
Sequestration of Drugs from Biomolecular and Biomimicking Environments: Spectroscopic and Calorimetric Studies

Abstract
:1. Introduction
2. Sequestration of Drugs from Biomolecular Assemblies
2.1. Sequestration of Small Molecules from DNA Using Micelles
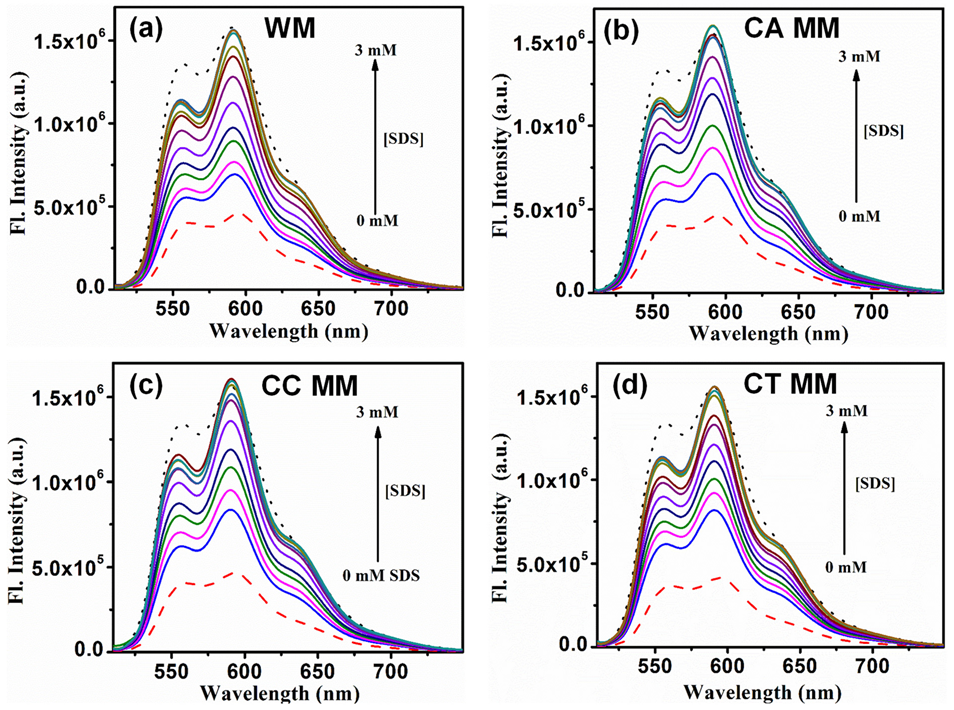
