggests that the protons with the chemical shifts 4.23, 4.57, and 5.11, respectively, are adjacent to each other and in long correlation with 153.5 ppm, further consecutive cyclization in alkyl carbonate anion took place, which further allowed the formation of 3-chloro-1,2-propylenecarbonate along with the release of [DBUH][Cl] salt (
Scheme 3b). Hence, similar to the previously reported cyclic carbonate synthesis, the equivalent interaction between 1,3-dichloro-2-propanol, DBU, and CO
2 results in a 3-chloro-1,2-propylenecarbonate, which formed in the process [
20]. This DBU mediated fixation of CO
2 in the form of 3-chloro-1,2-propylenecarbonate was carried out at room temperature and atmospheric pressure. Therefore, this method can be considered safer and sustainable compared to epichlorohydrin encompassed with high energy-consuming catalytic approaches. Even though both 1,3-dichloro-2-propanol and epichlorohydrin are derived from the hydro-chlorination of glycerol, the processing with epichlorohydrin in 3-chloro-1,2-propylenecarbonate synthesis is not safe considering its toxic and flammable nature.
After the synthesis of 3-chloro-1,2-propylenecarbonate, the reaction mixture was further explored in terms of its recovery via water and ethyl acetate-involved solvent extraction methods. In this case, after the separation of [DBUH][Cl] salt using ethyl acetate, water was further used to separate DMSO from 3-chloro-1,2-propylenecarbonate, which remained in the organic phase. After the removal of ethyl acetate, a 68% recovery of 3-chloro-1,2-propylenecarbonate was achieved. This represents that even though 3-chloro-1,2-propylenecarbonate is insoluble in water due to DMSO, a part of it remained in the aqueous phase. Further, a similar synthesis of 3-chloro-1,2-propylenecarbonate was carried out in 2-Me-THF solvent. During the bubbling of CO
2 and the simultaneous addition of DBU in the reaction mixture, containing 1,3-dichloro-2-propanol in a 2-Me-THF, it was observed that a white crystalline solid precipitate was forming and became separated in the reaction mixture. The
13C NMR analysis of the reaction mixture along with the white precipitate was carried out and the obtained spectra are shown in
Figure 4.
Figure 4 depicts that the 3-chloro-1,2-propylenecarbonate as well as [DBUH][Cl] salt were forming after an equivalent interaction between 1,3-dichloro-2-propanol, DBU, and CO
2 when 2-Me-THF was used as the solvent during the synthesis. Recently, Jupke et al. reported that 2-Me-THF has a higher CO
2 solubility in the reaction system than water under identical experimental conditions [
23]. Jessop and Matsuda et al. showed that 2-Me-THF has lower values of Kamlet-Taft parameters such as polarizability, π* (0.5–1.1), which further allowed for the higher solubility of hydrophobic CO
2 in 2-Me-THF than water [
24,
25]. Matsuda et al. also further reported that CO
2 expanded 2-Me-THF, and other bio-based solvents turned out to be an excellent solvent media for biotransformation. The author explained that with an increase in the CO
2 pressure, the polarizability (π*) value of the 2-Me-THF linearly decreased as a result of the higher solubility of CO
2, which further increased the transformation rate in this CO
2 expanded solvent system [
26].
Therefore, similar to DMSO, 2-Me-THF can be used as a solvent in the synthesis of 3-chloro-1,2-propylenecarbonate. In this case, 2-Me-THF is preferred more considering its renewable nature and this solvent is already referred to as an alternative to THF and other organic solvents [
27,
28]. After the completion of the reaction, 2-Me-THF was further removed by evaporation and [DBUH][Cl] salt was separated from 3-chloro-1,2-propylenecarbonate using ethyl acetate solvent and a filtration technique. Further, 93% of the 3-chloro-1,2-propylenecarbonate was recovered when ethyl acetate was removed from the organic phase. Hence, the use of 2-Me-THF in the synthesis not only facilitated the separation of components from the reaction mixture but also further allowed a higher level of recovery of 3-chloro-1,2-propylenecarbonate.
