
Overview of the Molecular Modalities and Signaling Pathways Intersecting with β-Amyloid and Tau Protein in Alzheimer’s Disease


Abstract
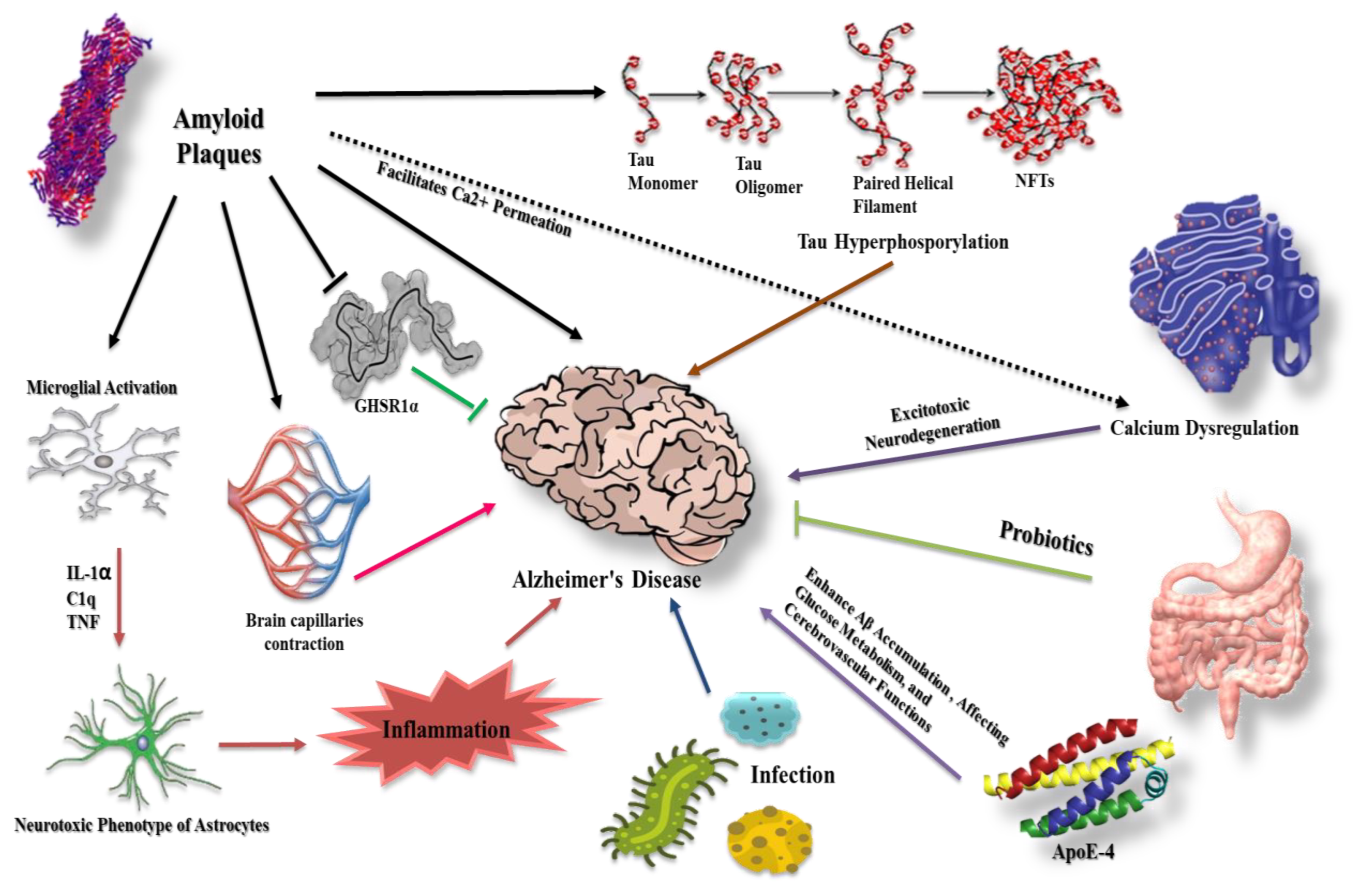
:1. Introduction
2. β-Amyloid in Alzheimer’s Disease
3. Tau Protein and Formation of Neurofibrillary Tangles
4. Prion-like Conformation of β-Amyloid and Tau Proteins
5. The Intervention of Glial Cells in β-Amyloid and Tau Protein Dysregulation
5.1. Astrocytes
5.2. Microglia
5.3. Cellular Cross Talk between Astrocyte and Microglia
6. Aβ Effect on the Cerebral Capillaries in Alzheimer’s Disease
7. Aβ Reaction with Hippocampal Ghrelin/GHSR1α in Alzheimer’s Disease
8. The Potential Role of Apolipoprotein in Alzheimer’s Pathogenesis
9. Calcium as a Playmaker in Alzheimer’s Disease
9.1. Calcium Channels
9.2. Intracellular Calcium
10. The Role of MicroRNA-137 in the Onset and Progression of Alzheimer’s Disease
11. Gut Microbiota and Alzheimer’s Disease
12. Infection and Alzheimer’s Disease
13. Conclusions

{kind=link}
| Drug | Action | References/Clinical Trials |
|---|---|---|
| ALZT-OP1a + ALZT-OP1b | Amyloid-related and antineuroinflammatory | NCT02547818, [161] |
| Plasma exchange with albumin 1 immunoglobulin | Removes amyloid | NCT01561053, [161] |
| AADvac-1 | Tau immunotherapy | [162,163], NCT02579252, NCT03174886 |
| ACI-35 | Tau immunotherapy | ISRCTN13033912, NCT04445831, [164] |
| ANAVEX2–73 | Anti-tau, anti-amyloid, and antineuroinflammatory | NCT03790709, [161] |
| GV-971 | Amyloid-related | NCT02293915, [161] |
| Crenezumab | Removes amyloid | NCT02670083, NCT03114657, NCT03491150, [161] |
| E2609 (elenbecestat) | Reduces amyloid production | NCT02956486, NCT03036280, [161] |
| BIIB076 | Tau immunotherapy | NCT03056729 |
| RG7345 | Tau immunotherapy | NCT02281786 |
| PNT001 | Tau immunotherapy | NCT04096287, NCT04677829 |
| Gantenerumab | Removes amyloid | NCT02294851, NCT02460094, NCT02658916, NCT03068468, NCT03352557, NCT03658135, NCT02051608, NCT01224106, NCT03444870, NCT03443973. |
| Solanezumab | Removes amyloid and prevent aggregation | NCT01760005, NCT02008357, [161] |
| Semorinemab | Extracellular tau | NCT02820896, NCT03289143, NCT03828747 |
| Bepranemab | Tau immunotherapy | NCT03464227, NCT03605082, NCT04185415, NCT04658199, NCT04867616 |
| JNJ-63733657 | Tau immunotherapy | NCT03375697, NCT03689153, NCT04619420. |
| Zagotenemab | Tau immunotherapy | NCT02754830, NCT03019536, NCT03518073 |
| Lu AF87908 | Tau immunotherapy | NCT04149860 |
| E2814 | Tau immunotherapy | NCT04231513 |
| Tilavonemab | Tau immunotherapy | NCT02494024, NCT03413319, NCT02985879, NCT03391765, NCT03744546, NCT02880956, NCT03712787. |
| Donanemab | Amyloid-related | NCT04437511 |
| Lecanemab | Blocks the formation of amyloid plaques in the brain | FDA-approved, NCT03887455 |
| Remternetug | Anti-amyloid, immunomodulator | NCT05463731 |