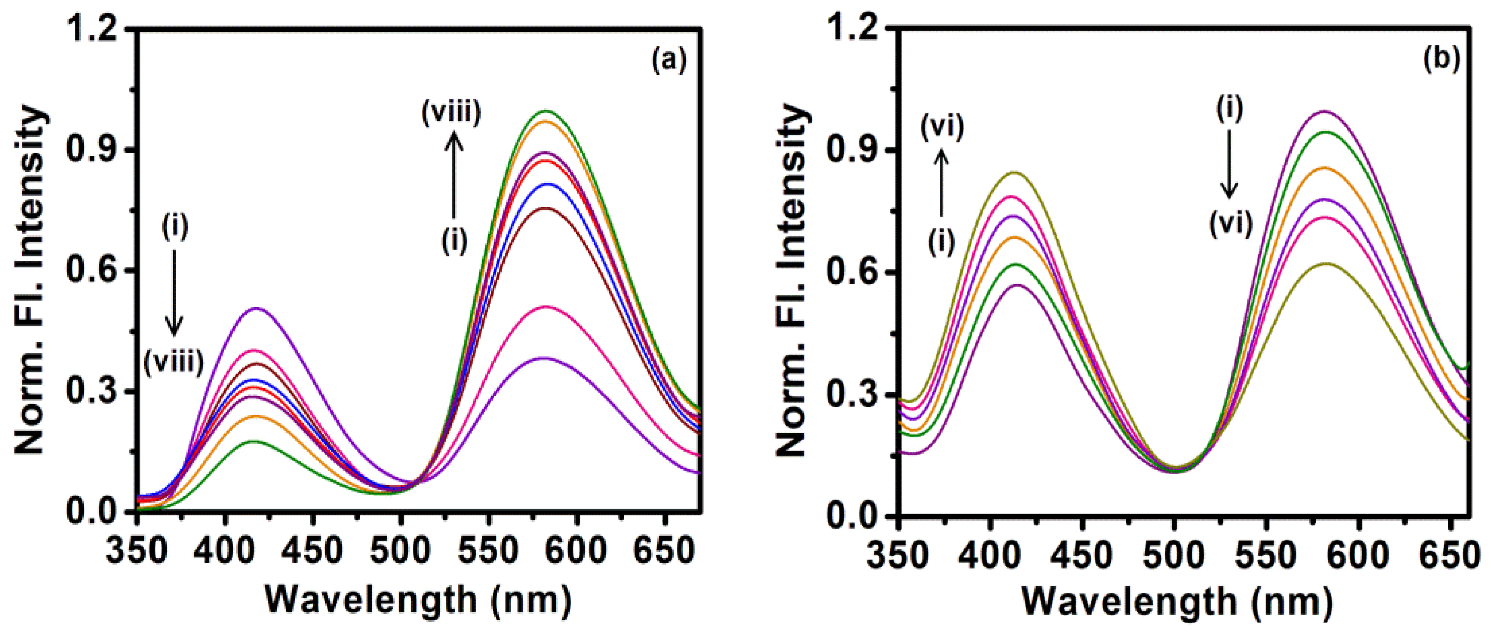
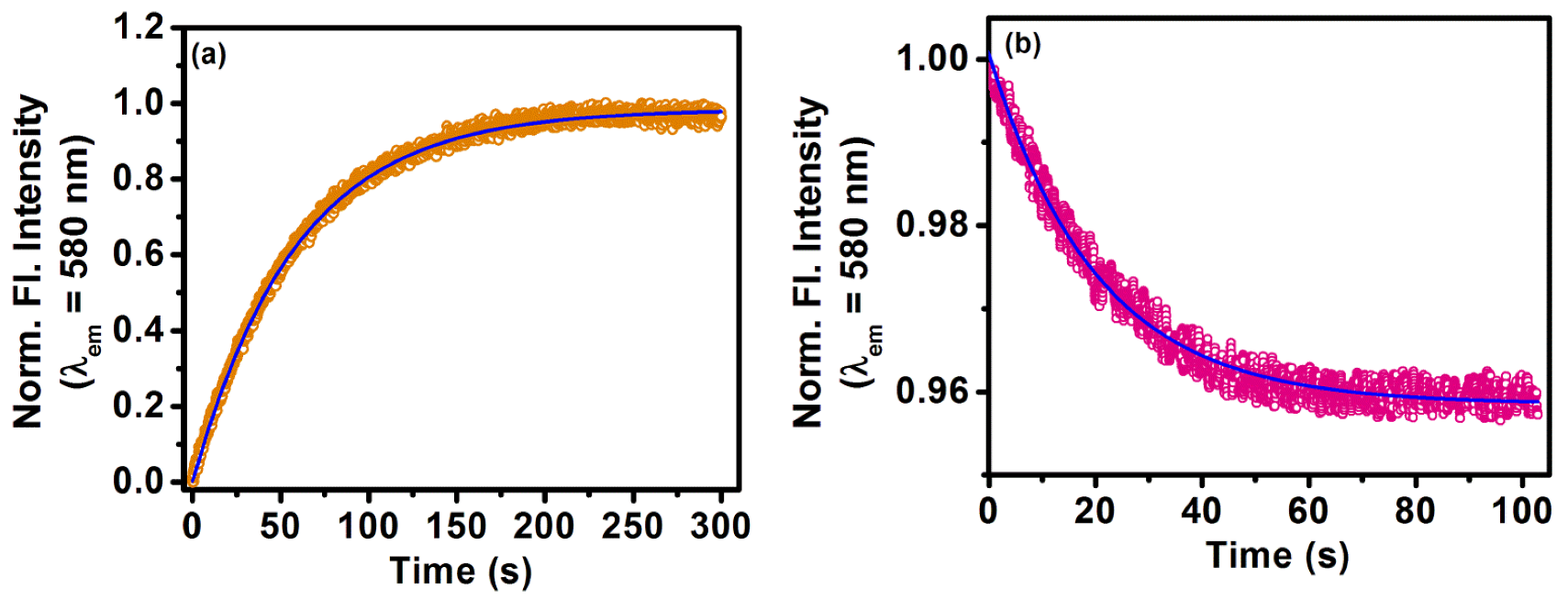
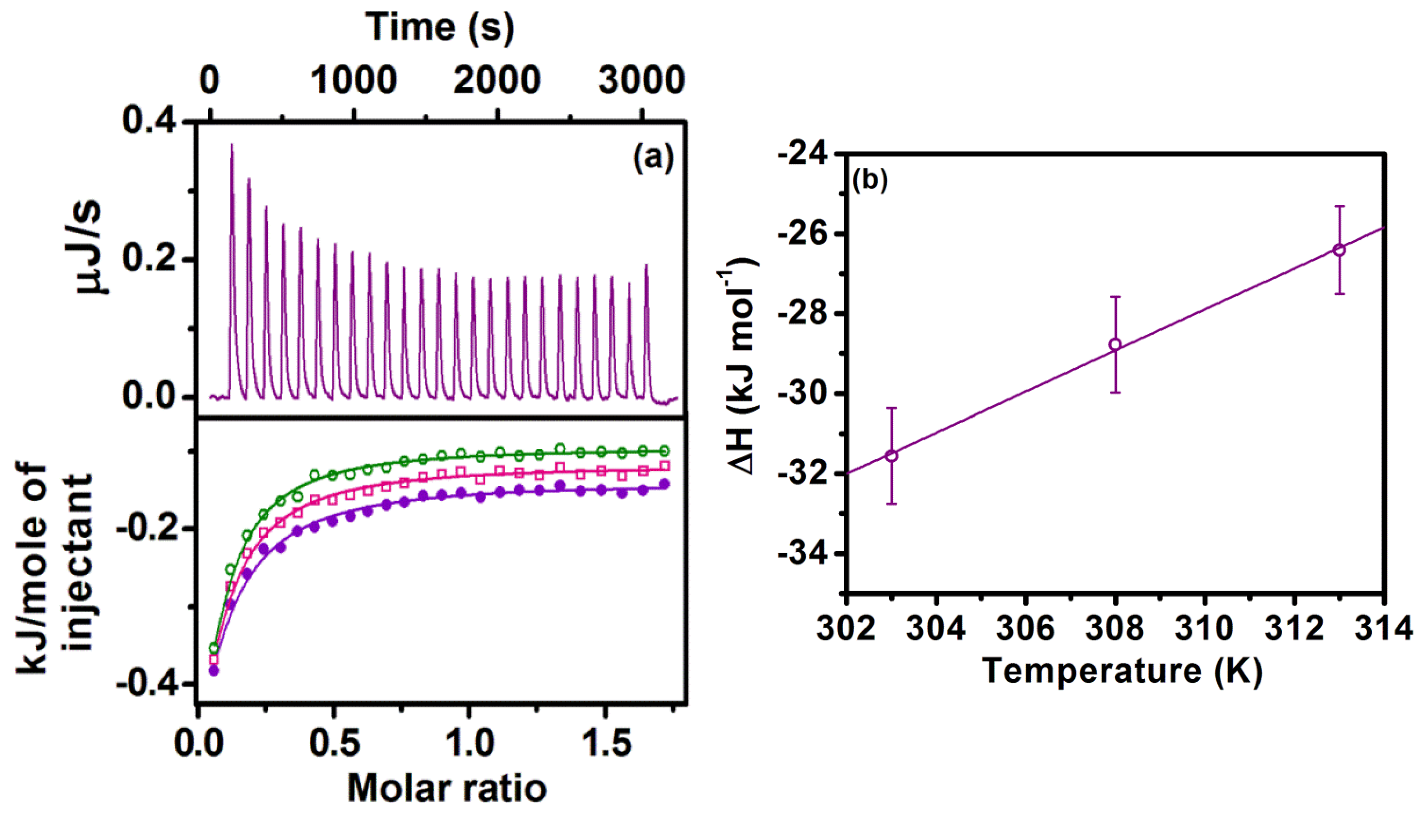
2.2. Sequestration of Small Drug Molecules from DNA Using Mixed Micelles
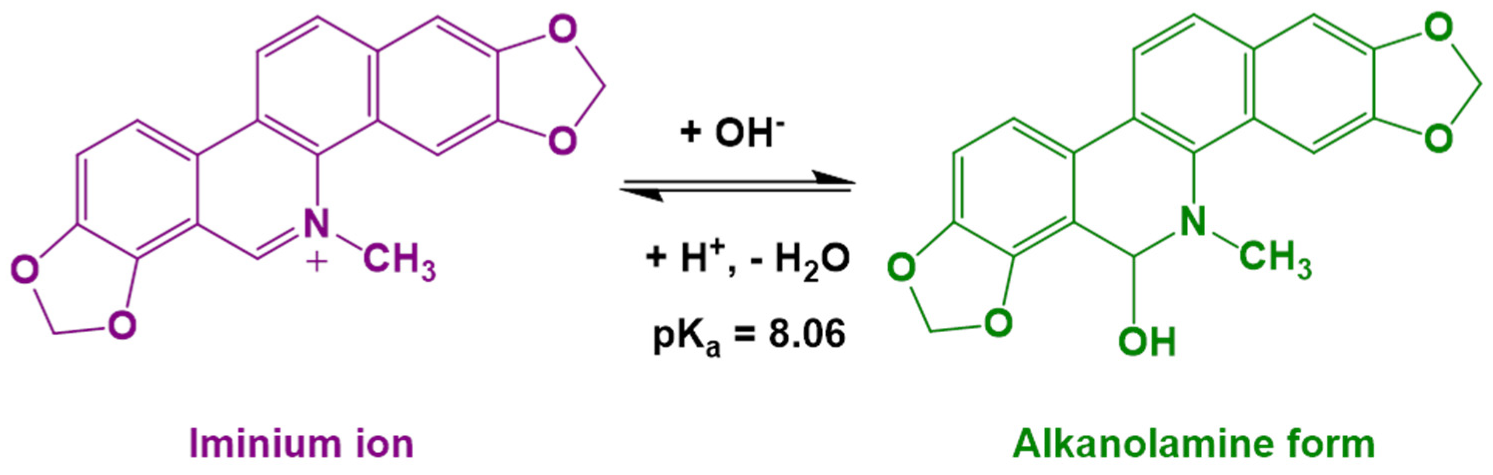