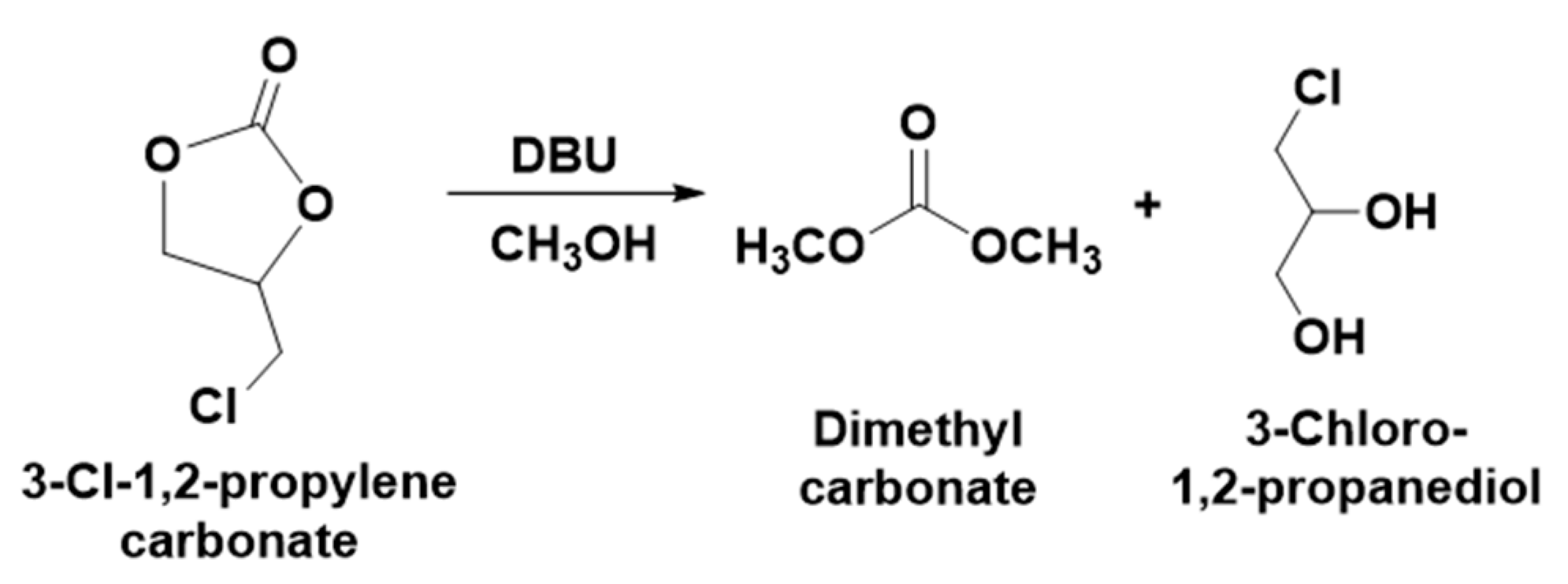
To valorize 3-chloro-1,2-propylenecarbonate, it was further explored in the base-catalyzed transesterification in methanol where DBU was used as a base and the synthesis was carried out at different temperatures. The current catalytic approaches for the synthesis of 3-chloro-1,2-propylenecarbonate from ECH are considered as only an ideal example to demonstrate the valorization of CO
2 upon the synthesis of cyclic carbonates. Hence, 3-chloro-1,2-propylenecarbonate remains underutilized and needs to be upgraded to value-added chemical entities considering the value of the active form of CO
2 in the molecule. The concept of the transesterification of 3-chloro-1,2-propylenecarbonate under alkaline conditions was designed based on the previous reports where the cyclic carbonates such as ethylene carbonates, catechol carbonate, etc. were used to synthesize various aliphatic carbonates [
13,
29]. As shown in
Scheme 4, the reaction of the alkaline transesterification involved the interaction of methanol with 3-chloro-1,2-propylenecarbonate and results in the formation of the DMC and 3-Chloro-1, 2-propanediol.