| Aducanumab | Anti-amyloid | FDA-approved, NCT02477800, [165] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hussein, W.; Sağlık, B.N.; Levent, S.; Korkut, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease. Molecules 2018, 23, 2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawas, C.; Gray, S.; Brookmeyer, R.; Fozard, J.; Zonderman, A. Age-specific incidence rates of Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 2000, 54, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2021, 17, 327–406. [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Yao, M.; Nguyen, T.V.; Pike, C.J. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Han, X.J.; Hu, Y.Y.; Yang, Z.J.; Jiang, L.P.; Shi, S.L.; Li, Y.R.; Guo, M.Y.; Wu, H.L.; Wan, Y.Y. Amyloid β-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 2017, 16, 4521–4528. [Google Scholar] [CrossRef] [Green Version]
- Barrantes, A.; Rejas, M.T.; Benítez, M.J.; Jiménez, J.S. Interaction between Alzheimer’s Abeta1-42 peptide and DNA detected by surface plasmon resonance. J. Alzheimer’s Dis. 2007, 12, 345–355. [Google Scholar] [CrossRef]
- Barghorn, S.; Davies, P.; Mandelkow, E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry 2004, 43, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Xia, Y.; Yu, G.; Shu, X.; Ge, H.; Zeng, K.; Wang, J.; Wang, X. Cleavage of GSK-3β by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK-3β activity induced by H2O2. J. Neurochem. 2013, 126, 234–242. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xiong, S.; Xie, C.; Davies, P.; Markesbery, W.R. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimer’s Dis. 2004, 6, 659–671. [Google Scholar] [CrossRef]
- Su, X.Y.; Wu, W.H.; Huang, Z.P.; Hu, J.; Lei, P.; Yu, C.H.; Zhao, Y.F.; Li, Y.M. Hydrogen peroxide can be generated by tau in the presence of Cu(II). Biochem. Biophys. Res. Commun. 2007, 358, 661–665. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yao, Y.; Cornec, A.S.; Oukoloff, K.; James, M.J.; Koivula, P.; Trojanowski, J.Q.; Smith, A.B., 3rd; Lee, V.M.; Ballatore, C.; et al. A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model. Mol. Neurodegener. 2018, 13, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaikkan, C.; Middleton, S.J.; Marco, A.; Pao, P.C.; Mathys, H.; Kim, D.N.; Gao, F.; Young, J.Z.; Suk, H.J.; Boyden, E.S.; et al. Gamma Entrainment Binds Higher-Order Brain Regions and Offers Neuroprotection. Neuron 2019, 102, 929–943.e928. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Molecular Structure of Aggregated Amyloid-β: Insights from Solid-State Nuclear Magnetic Resonance. Cold Spring Harb. Perspect. Med. 2016, 6, a024083. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Condello, C.; Stöehr, J. Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol. Dis. 2018, 109, 191–200. [Google Scholar] [CrossRef]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef]
- Pignataro, A.; Middei, S. Trans-Synaptic Spread of Amyloid-β in Alzheimer’s Disease: Paths to β-Amyloidosis. Neural Plast. 2017, 2017, 5281829. [Google Scholar] [CrossRef] [Green Version]
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, H.; Yoshimura, M.; Yamazaki, T.; Saido, T.C.; Ito, Y.; Yokofujita, J.; Okeda, R.; Ihara, Y. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am. J. Pathol. 1998, 152, 983–992. [Google Scholar] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, N.; Yue, T.; Liu, L. Matrine reverses the drug resistance of K562/ADM cells to ADM and VCR via promoting autophagy. Transl. Cancer Res. 2020, 9, 786–794. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, H.S.; Shin, K.Y.; Kim, E.M.; Kim, M.; Kim, H.S.; Park, C.H.; Jeong, Y.H.; Yoo, J.; Lee, J.P.; et al. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 2393–2404. [Google Scholar] [CrossRef] [Green Version]