2.3. Sequestration of Sanguinarine (SG) from the Liposome Membrane Using Cyclodextrin
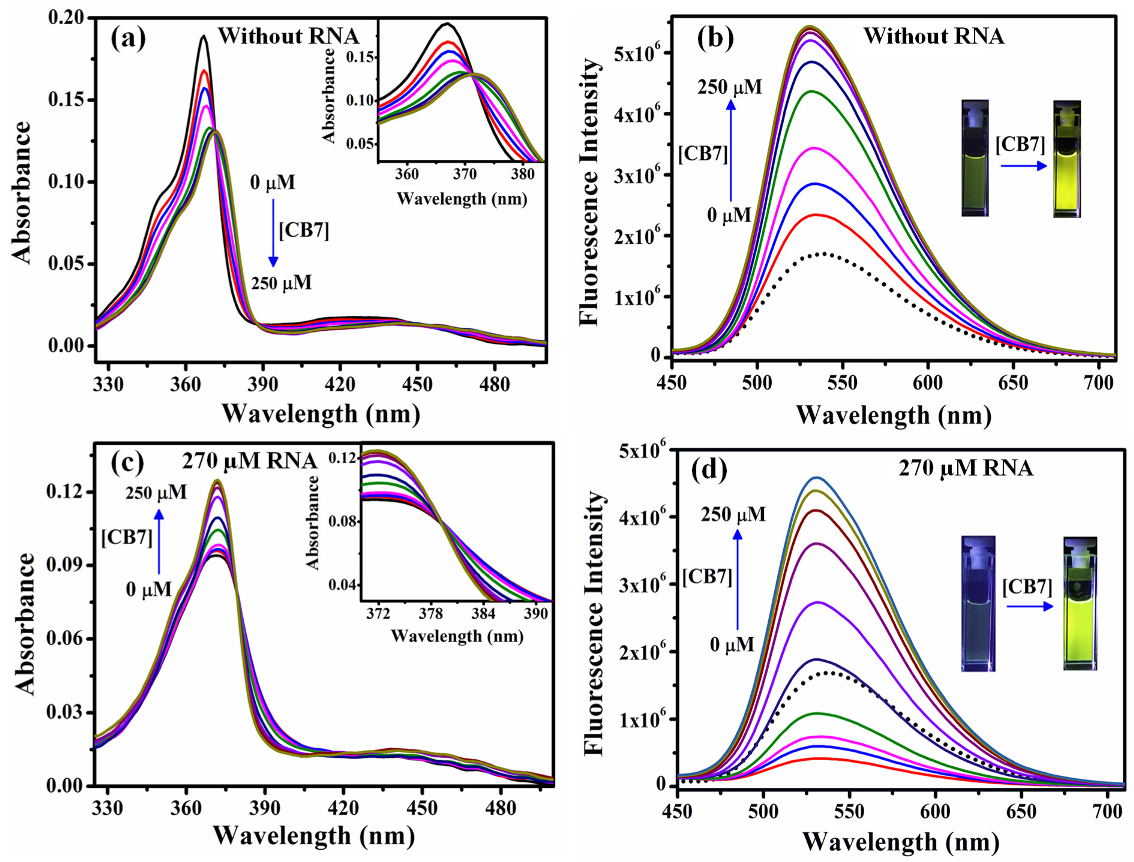
2.4. Sequestration of Cryptolepine Hydrate (CRYP) from RNA Using Cucurbit[7]uril
3. Conclusions
- (i)
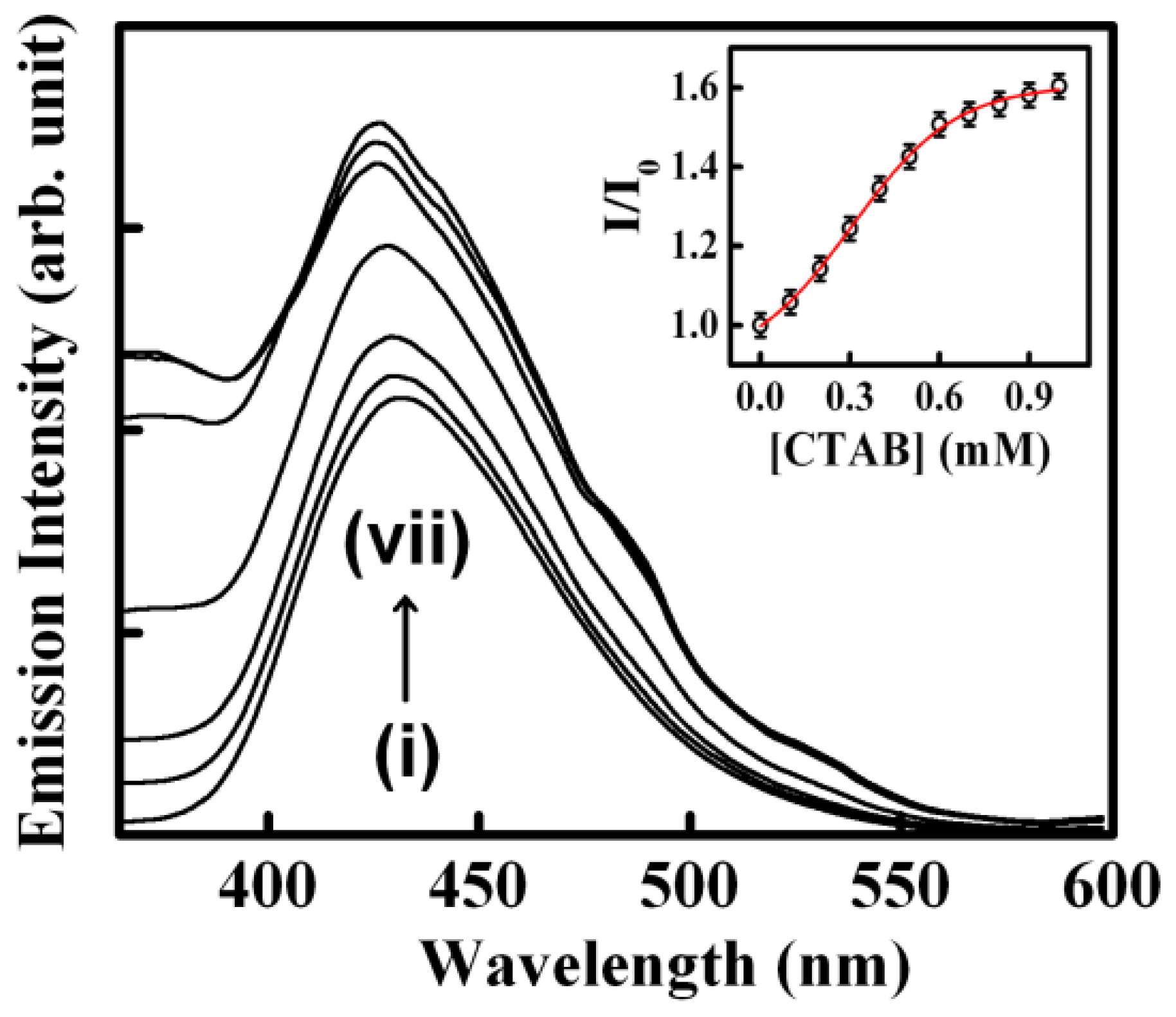
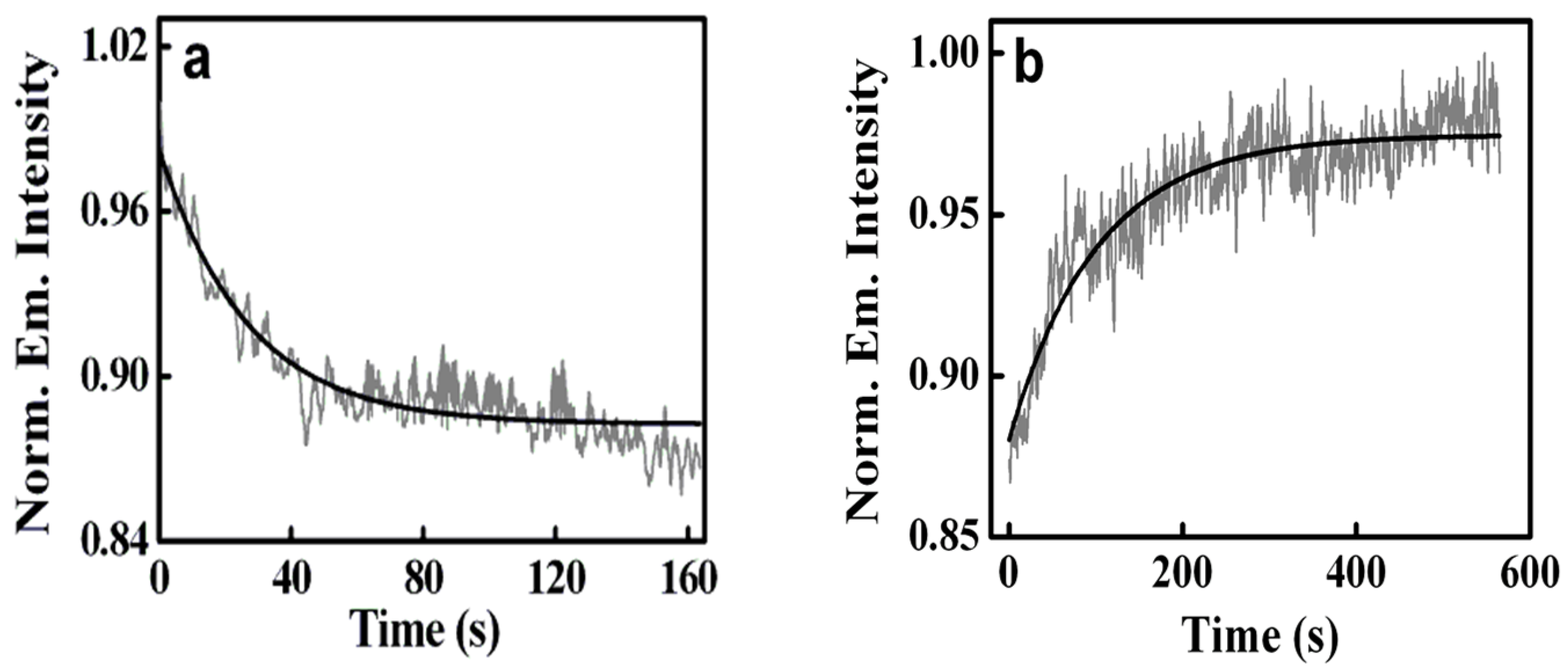
- The biological photosensitizer harmane (HM) strongly binds to the DNA duplex principally via the intercalation mode and can subsequently be sequestered using the cationic surfactant CTAB where the cationic surfactant acts as a hydrophobic sink for the drug molecules.
