
The Role of Astrocytes and Blood–Brain Barrier Disruption in Alzheimer’s Disease


Abstract
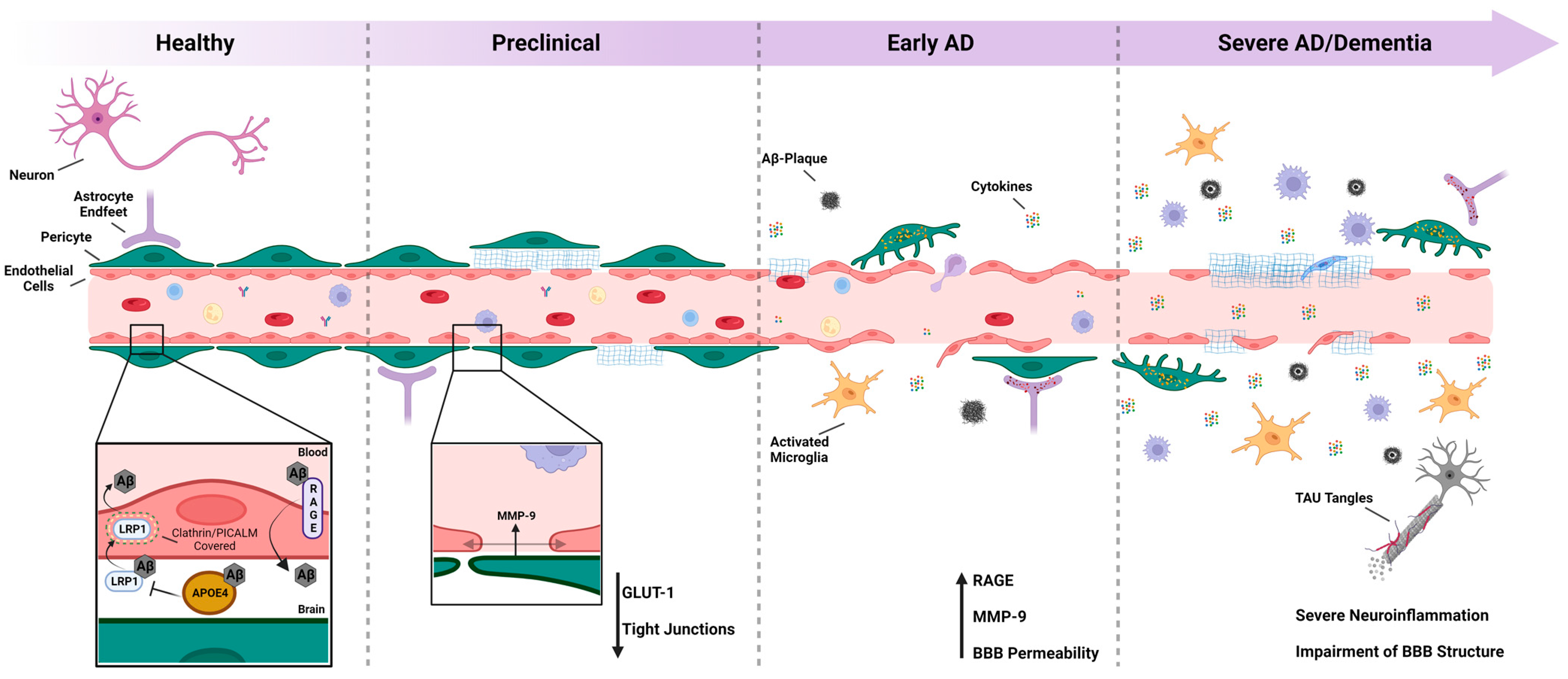
:1. Introduction
2. Pathogenesis of AD
3. Aβ Clearance through the BBB
4. Aβ Uptake through the BBB
5. The Role of Astrocytes in AD: Implications of Aβ and Tau Pathologies
6. The Role of Astrocytes in Synaptic Dysfunction in AD
7. Targeting the BBB and Reactive Astrocytes in AD: Promising Therapeutic Strategies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kunigelis, K.E.; Vogelbaum, M.A. Therapeutic Delivery to Central Nervous System. Neurosurg. Clin. N. Am. 2021, 32, 291–303. [Google Scholar] [CrossRef]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood–brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. The Role of the Blood Brain Barrier in Neurodegenerative Disorders and their Treatment. J. Alzheimer’s Dis. 2011, 24, 643–656. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Sikpa, D.; Whittingstall, L.; Savard, M.; Lebel, R.; Côté, J.; McManus, S.; Chemtob, S.; Fortin, D.; Lepage, M.; Gobeil, F. Pharmacological Modulation of Blood–Brain Barrier Permeability by Kinin Analogs in Normal and Pathologic Conditions. Pharmaceuticals 2020, 13, 279. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Robilliard, L.D.; Nicholson, L.F.B.; Graham, E.S.; O’Carroll, S.J. Bradykinin receptor-1 activation induces inflammation and increases the permeability of human brain microvascular endothelial cells. Cell Biol. Int. 2019, 44, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Eratne, D.; Loi, S.M.; Farrand, S.; Kelso, W.; Velakoulis, D.; Looi, J.C. Alzheimer’s disease: Clinical update on epidemiology, pathophysiology and diagnosis. Australas. Psychiatry 2018, 26, 347–357. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Drew, L. An age-old story of dementia. Nature 2018, 559, S2–S3. [Google Scholar] [CrossRef]
- Hung, A.; Liang, Y.; Chow, T.C.; Tang, H.; Wu, S.L.; Wai, M.; Yew, D. Mutated tau, amyloid and neuroinflammation in Alzheimer disease—A brief review. Prog. Histochem. Cytochem. 2016, 51, 1–8. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2019, 21, 21–35. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 2020, 167, 382–394. [Google Scholar] [CrossRef]
- Sigurdsson, E.M.; Knudsen, E.; Asuni, A.; Fitzer-Attas, C.; Sage, D.; Quartermain, D.; Goni, F.; Frangione, B.; Wisniewski, T. An Attenuated Immune Response Is Sufficient to Enhance Cognition in an Alzheimer’s Disease Mouse Model Immunized with Amyloid-β Derivatives. J. Neurosci. 2004, 24, 6277–6282. [Google Scholar] [CrossRef] [PubMed]
- Kurz, C.; Walker, L.; Rauchmann, B.; Perneczky, R. Dysfunction of the blood–brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol. Appl. Neurobiol. 2021, 48, e12782. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Zlokovic, B.V. Role of the Blood-Brain Barrier in the Pathogenesis of Alzheimers Disease. Curr. Alzheimer Res. 2007, 4, 191–197. [Google Scholar] [CrossRef]
- Boutajangout, A.; Wisniewski, T. Tau-Based Therapeutic Approaches for Alzheimer’s Disease—A Mini-Review. Gerontology 2014, 60, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef]