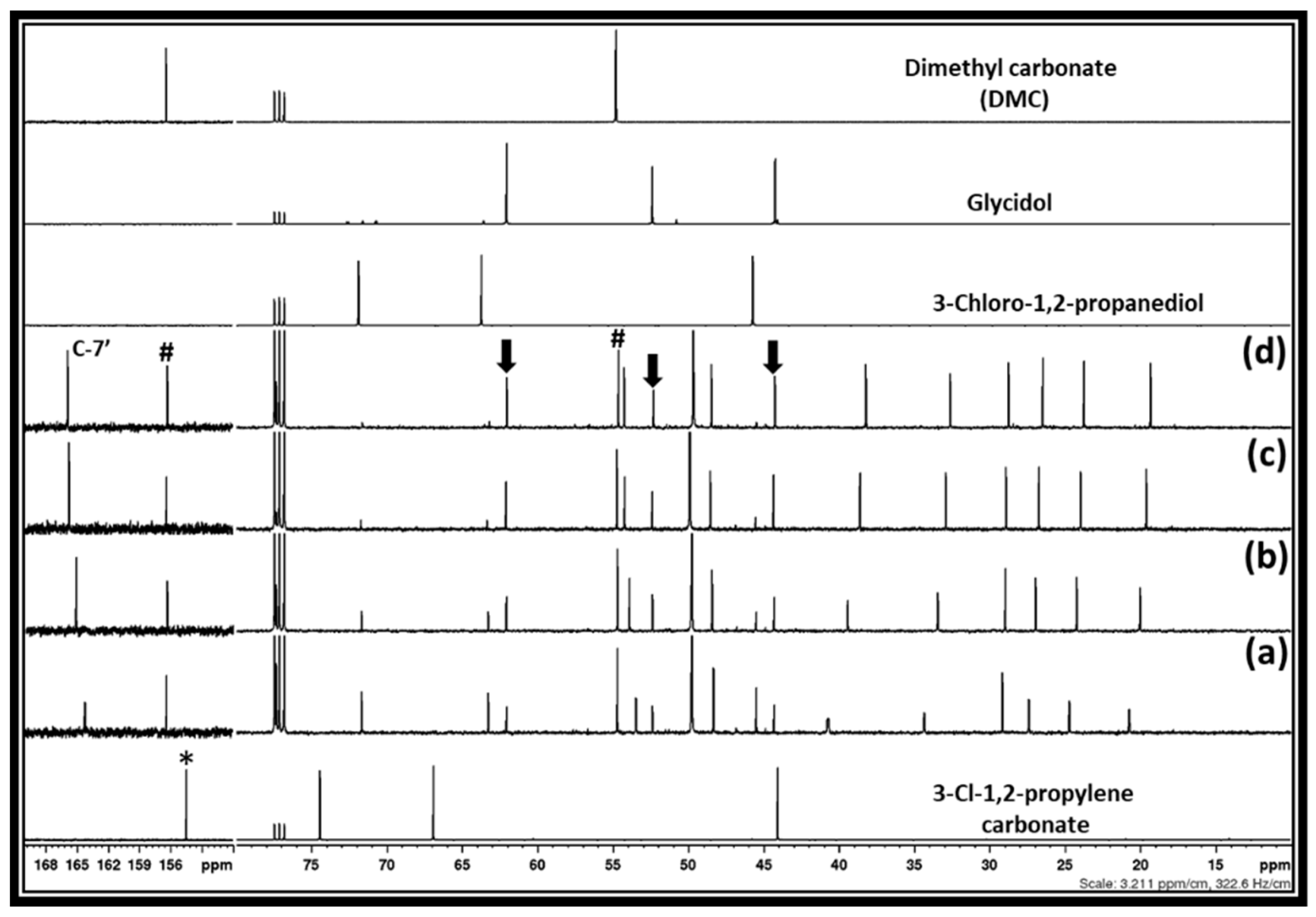
After the interaction of the equivalent amounts of DBU and 3-chloro-1,2-propylenecarbonate in methanol for 30 min, it was observed that the expected products such as DMC and 3-chloro-1, 2-propanediol formed in the reaction mixture (
Figure 5b). However, besides the signal for these chemical species, new signals at 44.3, 52.4, and 62.1 ppm were also observed. The reaction mixture was stirred for various reaction times such as 2, 6 and 18 h and it was observed that these newly observed signals belong to unknown chemical species, the amounts of which gradually increased. Simultaneously, the signal belonging to 3-chloro-1, 2-propanediol steadily decreased as the reaction time increased. Besides that, it was also observed that the chemical shifts for the carbon atoms of DBU, especially at positions six and nine, respectively, were shielded, whereas the carbon atom at position seven became de-shielded under the given reaction period. Considering the changes in the chemical shifts for the carbon atoms in the DBU molecule, it is evident that the [DBUH]
+ cation is gradually forming in the reaction mixture.