- (ii)
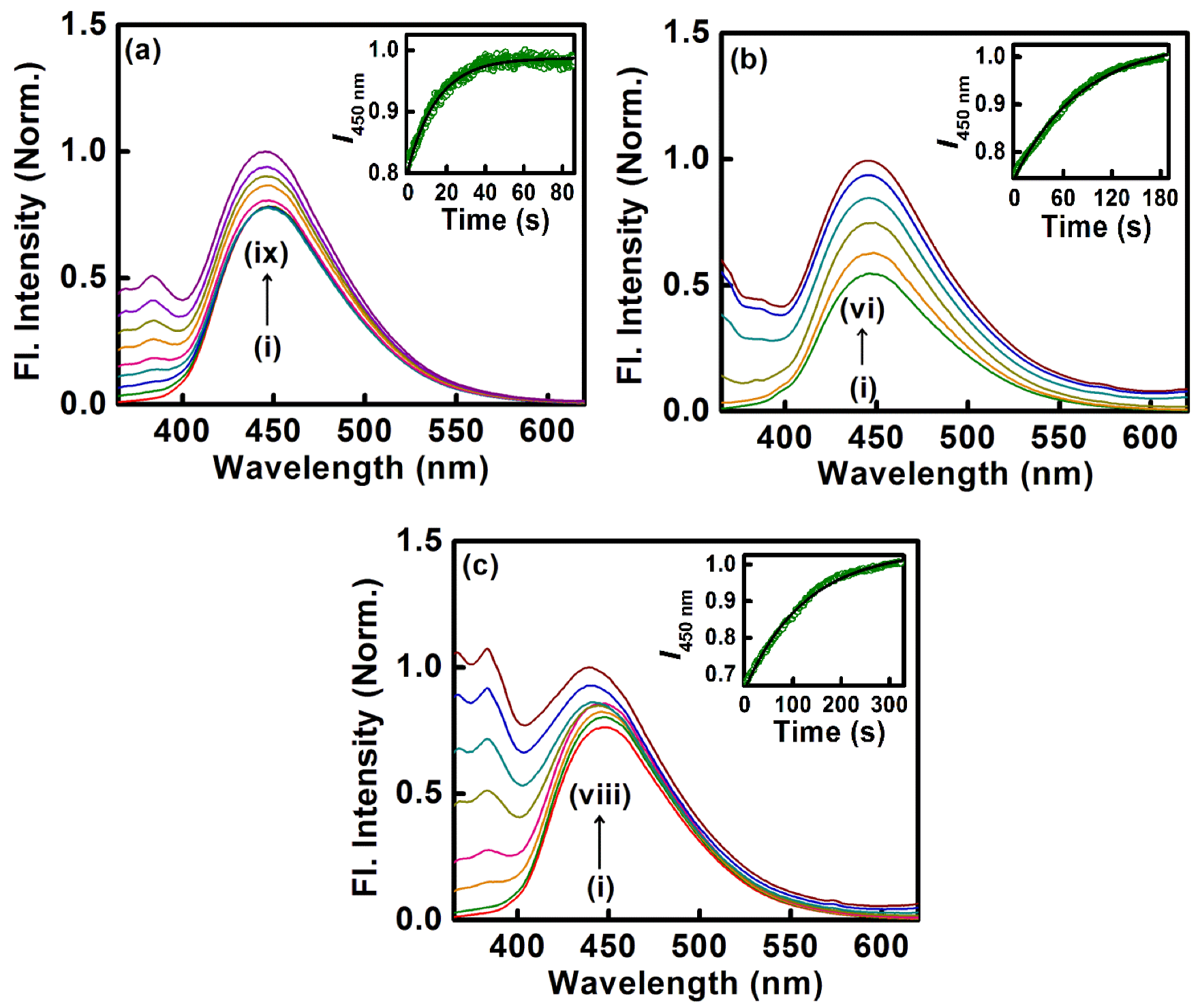
- Another biological photosensitizer, norharmane (NHM), which binds to the DNA duplex, has also been shown to have been sequestered with the aid of cationic surfactants, namely DTAB, TTAB, and CTAB, for which the rate of sequestration of the bound drug is found to be tunable with variations in the chain length of the surfactants.
- (iii)
- The deintercalation of the anticancer drug epirubicin hydrochloride bound to well-matched and mismatched DNA has been shown using mixed micellar assemblies consisting of the anionic surfactant SDS and non-ionic surfactant P123. It was also noticed that the mixed micelles did not alter the native structure of DNA.
- (iv)
- The dissociation of the alkaloid sanguinarine cation bound to negatively charged DMPG liposomes using molecular guest β-cyclodextrin shows the application of host–guest chemistry in this context.
- (v)
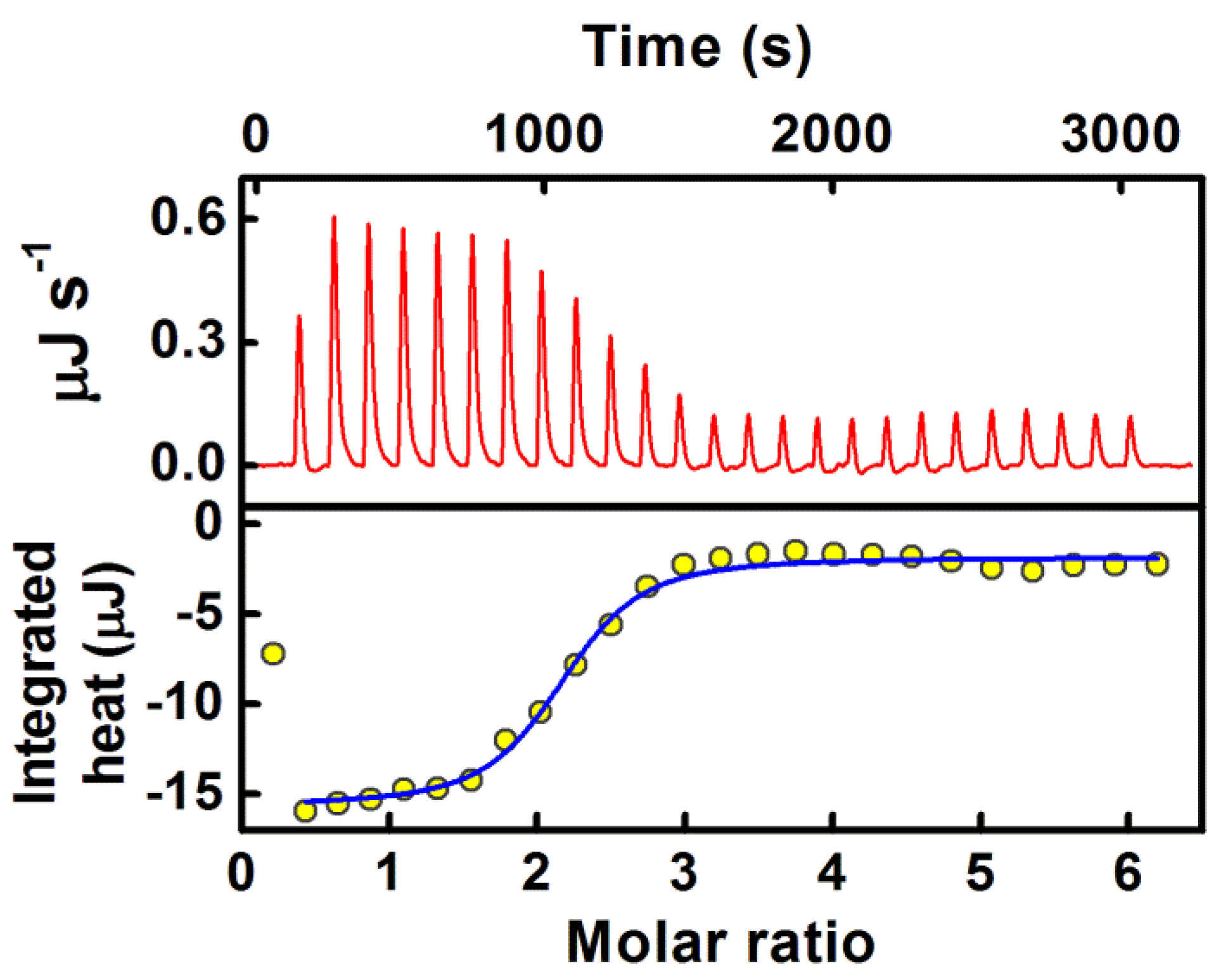
- The application of host–guest chemistry in the sequestration of bound drug molecules is further demonstrated in the context of the deintercalation of the antimalarial drug cryptolepine hydrate (CRYP) from the RNA duplex using cucurbit[7]uril hydrate.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asselah, T.; Durantel, D.; Pasmant, E.; Lau, G.; Schinazi, R.F. COVID-19: Discovery, diagnostics and drug development. J. Hepatol. 2020, 74, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.I.; Correia, I.; Seagal, J.; DeGoey, D.A.; Schrimpf, M.R.; Hardee, D.J.; Noey, E.L.; Kati, W.M. Antiviral Drug Discovery for the Treatment of COVID-19 Infections. Viruses 2022, 14, 961. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Kumar, G.S. Targeting the Heme Proteins Hemoglobin and Myoglobin by Janus Green Blue and Study of the Dye–Protein Association by Spectroscopy and Calorimetry. RSC Adv. 2014, 4, 42706–42715. [Google Scholar] [CrossRef]
- Hazra, S.; Kumar, G.S. Structural and Thermodynamic Studies on the Interaction of Iminium and Alkanolamine Forms of Sanguinarine with Hemoglobin. J. Phys. Chem. B 2014, 118, 3771–3784. [Google Scholar] [CrossRef]
- Singh, P.; Chowdhury, P.K. Unravelling the Intricacy of the Crowded Environment through Tryptophan Quenching in Lysozyme. J. Phys. Chem. B 2017, 121, 4687–4699. [Google Scholar] [CrossRef]
- Kundu, N.; Roy, A.; Banik, D.; Sarkar, N. Unveiling the Mode of Interaction of Berberine Alkaloid in Different Supramolecular Confined Environments: Interplay of Surface Charge between NanoConfined Charged Layer and DNA. J. Phys. Chem. B 2016, 120, 1106–1120. [Google Scholar] [CrossRef]
- Das, S.; Ghosh, P.; Koley, S.; Roy, A.S. Binding of naringin and naringenin with hen egg white lysozyme: A spectroscopic investigation and molecular docking study. Spectrochim. Acta Part A 2018, 192, 211–221. [Google Scholar] [CrossRef]
- Sarkar, S.; Roy, S.; Singh, P.C. Groove Switching of Hydroxychloroquine Modulates the Efficacy of Binding and Induced Stability to DNA. J. Phys. Chem. B 2021, 125, 6889–6896. [Google Scholar] [CrossRef]
- Sarkar, S.; Bisoi, A.; Singh, P.C. Spectroscopic and Molecular Dynamics Aspect of Antimalarial Drug Hydroxychloroquine Binding with Human Telomeric G-Quadruplex. J. Phys. Chem. B 2022, 126, 5241–5249. [Google Scholar] [CrossRef]
- Mukherjee, A.; Saurabh, S.; Olive, E.; Jang, Y.H.; Lansac, Y. Protamine Binding Site on DNA: Molecular Dynamics Simulations and Free Energy Calculations with Full Atomistic Details. J. Phys. Chem. B 2021, 125, 3032–3044. [Google Scholar] [CrossRef]
- Punihaole, D.; Workman, R.J.; Upadhyay, S.; Van Bruggen, C.; Schmitz, A.J.; Reineke, T.M.; Frontiera, R.R. New Insights into Quinine-DNA Binding Using Raman Spectroscopy and Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 9840–9851. [Google Scholar] [CrossRef]