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Kolb, B.; Whishaw, I.Q. Fundamentals of Human Neuropsychology; Macmillan: New York, NY, USA, 2009. [Google Scholar]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Bajetto, A.; Bonavia, R.; Barbero, S.; Schettini, G. Characterization of chemokines and their receptors in the central nervous system: Physiopathological implications. J. Neurochem. 2002, 82, 1311–1329. [Google Scholar] [CrossRef]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 2017, 27, 795–808. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Zhu, X.H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative imaging of energy expenditure in human brain. NeuroImage 2012, 60, 2107–2117. [Google Scholar] [CrossRef] [Green Version]
- Asllani, I.; Habeck, C.; Scarmeas, N.; Borogovac, A.; Brown, T.R.; Stern, Y. Multivariate and univariate analysis of continuous arterial spin labeling perfusion MRI in Alzheimer’s disease. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2008, 28, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Mavrikaki, M.; Ullrich, C.; Albarran-Zeckler, R.; Brantley, A.F.; Smith, R.G. Hippocampal Dopamine/DRD1 Signaling Dependent on the Ghrelin Receptor. Cell 2015, 163, 1176–1190. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.M.; Noble, E.E.; Reiner, D.J.; Liu, C.M.; Suarez, A.N.; Konanur, V.R.; Hayes, M.R.; Kanoski, S.E. Hippocampus ghrelin receptor signaling promotes socially-mediated learned food preference. Neuropharmacology 2018, 131, 487–496. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Diano, S.; Farr, S.A.; Benoit, S.C.; McNay, E.C.; da Silva, I.; Horvath, B.; Gaskin, F.S.; Nonaka, N.; Jaeger, L.B.; Banks, W.A.; et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 2006, 9, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Guo, L.; Sui, S.; Driskill, C.; Phensy, A.; Wang, Q.; Gauba, E.; Zigman, J.M.; Swerdlow, R.H.; Kroener, S.; et al. Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a role in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6278. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, Y.; Zhang, W. The growth hormone secretagogue receptor: Its intracellular signaling and regulation. Int. J. Mol. Sci. 2014, 15, 4837–4855. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.O.; Shin, S.J.; Park, J.Y.; Ku, B.K.; Song, J.S.; Kim, J.J.; Jeon, S.G.; Lee, S.M.; Moon, M. MK-0677, a Ghrelin Agonist, Alleviates Amyloid Beta-Related Pathology in 5XFAD Mice, an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1800. [Google Scholar] [CrossRef] [Green Version]
- Eslami, M.; Sadeghi, B.; Goshadrou, F. Chronic ghrelin administration restores hippocampal long-term potentiation and ameliorates memory impairment in rat model of Alzheimer’s disease. Hippocampus 2018, 28, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Cha, M.Y.; Mook-Jung, I. Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J. Alzheimer’s Dis. JAD 2014, 41, 233–241. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef] [Green Version]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miners, J.S.; Van Helmond, Z.; Chalmers, K.; Wilcock, G.; Love, S.; Kehoe, P.G. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2006, 65, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.W.; Kovacina, K.S.; Craft, S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Schneider-Axmann, T.; Lucassen, P.J.; Bayer, T.A.; Wirths, O. Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol. 2010, 119, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef]