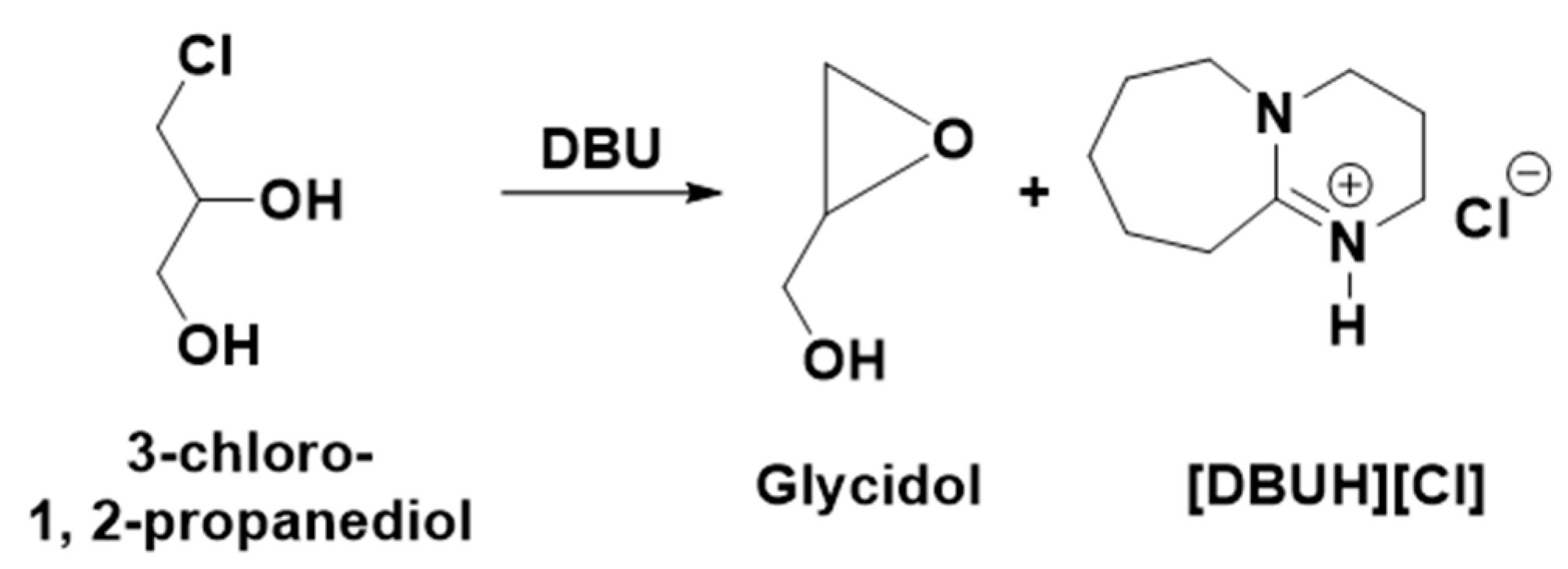
It was previously reported that the synthesis of epoxides such as epichlorohydrin and glycidol from the hydro-chlorinated analogs of glycerol such as 1,3-dichloro-2-propanol and 3-Chloro-1, 2-propanediol, respectively, is a base-catalyzed reaction where an equivalent interaction of the base with either of these chlorinated species results in the formation of the corresponding epoxides [
5,
7]. In the present work, since the transesterification of 3-chloro-1,2-propylenecarbonate was carried out with an equivalent amount of DBU, the possibility is that the in situ-formed 3-chloro-1, 2-propanediol can transform further to an oxiranic function-comprising molecule, i.e., glycidol through the release of a Cl atom with a DBU base (
Scheme 5). To confirm the glycidol formation, the NMR spectra of the reaction mixture after the 18 h and commercially available glycidol were compared and it was observed that identical signals related to glycidol (shown by downward arrow) were observed. Since the signal related to the [DBUH]
+ cation was also observed, this also confirmed that the formation of glycidol has occurred through the formation of [DBUH][Cl] salt in the reaction composition. Hence, the base-catalyzed transesterification of 3-chloro-1,2-propylenecarbonate in methanol results in the formation of DMC and glycidol along with [DBUH][Cl]. Overall, the dechlorination of the glycerol-derived 1,3-dichloro-2-propanol has occurred during the synthesis of DMC as well as glycidol, which not only facilitates the uptake of CO
2 but also allowed for the synthesis of industrially important value-added chemicals. Besides that, the DBU molecule not only assisted in the efficient CO
2 capture and served as a base catalyst in DMC synthesis but also performed as a reservoir for the chloride ion through the formation of its non-volatile and thermally stable chloride salt.