- Swain, B.C.; Mukherjee, S.K.; Rout, J.; Sakshi; Mishra, P.P.; Mukherjee, M.; Tripathy, U. A spectroscopic and computational intervention of interaction of lysozyme with 6-mercaptopurine. Anal. Bioanal. Chem. 2020, 412, 2565–2577. [Google Scholar] [CrossRef]
- Pramanik, U.; Kongasseri, A.A.; Shekhar, S.; Mathew, A.; Yadav, R.; Mukherjee, S. Structural Compactness in Hen Egg White Lysozyme Induced by Bisphenol S: A Spectroscopic and Molecular Dynamics Simulation Approach. ChemPhysChem 2021, 22, 1745–1753. [Google Scholar] [CrossRef]
- Li, M.; Cong, Y.; Qi, Y.; Zhang, J.Z.H. Binding of berberine derivates to G-quadruplex: Insight from a computational study. Phys. Chem. Chem. Phys. 2023, 25, 10741–10748. [Google Scholar] [CrossRef]
- Dandekar, B.R.; Sinha, S.; Mondal, J. Role of molecular dynamics in optimising ligand discovery: Case study with novel inhibitor search for peptidyl t-RNA hydrolase. Chem. Phys. Impact 2021, 3, 100048–100057. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, R. Structural Basis of the Potential Binding Mechanism of Remdesivir to SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. [Google Scholar] [CrossRef]
- Walters, S.M.; Bolinski, R.S.; Almirol, E.; Grundy, S.; Fletcher, S.; Schneider, J.; Freidman, S.R.; Ouellet, L.J.; Ompad, D.C.; Jenkins, W.; et al. Structural and community changes during COVID-19 and their effects on overdose precursors among rural people who use drugs: A mixed-methods analysis. Addict. Sci. Clin. Pract. 2022, 17, 24–39. [Google Scholar] [CrossRef]
- Di Nunno, N.; Esposito, M.; Argo, A.; Salerno, M.; Sessa, F. Pharmacogenetics and Forensic Toxicology: A New Step towards a Multidisciplinary Approach. Toxics 2021, 9, 292. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, S.; Lin, W.; Wu, N.; Wang, Y.; Chen, M.; Xie, X.-Q.; Feng, Z. Computational Systems Pharmacology-Target Mapping for Fentanyl-Laced Cocaine Overdose. ACS Chem. Neurosci. 2019, 10, 3486–3499. [Google Scholar] [CrossRef]
- Li, J.; Kataoka, K. Chemo-physical Strategies to Advance the in Vivo Functionality of Targeted Nanomedicine: The Next Generation. J. Am. Chem. Soc. 2021, 143, 538–559. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.; Ke, W.; Dirisala, A.; Toh, K.; Tanaka, M.; Li, J. Stealth and pseudo-stealth nanocarriers. Adv. Drug Deliv. Rev. 2023, 198, 114895–114910. [Google Scholar] [CrossRef]
- Cui, X.; Mao, S.; Liu, M.; Yuan, H.; Du, Y. Mechanism of surfactant micelle formation. Langmuir 2008, 24, 10771–10775. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.; Ghandehari, H. Polymeric Conjugates for Drug Delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manet, S.; Karpichev, Y.; Dedovets, D.; Oda, R. Effect of Hofmeister and Alkylcarboxylate Anionic Counterions on the Krafft Temperature and Melting Temperature of Cationic Gemini Surfactants. Langmuir 2013, 29, 3518–3526. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.; Crothers, D.M. Studies of the binding of actinomycin and related compounds to DNA. J. Mol. Biol. 1968, 35, 251–290. [Google Scholar] [CrossRef]
- Westerlund, F.; Wilhelmsson, L.M.; Norden, B.; Lincoln, P. Micelle-sequestered dissociation of cationic DNA–intercalated drugs: Unexpected surfactant-induced rate enhancement. J. Am. Chem. Soc. 2003, 125, 3773–3779. [Google Scholar] [CrossRef] [Green Version]
- Westerlund, F.; Nordell, P.; Nordén, B.; Lincoln, P. Kinetic Characterization of an Extremely Slow DNA Binding Equilibrium. J. Phys. Chem. B 2007, 30, 9132–9137. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N. Exploring the Strength, Mode, Dynamics, and Kinetics of Binding Interaction of a Cationic Biological Photosensitizer with DNA: Implication on Dissociation of the Drug–DNA Complex via Detergent Sequestration. J. Phys. Chem. B 2011, 115, 11938–11949. [Google Scholar] [CrossRef]
- Patra, A.; Hazra, S.; Kumar, G.S.; Mitra, R.K. Entropy Contribution toward Micelle-Driven Deintercalation of Drug-DNA Complex. J. Phys. Chem. B 2014, 118, 901–908. [Google Scholar] [CrossRef]