- Bell, R.D.; Sagare, A.P.; E Friedman, A.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport Pathways for Clearance of Human Alzheimer’s Amyloid β-Peptide and Apolipoproteins E and J in the Mouse Central Nervous System. J. Cereb. Blood Flow Metab. 2006, 27, 909–918. [Google Scholar] [CrossRef]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; A Winkler, E.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Tai, L.M.; Mehra, S.; Shete, V.; Estus, S.; Rebeck, G.W.; Bu, G.; LaDu, M.J. Soluble apoE/Aβ complex: Mechanism and therapeutic target for APOE4-induced AD risk. Mol. Neurodegener. 2014, 9, 2. [Google Scholar] [CrossRef]
- Nelson, A.; Sagare, A.; Zlokovic, B. Blood–Brain Barrier Transport of Alzheimer’s Amyloid β-Peptide. In Developing Therapeutics for Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 2016; pp. 251–270. [Google Scholar] [CrossRef]
- Narayan, P.; Orte, A.; Clarke, R.W.; Bolognesi, B.; Hook, S.; Ganzinger, K.A.; Meehan, S.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β1−40 peptide. Nat. Struct. Mol. Biol. 2011, 19, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Meehan, S.; Carver, J.A.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. Amyloid-β Oligomers are Sequestered by both Intracellular and Extracellular Chaperones. Biochemistry 2012, 51, 9270–9276. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Cirrito, J.R.; Parsadanian, M.; May, P.C.; A O’Dell, M.; Taylor, J.W.; Harmony, J.A.; Aronow, B.J.; Bales, K.R.; Paul, S.M.; et al. ApoE and Clusterin Cooperatively Suppress Aβ Levels and Deposition: Evidence that ApoE Regulates Extracellular Aβ Metabolism In Vivo. Neuron 2004, 41, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; I Brooks, A.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.; Schönknecht, P.; Henze, M.; Seidl, U.; Haberkorn, U.; Schröder, J. Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res. Neuroimaging 2007, 155, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Deane, R.; Chow, N.; Long, X.; Sagare, A.; Singh, I.; Streb, J.W.; Guo, H.; Rubio, A.; Van Nostrand, W.; et al. SRF and myocardin regulate LRP-mediated amyloid-β clearance in brain vascular cells. Nature 2008, 11, 143–153. [Google Scholar] [CrossRef]
- Li, M.; Shang, D.-S.; Zhao, W.-D.; Tian, L.; Li, B.; Fang, W.-G.; Zhu, L.; Man, S.-M.; Chen, Y.-H. Amyloid β Interaction with Receptor for Advanced Glycation End Products Up-Regulates Brain Endothelial CCR5 Expression and Promotes T Cells Crossing the Blood-Brain Barrier. J. Immunol. 2009, 182, 5778–5788. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Vogelgesang, S.; Kroemer, H.K. MDR1–P-glycoprotein (ABCB1)-Mediated Disposition of Amyloid-? Peptides: Implications for the Pathogenesis and Therapy of Alzheimer’s Disease. Clin. Pharmacol. Ther. 2010, 88, 441–443. [Google Scholar] [CrossRef]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef]
- Ding, Y.; Zhong, Y.; Baldeshwiler, A.; Abner, E.L.; Bauer, B.; Hartz, A.M.S. Protecting P-glycoprotein at the blood–brain barrier from degradation in an Alzheimer’s disease mouse model. Fluids Barriers CNS 2021, 18, 10. [Google Scholar] [CrossRef]
- Chai, A.B.; Hartz, A.M.S.; Gao, X.; Yang, A.; Callaghan, R.; Gelissen, I.C. New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models. Int. J. Mol. Sci. 2020, 22, 246. [Google Scholar] [CrossRef]