The synthesis of DMC and glycidol from 3-chloro-1,2-propylenecarbonate was further carried out at higher temperatures such as 35 and 50 °C, where the rate of formation of glycidol increased with the temperature and the complete conversion of the in situ-formed 3-Chloro-1,2-propanediol to glycidol took place in 2 h and 30 min, respectively (supporting information
Figure S4a,b). Hence, in this synthesis, the applied temperatures significantly influenced glycidol synthesis levels, whereas the rate of DMC formation remained unaltered.
Using the distillation method, 92% of DMC was recovered along with methanol from the reaction mixture, whereas the remaining glycidol and [DBUH][Cl] salt were separated using solvent extraction. In the case of solvent extraction initially, 2-Me-THF was added in a mixture of [DBUH][Cl] salt and glycidol in order to remove glycidol selectively from the mixture. However, after the addition of 2-Me-THF, no solid [DBUH][Cl] salt precipitated out from the mixture. On the other hand, the turbid solution was obtained after 1 h and the transparent viscous liquid settled at the bottom of the flask. The separation of [DBUH][Cl] from the glycidol is not possible, probably as a result of the formation of a deep eutectic mixture through hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD) interactions. In this case, considering previous reports regarding the compositions of various deep eutectic solvents (DES), the chloride anion containing ionic liquids such as choline chloride and a hydroxyl group comprised of molecules such as glycerol, ethylene glycol, etc., performed as a HBD and HBA, respectively to form a DES [
30]. Sato et al. also showed that a similar hydrogen bonding interaction between the –OH group of glycidol and the Cl
− anion of the quaternary alkylammonium salt was established and resulted in the formation of a binding complex [
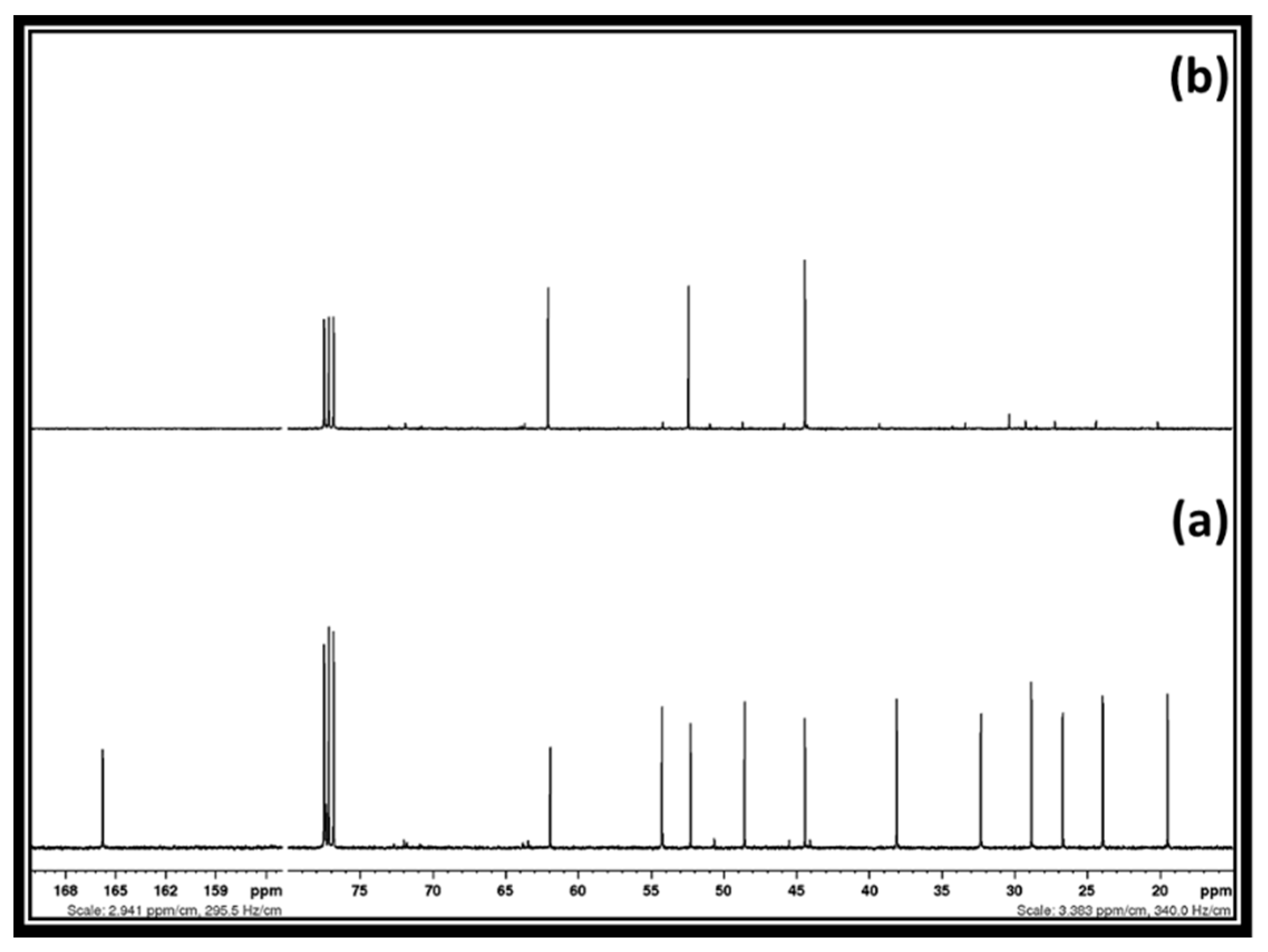