- Kundu, P.; Ghosh, S.; Chattopadhyay, N. Exploration of the Binding Interaction of a Potential Nervous System Stimulant with Calf-Thymus DNA and Dissociation of the Drug-DNA Complex by Detergent Sequestration. Phys. Chem. Chem. Phys. 2015, 17, 17699–17709. [Google Scholar] [CrossRef]
- Mukherjee, A.; Ghosh, S.; Pal, M.; Singh, B. Deciphering the Effective Sequestration of DNA Bounded Bioactive Small Mole-cule Safranin-O by Non-ionic Surfactant TX-114 and Diminution its Cytotoxicity. J. Mol. Liq. 2019, 289, 111116–111124. [Google Scholar] [CrossRef]
- Enache, M.; Ionescu, S.; Volanschi, E. Studies on the anticancer drug mitoxantrone–DNA–sodium dodecyl sulfate system. J. Mol. Liq. 2015, 208, 333–341. [Google Scholar] [CrossRef]
- Marson, D.; Laurini, E.; Fermeglia, M.; Smith, D.K.; Pricl, S. Mallard Blue Binding to Heparin, Its SDS Micelle-Driven de-Complexation, and Interaction with Human Serum Albumin: A Combined Experimental/Modeling Investigation. Fluid Phase Equilib. 2018, 470, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Interaction of an anti-cancer photosensitizer with a genomic DNA: From base pairspecificity and thermodynamic landscape to tuning the rate of detergent-sequestered dissociation. J. Colloid Interface Sci. 2016, 470, 211–220. [Google Scholar] [CrossRef]
- Mora, A.K.; Singh, P.K.; Nath, S. Controlled Sequestration of DNA Intercalated Drug by Polymer-Surfactant Supramolecular Assemblies. J. Phys. Chem. B 2016, 120, 4143–4151. [Google Scholar] [CrossRef]
- Kalel, R.; Mora, A.K.; Patro, B.S.; Palit, D.K.; Nath, S. Synergistic Enhancement in the Drug Sequestration Power and Reduction in the Cytotoxicity of Surfactants. Phys. Chem. Chem. Phys. 2017, 19, 25446–25455. [Google Scholar] [CrossRef]
- Khamari, L.; Pramanik, S.; Shekhar, S.; Mahato, P.; Mukherjee, S. Preferential Binding of Epirubicin Hydrochloride with Single Nucleotide Mismatched DNA and Subsequent Sequestration by a Mixed Micelle. J. Phys. Chem. B 2021, 125, 11660–11672. [Google Scholar] [CrossRef]
- Mora, A.K.; Basu, A.; Kalel, R.; Nath, S. Polymer-assisted Drug Sequestration from Plasma Protein by a Surfactant with Curtailed Denaturing Capacity. Phys. Chem. Chem. Phys. 2019, 21, 7127–7136. [Google Scholar] [CrossRef]
- Maurya, N.; Parray, Z.A.; Maurya, J.K.; Islam, A.; Patel, R. Ionic Liquid Green Assembly-Mediated Migration of Piperine from Calf-Thymus DNA: A New Possibility of the Tunable Drug Delivery System. ACS Omega 2019, 4, 21005–21017. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Adhikari, C.; Nayak, D.; Chakraborty, A. First Evidence of the Liposome-Mediated Deintercalation of Anticancer Drug Doxorubicin from the Drug-DNA Complex: A Spectroscopic Approach. Langmuir 2016, 32, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Adhikari, C.; Chakraborty, A. Lipoplex-mediated Deintercalation of Doxorubicin from Calf Thymus DNA-doxorubicin Complex. Langmuir 2016, 32, 8889–8899. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Adhikari, C.; Chakraborty, A. Interaction of Different Divalent Metal Ions with Lipid Bilayer: Impact on the Encapsulation of Doxorubicin by Lipid Bilayer and Lipoplex Mediated Deintercalation. J. Phys. Chem. B 2017, 121, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Bose, D.; Mahata, A.; Ghosh, D.; Chattopadhyay, N. Differential interaction of β-cyclodextrin with lipids of varying surface charges: A spectral deciphering using a cationic phenazinium dye. J. Phys. Chem. B 2010, 114, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Jana, B.; Ghosh, S.; Chattopadhyay, N. Competitive binding of nile red between lipids and β-cyclodextrin. J. Photochem. Photobiol. B 2013, 126, 1–10. [Google Scholar] [CrossRef]