- van Assema, D.M.E.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.I.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood–brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2011, 135, 181–189. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Wei, S.; Shang, S.; Dang, L.; Gao, F.; Gao, Y.; Gao, L.; Chen, C.; Huo, K.; Wang, J.; Wang, J.; et al. Blood Triglyceride and High-Density Lipoprotein Levels Are Associated with Plasma Amyloid-β Transport: A Population-Based Cross-Sectional Study. J. Alzheimer’s Dis. 2021, 84, 303–314. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Aβ accumulation in a mouse model of Alzheimer’s disease via modulation of β- and γ-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Parpura, V.; Verkhratsky, A. Homeostatic function of astrocytes: Ca2+ and Na+ signalling. Transl. Neurosci. 2012, 3, 334–344. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Araujo, A.P.B.; Melo, H.M.; da Silva, G.S.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Aβ Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Bégard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef]
- Subramanian, J.; Savage, J.C.; Tremblay, M. Synaptic Loss in Alzheimer’s Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Front. Cell. Neurosci. 2020, 14, 592607. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Yang, F.; Zhu, W.; Cai, M.; Li, X.-T.; Zhang, J.-S.; Yu, Z.-H.; Zhang, W.; Cai, D.-F. Bilobalide inhibits inflammation and promotes the expression of Aβ degrading enzymes in astrocytes to rescue neuronal deficiency in AD models. Transl. Psychiatry 2021, 11, 542. [Google Scholar] [CrossRef]
- Kuwar, R.; Rolfe, A.; Di, L.; Blevins, H.; Xu, Y.; Sun, X.; Bloom, G.S.; Zhang, S.; Sun, D. A Novel Inhibitor Targeting NLRP3 Inflammasome Reduces Neuropathology and Improves Cognitive Function in Alzheimer’s Disease Transgenic Mice. J. Alzheimer’s Dis. 2021, 82, 1769–1783. [Google Scholar] [CrossRef]
- Yue, Q.; Zhou, X.; Zhang, Z.; Hoi, M.P.M. Murine Beta-Amyloid (1–42) Oligomers Disrupt Endothelial Barrier Integrity and VEGFR Signaling via Activating Astrocytes to Release Deleterious Soluble Factors. Int. J. Mol. Sci. 2022, 23, 1878. [Google Scholar] [CrossRef]
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; E Bennett, R.; Hudry, E.; Hyman, B.T. APOE4 derived from astrocytes leads to blood–brain barrier impairment. Brain 2021, 145, 3582–3593. [Google Scholar] [CrossRef]
- Kim, H.; Leng, K.; Park, J.; Sorets, A.G.; Kim, S.; Shostak, A.; Embalabala, R.J.; Mlouk, K.; Katdare, K.A.; Rose, I.V.L.; et al. Reactive astrocytes transduce inflammation in a blood-brain barrier model through a TNF-STAT3 signaling axis and secretion of alpha 1-antichymotrypsin. Nat. Commun. 2022, 13, 6581. [Google Scholar] [CrossRef]
- Pandit, R.; Leinenga, G.; Götz, J. Repeated ultrasound treatment of tau transgenic mice clears neuronal tau by autophagy and improves behavioral functions. Theranostics 2019, 9, 3754–3767. [Google Scholar] [CrossRef] [PubMed]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef]