- Koffie, R.M.; Hashimoto, T.; Tai, H.C.; Kay, K.R.; Serrano-Pozo, A.; Joyner, D.; Hou, S.; Kopeikina, K.J.; Frosch, M.P.; Lee, V.M.; et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain 2012, 135, 2155–2168. [Google Scholar] [CrossRef] [Green Version]
- Rannikmäe, K.; Kalaria, R.N.; Greenberg, S.M.; Chui, H.C.; Schmitt, F.A.; Samarasekera, N.; Al-Shahi Salman, R.; Sudlow, C.L. APOE associations with severe CAA-associated vasculopathic changes: Collaborative meta-analysis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflamm. 2014, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef]
- Craft, S. Alzheimer disease: Insulin resistance and AD—Extending the translational path. Nat. Rev. Neurol. 2012, 8, 360–362. [Google Scholar] [CrossRef] [Green Version]
- Ekblad, L.L.; Johansson, J.; Helin, S.; Viitanen, M.; Laine, H.; Puukka, P.; Jula, A.; Rinne, J.O. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology 2018, 90, e1150–e1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, X.; Zhao, L. Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer’s Disease Risk Reduction and Early Intervention. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 6665–6681. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e115. [Google Scholar] [CrossRef] [Green Version]
- Goodison, W.V.; Frisardi, V.; Kehoe, P.G. Calcium channel blockers and Alzheimer’s disease: Potential relevance in treatment strategies of metabolic syndrome. J. Alzheimer’s Dis. JAD 2012, 30 (Suppl. S2), S269–S282. [Google Scholar] [CrossRef]
- Nimmrich, V.; Eckert, A. Calcium channel blockers and dementia. Br. J. Pharmacol. 2013, 169, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Yang, C.H.; Lee, S.Y.; Lee, S.-H.; Kim, Y.; Ho, W.-K. Voltage-gated calcium channels contribute to spontaneous glutamate release directly via nanodomain coupling or indirectly via calmodulin. Prog. Neurobiol. 2022, 208, 102182. [Google Scholar] [CrossRef]
- Robinson, D.M.; Keating, G.M. Memantine: A review of its use in Alzheimer’s disease. Drugs 2006, 66, 1515–1534. [Google Scholar] [CrossRef]
- Suh, H.-W.; Kim, J.; Kwon, O.; Cho, S.-H.; Kim, J.W.; Kwak, H.-Y.; Kim, Y.; Lee, K.M.; Chung, S.-Y.; Lee, J.-H. Neurocircuitry of acupuncture effect on cognitive improvement in patients with mild cognitive impairment using magnetic resonance imaging: A study protocol for a randomized controlled trial. Trials 2019, 20, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, B.; Kennelly, S.; Dwyer, S.; Cregg, F.; Walsh, C.; Coen, R.; Kenny, R.A.; Howard, R.; Murphy, C.; Adams, J.; et al. NILVAD protocol: A European multicentre double-blind placebo-controlled trial of nilvadipine in mild-to-moderate Alzheimer’s disease. BMJ Open 2014, 4, e006364. [Google Scholar] [CrossRef] [PubMed]