31]. Therefore, the hydrogen bonding interaction possibly does not allow the precipitation of [DBUH][Cl] salt in 2-Me-THF. In order to trigger the separation of these two chemical species, the brine solution was added to their mixture, followed by the addition of 2-Me-THF to extract glycidol. In this case, NaCl in the brine solution possibly disturbed the hydrogen bonding between [DBUH][Cl] salt and glycidol and allowed the transfer of the latter to the 2-Me-THF phase. After the evaporation of 2-Me-THF from the organic phase, an 89% recovery of the pure form of the glycidol was achieved. The 13C NMR spectra of the recovered glycidol is shown in
Figure 6. Water was removed from the aqueous phase and dry methanol was added to precipitate and separate NaCl from the [DBUH][Cl] salt. The alkaline alcoholic solution (NaOH in methanol) was further used to recover the molecular DBU from its chloride salt. In this context, 83% of the pure form of the DBU was obtained and the spectra of the recovered DBU is shown in supporting information
Figure S5.
Hence, in this overall reaction approach, the hydro-chlorinated derivative of glycerol, i.e., 1,3-dichloro-2-propanol, was further used for the CO2 capture and its further valorization was demonstrated to synthesize DMC along with the glycidol. The specialty of this work is that DBU superbase was applied for various tasks where it performed as an efficient, selective, and recoverable base catalyst along with CO2 capturing as well as a dechlorinating agent in the synthesis. Apart from this, we introduce a new alternative and simultaneous synthetic approach to produce DMC and glycidol compared to the existing various individual catalytic approaches. In terms of the solvent system, 2-Me-THF emerged as a new and renewable solvent media for the CO2 activation to value-added chemicals, which was previously limited to CO2 capture in the form of expanded liquids.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