- Epelbaum, S.; Burgos, N.; Canney, M.; Matthews, D.; Houot, M.; Santin, M.D.; Desseaux, C.; Bouchoux, G.; Stroer, S.; Martin, C.; et al. Pilot study of repeated blood-brain barrier disruption in patients with mild Alzheimer’s disease with an implantable ultrasound device. Alzheimers Res. Ther. 2022, 14, 40. [Google Scholar] [CrossRef]
- Rezai, A.R.; Ranjan, M.; D’Haese, P.F.; Haut, M.W.; Carpenter, J.; Najib, U.; Mehta, R.I.; Chazen, J.L.; Zibly, Z.; Yates, J.R.; et al. Noninvasive hippocampal blood-brain barrier opening in Alzheimer’s disease with focused ultrasound. Proc. Natl. Acad. Sci. USA 2020, 117, 9180–9182. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.I.; Carpenter, J.S.; Mehta, R.I.; Haut, M.W.; Wang, P.; Ranjan, M.; Najib, U.; D’haese, P.-F.; Rezai, A.R. Ultrasound-mediated blood–brain barrier opening uncovers an intracerebral perivenous fluid network in persons with Alzheimer’s disease. Fluids Barriers CNS 2023, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Lazic, A.; Balint, V.; Ninkovic, D.S.; Peric, M.; Stevanovic, M. Reactive and Senescent Astroglial Phenotypes as Hallmarks of Brain Pathologies. Int. J. Mol. Sci. 2022, 23, 4995. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Xu, C.; Zhang, H.; Lin, C. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.-A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kam, T.-I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.-H.; Song, J.-J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.C.; Lombardero, L.; Rodríguez-Puertas, R.; de Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological ablation of astrocytes reduces Aβ degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 73. [Google Scholar] [CrossRef]
- Kelly, M. Exercise-Induced Modulation of Neuroinflammation in Models of Alzheimer’s Disease. Brain Plast. 2018, 4, 81–94. [Google Scholar] [CrossRef]
- Ribarič, S. Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 3245. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Moreno, J.; Satorres, E.; Soria-Urios, G.; Meléndez, J.C. Cognitive Stimulation in Moderate Alzheimer’s Disease. J. Appl. Gerontol. 2022, 41, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Stekic, A.; Zeljkovic, M.; Kontic, M.Z.; Mihajlovic, K.; Adzic, M.; Stevanovic, I.; Ninkovic, M.; Grkovic, I.; Ilic, T.V.; Nedeljkovic, N.; et al. Intermittent Theta Burst Stimulation Ameliorates Cognitive Deficit and Attenuates Neuroinflammation via PI3K/Akt/mTOR Signaling Pathway in Alzheimer’s-Like Disease Model. Front. Aging Neurosci. 2022, 14, 889983. [Google Scholar] [CrossRef] [PubMed]
- Luzi, F.; Savickas, V.; Taddei, C.; Hader, S.; Singh, N.; Gee, A.D.; Bongarzone, S. Radiolabeling of [11C]FPS-ZM1, a receptor for advanced glycation end products-targeting positron emission tomography radiotracer, using a [11C]CO2-to-[11C]CO chemical conversion. Futur. Med. Chem. 2020, 12, 511–521. [Google Scholar] [CrossRef]
- Barca, C.; Foray, C.; Hermann, S.; Döring, C.; Schäfers, M.; Jacobs, A.H.; Zinnhardt, B. Characterization of the inflammatory post-ischemic tissue by full volumetric analysis of a multimodal imaging dataset. Neuroimage 2020, 222, 117217. [Google Scholar] [CrossRef]