- Anekonda, T.S.; Quinn, J.F.; Harris, C.; Frahler, K.; Wadsworth, T.L.; Woltjer, R.L. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol. Dis. 2011, 41, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriguchi, S.; Shioda, N.; Yamamoto, Y.; Tagashira, H.; Fukunaga, K. The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: Enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J. Neurochem. 2012, 121, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Khashwji, H.; Estrada, T.; LaFerla, F.M. ST101 induces a novel 17kDa APP cleavage that precludes Aβ generation in vivo. Ann. Neurol. 2011, 69, 831–844. [Google Scholar] [CrossRef]
- Malenka, R.C. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 1994, 78, 535–538. [Google Scholar] [CrossRef]
- Dreses-Werringloer, U.; Lambert, J.C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Chandakkar, P.; Zhao, H.; Blanc, L.; Ruiz, S.; Marambaud, P. CALHM1 ion channel elicits amyloid-β clearance by insulin-degrading enzyme in cell lines and in vivo in the mouse brain. J. Cell Sci. 2015, 128, 2330–2338. [Google Scholar] [CrossRef] [Green Version]
- Emptage, N.J.; Reid, C.A.; Fine, A. Calcium Stores in Hippocampal Synaptic Boutons Mediate Short-Term Plasticity, Store-Operated Ca2+ Entry, and Spontaneous Transmitter Release. Neuron 2001, 29, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.C. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 2007, 6, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Siebert, A.P.; Cheung, K.-H.; Lee, R.J.; Johnson, B.; Cohen, A.S.; Vingtdeux, V.; Marambaud, P.; Foskett, J.K. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl. Acad. Sci. USA 2012, 109, E1963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimer’s Dis. Park. 2017, 7, 374. [Google Scholar] [CrossRef]
- Smith, I.F.; Green, K.N.; LaFerla, F.M. Calcium dysregulation in Alzheimer’s disease: Recent advances gained from genetically modified animals. Cell Calcium 2005, 38, 427–437. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; de Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins Are Enriched in Endoplasmic Reticulum Membranes Associated with Mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- Honarnejad, K.; Herms, J. Presenilins: Role in calcium homeostasis. Int. J. Biochem. Cell Biol. 2012, 44, 1983–1986. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 Mutations Increase Levels of Ryanodine Receptors and Calcium Release in PC12 Cells and Cortical Neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.-H.; Mei, L.; Mak, D.-O.D.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-Function Enhancement of IP3 Receptor Modal Gating by Familial Alzheimer’s Disease—Linked Presenilin Mutants in Human Cells and Mouse Neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [Green Version]
- Rybalchenko, V.; Hwang, S.Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Unni, V.K.; Zakharenko, S.S.; Zablow, L.; DeCostanzo, A.J.; Siegelbaum, S.A. Calcium release from presynaptic ryanodine-sensitive stores is required for long-term depression at hippocampal CA3-CA3 pyramidal neuron synapses. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 9612–9622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baracaldo-Santamaría, D.; Avendaño-Lopez, S.S.; Ariza-Salamanca, D.F.; Rodriguez-Giraldo, M.; Calderon-Ospina, C.A.; González-Reyes, R.E.; Nava-Mesa, M.O. Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 9067. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Yang, B.; Liu, C.; Liang, G.; Eckenhoff, M.F.; Liu, W.; Pickup, S.; Meng, Q.; Tian, Y.; Li, S.; et al. Long-term dantrolene treatment reduced intraneuronal amyloid in aged Alzheimer triple transgenic mice. Alzheimer Dis. Assoc. Disord. 2015, 29, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ono, K.; Dickstein, D.L.; Arrieta-Cruz, I.; Zhao, W.; Qian, X.; Lamparello, A.; Subnani, R.; Ferruzzi, M.; Pavlides, C.; et al. Carvedilol as a potential novel agent for the treatment of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 2321.e1–2321.e12. [Google Scholar] [CrossRef] [Green Version]
- Amin, N.D.; Bai, G.; Klug, J.R.; Bonanomi, D.; Pankratz, M.T.; Gifford, W.D.; Hinckley, C.A.; Sternfeld, M.J.; Driscoll, S.P.; Dominguez, B.; et al. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 2015, 350, 1525–1529. [Google Scholar] [CrossRef] [Green Version]
- Siegert, S.; Seo, J.; Kwon, E.J.; Rudenko, A.; Cho, S.; Wang, W.; Flood, Z.; Martorell, A.J.; Ericsson, M.; Mungenast, A.E.; et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat. Neurosci. 2015, 18, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Biophys. Acta 2003, 1632, 16–30. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- Lone, M.A.; Hülsmeier, A.J.; Saied, E.M.; Karsai, G.; Arenz, C.; von Eckardstein, A.; Hornemann, T. Subunit composition of the mammalian serine-palmitoyltransferase defines the spectrum of straight and methyl-branched long-chain bases. Proc. Natl. Acad. Sci. USA 2020, 117, 15591–15598. [Google Scholar] [CrossRef] [PubMed]
- Rotthier, A.; Auer-Grumbach, M.; Janssens, K.; Baets, J.; Penno, A.; Almeida-Souza, L.; Van Hoof, K.; Jacobs, A.; De Vriendt, E.; Schlotter-Weigel, B.; et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am. J. Hum. Genet. 2010, 87, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-G. Research Progress in the Pathogenesis of Alzheimer’s Disease. Chin. Med. J. 2018, 131, 1618. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Dao, D.T.; Terrillion, C.E.; Arad, M.; Smith, R.J.; Soldatov, N.M.; Gould, T.D. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog. Neurobiol. 2012, 99, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Xu, B.; Chen, J.; Sui, Y.; Ren, L.; Li, J.; Zhang, H.; Guo, L.; Sun, X. Micro-RNA-137 Inhibits Tau Hyperphosphorylation in Alzheimer’s Disease and Targets the CACNA1C Gene in Transgenic Mice and Human Neuroblastoma SH-SY5Y Cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 5635–5644. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Zhong, X.; Mei, H. A Systematic Investigation into Aging Related Genes in Brain and Their Relationship with Alzheimer’s Disease. PLoS ONE 2016, 11, e0150624. [Google Scholar] [CrossRef] [Green Version]
- Cryan, J.F.; Dinan, T.G. More than a gut feeling: The microbiota regulates neurodevelopment and behavior. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2015, 40, 241–242. [Google Scholar] [CrossRef] [Green Version]
- Akbari, E.; Asemi, Z.; Daneshvar Kakhaki, R.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef] [Green Version]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, N.; Agis-Balboa, R.C.; Walter, J.; Sananbenesi, F.; Fischer, A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimer’s Dis. JAD 2011, 26, 187–197. [Google Scholar] [CrossRef]
- Nankova, B.B.; Agarwal, R.; MacFabe, D.F.; La Gamma, E.F. Enteric bacterial metabolites propionic and butyric acid modulate gene expression, including CREB-dependent catecholaminergic neurotransmission, in PC12 cells—Possible relevance to autism spectrum disorders. PLoS ONE 2014, 9, e103740. [Google Scholar] [CrossRef]
- Tong, Y.; Bai, L.; Gong, R.; Chuan, J.; Duan, X.; Zhu, Y. Shikonin Protects PC12 Cells Against β-amyloid Peptide-Induced Cell Injury Through Antioxidant and Antiapoptotic Activities. Sci. Rep. 2018, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Sugahara, H.; Shimada, K.; Mitsuyama, E.; Kuhara, T.; Yasuoka, A.; Kondo, T.; Abe, K.; Xiao, J.Z. Therapeutic potential of Bifidobacterium breve strain A1 for preventing cognitive impairment in Alzheimer’s disease. Sci. Rep. 2017, 7, 13510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Bhatt, D.P.; Geiger, J.D.; Rosenberger, T.A. Acetate supplementation modulates brain adenosine metabolizing enzymes and adenosine A2Areceptor levels in rats subjected to neuroinflammation. J. Neuroinflamm. 2014, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kapila, R.; Dass, G.; Kapila, S. Improvement in Th1/Th2 immune homeostasis, antioxidative status and resistance to pathogenic E. coli on consumption of probiotic Lactobacillus rhamnosus fermented milk in aging mice. Age 2014, 36, 9686. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Vinolo, M.A.R.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [Green Version]
- Bourassa, M.W.; Alim, I.; Bultman, S.J.; Ratan, R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016, 625, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Varela, R.B.; Valvassori, S.S.; Lopes-Borges, J.; Mariot, E.; Dal-Pont, G.C.; Amboni, R.T.; Bianchini, G.; Quevedo, J. Sodium butyrate and mood stabilizers block ouabain-induced hyperlocomotion and increase BDNF, NGF and GDNF levels in brain of Wistar rats. J. Psychiatr. Res. 2015, 61, 114–121. [Google Scholar] [CrossRef]
- Sharma, R.; Padwad, Y. Probiotic bacteria as modulators of cellular senescence: Emerging concepts and opportunities. Gut Microbes 2020, 11, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, F.; Augello, F.R.; Palumbo, P.; Bonfili, L.; Artone, S.; Altamura, S.; Sheldon, J.M.; Latella, G.; Cifone, M.G.; Eleuteri, A.M.; et al. Bacterial Lysate from the Multi-Strain Probiotic SLAB51 Triggers Adaptative Responses to Hypoxia in Human Caco-2 Intestinal Epithelial Cells under Normoxic Conditions and Attenuates LPS-Induced Inflammatory Response. Int. J. Mol. Sci. 2023, 24, 8134. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Tsuzuki, Y.; Hokari, R.; Komoto, S.; Kurihara, C.; Kawaguchi, A.; Nagao, S.; Miura, S. Anti-inflammatory effects of the genus Bifidobacterium on macrophages by modification of phospho-I kappaB and SOCS gene expression. Int. J. Exp. Pathol. 2009, 90, 131–140. [Google Scholar] [CrossRef]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [Green Version]
- Sjogren, T.; Sjogren, H.; Lindgren, A.G. Morbus Alzheimer and morbus Pick; a genetic, clinical and patho-anatomical study. Acta Psychiatr. Neurol. Scand. Suppl. 1952, 82, 1–152. [Google Scholar]