- Zhou, R.; Ji, B.; Kong, Y.; Qin, L.; Ren, W.; Guan, Y.; Ni, R. PET Imaging of Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2021, 12, 739130. [Google Scholar] [CrossRef]
- Livingston, N.R.; Calsolaro, V.; Hinz, R.; Nowell, J.; Raza, S.; Gentleman, S.; Tyacke, R.J.; Myers, J.; Venkataraman, A.V.; Perneczky, R.; et al. Relationship between astrocyte reactivity, using novel 11C-BU99008 PET, and glucose metabolism, grey matter volume and amyloid load in cognitively impaired individuals. Mol. Psychiatry 2022, 27, 2019–2029. [Google Scholar] [CrossRef]
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.-T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 2022, 27, 4781–4789. [Google Scholar] [CrossRef]


{kind=link}
| Title | Goal | Status | Reference |
|---|---|---|---|
| Selective PET Imaging of Astrocytes and Microglia in Alzheimer’s Disease | Creating a novel PET imaging agent with the ability to specifically target astrocytes for the early detection of astrogliosis, starting from the stage of mild cognitive impairment (MCI) | In progress | ClinicalTrials.gov ID: NCT05582200 |
| First-in-human Evaluation of EAAT2 PET Tracer in Healthy and Alzheimer’s Diseased Brain | To develop a new PET imaging agent that will selectively target the astrocytic glutamate transporter, identifying early changes in astrocytes of patients with AD | In progress | ClinicalTrials.gov ID: NCT05374278 |
| Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound | To use magnetic resonance-guided focused ultrasound to open the BBB in patients with mild-to-moderate AD, evaluate the safety of this procedure, and measure the influence on amyloid markers of AD | Finished | [69] |
| Blood–brain Barrier Opening in Alzheimer’ Disease (BOREAL1) | To use an extra-dural ultrasound emitter to open the BBB in AD patients, evaluate the safety of this procedure, and measure Aβ and tau through PET-MRI imaging | Finished | [70] |
| Noninvasive hippocampal blood−brain barrier opening ;in Alzheimer’s disease with focused ultrasound | To evaluate the safety, feasibility, and reversibility on the use of magnetic resonance-guided low-intensity focused ultrasound to open the BBB in the hippocampus and entorhinal cortex in patients with early AD | Finished | [71] |
| ExAblate Blood–brain barrier (BBB) Disruption for the Treatment of Alzheimer’s Disease | To use focused ultrasound in combination with an intravenously administered microbubble contrast agent to open the BBB in the frontal, parietal, and medial lobes of patients with early AD | Finished | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, J.V.R.; Batista, C.; Diniz, L.P.; Mendes, F.A. The Role of Astrocytes and Blood–Brain Barrier Disruption in Alzheimer’s Disease. Neuroglia 2023, 4, 209-221. https://doi.org/10.3390/neuroglia4030015
Cruz JVR, Batista C, Diniz LP, Mendes FA. The Role of Astrocytes and Blood–Brain Barrier Disruption in Alzheimer’s Disease. Neuroglia. 2023; 4(3):209-221. https://doi.org/10.3390/neuroglia4030015
Chicago/Turabian StyleCruz, João Victor R., Carolina Batista, Luan Pereira Diniz, and Fabio A. Mendes. 2023. "The Role of Astrocytes and Blood–Brain Barrier Disruption in Alzheimer’s Disease" Neuroglia 4, no. 3: 209-221. https://doi.org/10.3390/neuroglia4030015