- Zhou, R.; Yang, G.; Shi, Y. Dominant negative effect of the loss-of-function γ-secretase mutants on the wild-type enzyme through heterooligomerization. Proc. Natl. Acad. Sci. USA 2017, 114, 12731–12736. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63.e53. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Du, S.; Johnson, J.L.; Tung, H.Y.; Landers, C.T.; Liu, Y.; Seman, B.G.; Wheeler, R.T.; Costa-Mattioli, M.; Kheradmand, F.; et al. Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat. Commun. 2019, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Kamer, A.R.; Pirraglia, E.; Tsui, W.; Rusinek, H.; Vallabhajosula, S.; Mosconi, L.; Yi, L.; McHugh, P.; Craig, R.G.; Svetcov, S.; et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging 2015, 36, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Poole, S.; Singhrao, S.K.; Chukkapalli, S.; Rivera, M.; Velsko, I.; Kesavalu, L.; Crean, S. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE−/− mice brains. J. Alzheimer’s Dis. JAD 2015, 43, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R.; et al. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Urnowey, S.; Ansai, T.; Bitko, V.; Nakayama, K.; Takehara, T.; Barik, S. Temporal activation of anti- and pro-apoptotic factors in human gingival fibroblasts infected with the periodontal pathogen, Porphyromonas gingivalis: Potential role of bacterial proteases in host signalling. BMC Microbiol. 2006, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouveia Roque, C.; Chung, K.M.; McCurdy, E.P.; Jagannathan, R.; Randolph, L.K.; Herline-Killian, K.; Baleriola, J.; Hengst, U. CREB3L2-ATF4 heterodimerization defines a transcriptional hub of Alzheimer’s disease gene expression linked to neuropathology. Sci. Adv. 2023, 9, eadd2671. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [Green Version]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Zilka, N.; Kovacech, B.; Skrabana, R.; Vince-Kazmerova, Z.; Katina, S.; Fialova, L.; Prcina, M.; et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017, 16, 123–134. [Google Scholar] [CrossRef]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Kovacech, B.; Smolek, T.; Katina, S.; Fialova, L.; Prcina, M.; Parrak, V.; Dal-Bianco, P.; et al. FUNDAMANT: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; López-Deber, M.P.; Reis, P.; Hickman, D.T.; Adolfsson, O.; Chuard, N.; Ndao, D.M.; et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elshazly, A.M.; Sinanian, M.M.; Elimam, D.M.; Zakaria, S. Overview of the Molecular Modalities and Signaling Pathways Intersecting with β-Amyloid and Tau Protein in Alzheimer’s Disease. Neuroglia 2023, 4, 191-208. https://doi.org/10.3390/neuroglia4030014
Elshazly AM, Sinanian MM, Elimam DM, Zakaria S. Overview of the Molecular Modalities and Signaling Pathways Intersecting with β-Amyloid and Tau Protein in Alzheimer’s Disease. Neuroglia. 2023; 4(3):191-208. https://doi.org/10.3390/neuroglia4030014
Chicago/Turabian StyleElshazly, Ahmed M., Melanie M. Sinanian, Diaaeldin M. Elimam, and Sherin Zakaria. 2023. "Overview of the Molecular Modalities and Signaling Pathways Intersecting with β-Amyloid and Tau Protein in Alzheimer’s Disease" Neuroglia 4, no. 3: 191-208. https://doi.org/10.3390/neuroglia4030014