- Kundu, P.; Ghosh, S.; Jana, B.; Chattopadhyay, N. Binding interaction of differently charged fluorescent probes with egg yolk phosphatidylcholine and the effect of β- cyclodextrin on the lipidprobe complexes: A fluorometric investigation. Spectrochim. Acta Part A 2015, 142, 15–24. [Google Scholar] [CrossRef]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Association and sequestered dissociation of an anticancer drug from liposome membrane: Role of hydrophobic hydration. Colloids Surf. B 2018, 170, 36–44. [Google Scholar] [CrossRef]
- Nandy, A.; Shekhar, S.; Paul, B.K.; Mukherjee, S. Exploring the Nucleobase-Specific Hydrophobic Interaction of Cryptolepine Hydrate with RNA and Its Subsequent Sequestration. Langmuir 2021, 37, 11176–11187. [Google Scholar] [CrossRef]
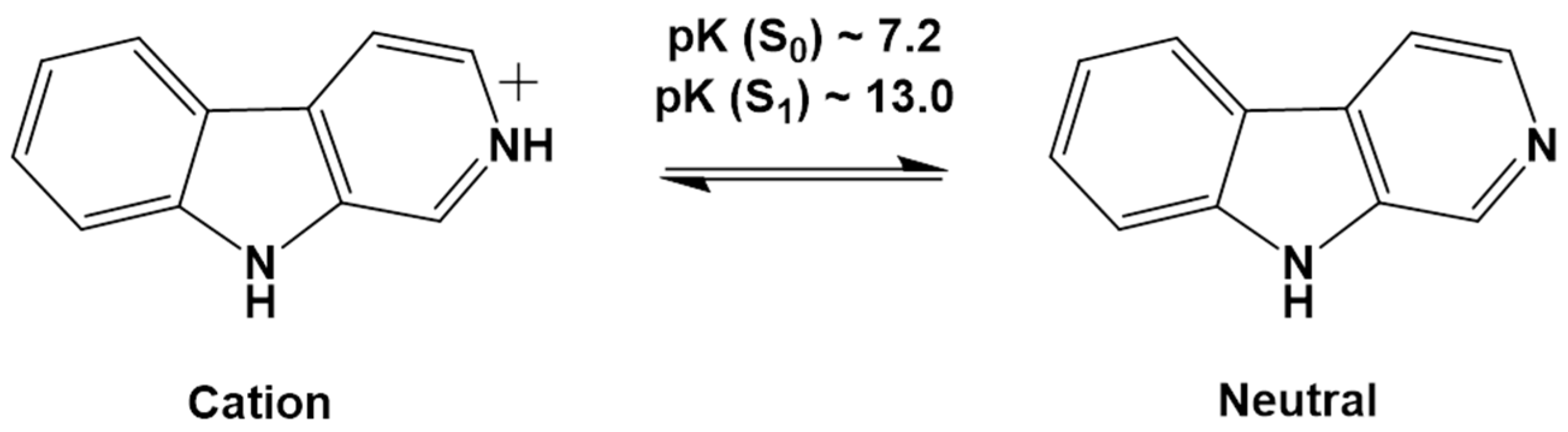
- Coronilla, A.S.; Carmona, C.; Munoz, M.A.; Balon, M. Ground and Singlet Excited State Pyridinic Protonation of N9-Methylbetacarboline in Water-N,N-Dimethylformamide Mixtures. J. Fluoresc. 2009, 19, 1025–1035. [Google Scholar] [CrossRef]
- Wolfbeis, O.S.; Furlinger, E.; Wintersteiger, R. Solvent- and pH-Dependence of the Absorption and Fluorescence Spectra of Harman: Detection of Three Ground State and Four Excited State Species. Montash. Chem. 1982, 113, 509–517. [Google Scholar] [CrossRef]
- Balon, M.; Munoz, M.A.; Guardado, P.; Hidalgo, J.; Carmona, C. Photophysics and Photochemistry of Betacarbolines. Trends Photochem. Photobiol. 1994, 3, 117–138. [Google Scholar]
- Tuveson, R.W. Using Bacterial Mutants and Transforming DNA to define Phototoxic Mechanisms. In Light-Activated Pesticides; Heitz, J.R., Downum, K.R., Eds.; ACS Symposium Series No. 339; American Chemical Society: Washington, DC, USA, 1987; pp. 192–205. [Google Scholar]
- Braestrup, C.; Nielsen, M.; Olsen, C.E. Urinary and brain β-carboline-3-carboxylates as potent inhibitors of brain benzodiazepine receptors. Proc. Natl. Acad. Sci. USA 1980, 77, 2288–2292. [Google Scholar] [CrossRef]
- Reyman, D.; Pardo, A.; Poyato, J.M.L. Phototautomerism of β-Carboline. J. Phys. Chem. 1994, 98, 10408–10411. [Google Scholar] [CrossRef]
- Hayashi, K.; Nagao, M.; Sugimura, T. Interactions of norharman and harman with DNA. Nucleic Acid Res. 1977, 4, 3679–3685. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, M.M.; Arnbjerg, J.; Denofrio, M.P.; Erra-Balsells, R.; Ogilby, P.R.; Cabrarizo, F.M. One- and Two-Photon Excitation of β-Carbolines in Aqueous Solution: pH-Dependent Spectroscopy, Photochemistry, and Photophysics. J. Phys. Chem. A 2009, 113, 6648–6656. [Google Scholar] [CrossRef]
- Chen, Q.; Li, D.; Zhao, Y.; Yang, H.; Zhu, Q.; Xu, J. Interaction of a novel red-region fluorescent probe, Nile Blue, with DNA and its application to nucleic acids assay. Analyst 1999, 124, 901–906. [Google Scholar] [CrossRef] [Green Version]
- Gabbay, E.J.; Grier, D.; Fingerle, R.E.; Reimer, R.; Levy, R.; Pearce, S.W.; Wilson, W.D. Interaction specificity of the anthracyclines with deoxyribonucleic acid. Biochemistry 1976, 15, 2062–2070. [Google Scholar] [CrossRef]
- Wilson, D.W.; Grier, D.; Reimer, R.; Bauman, J.D.; Preston, J.F.; Gabbay, E.J. Structure–activity relationship of daunorubicin and its peptide derivatives. J. Med. Chem. 1976, 19, 381–384. [Google Scholar] [CrossRef]
- Fox, K.R.; Brassett, C.; Waring, M.J. Kinetics of dissociation of nogalamycin from DNA: Comparison with other anthracycline antibiotics. Biochim. Biophys. Acta 1985, 840, 383–392. [Google Scholar] [CrossRef]
- Wakelin, L.P.G.; Atwell, G.J.; Rewcastle, G.W.; Denny, W.A. Relationships between DNA-binding kinetics and biological activity for the 9-aminoacridine-4-carboxamide class of antitumor agents. J. Med. Chem. 1987, 30, 855–861. [Google Scholar] [CrossRef]
- White, R.J.; Phillips, D.R. Drug-DNA dissociation kinetics in vitro transcription and sodium dodecyl sulphate sequestration. Biochem. Pharmacol. 1989, 38, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.R.; Greif, P.C.; Boston, R.C. Daunomycin-DNA dissociation kinetics. Mol. Pharmacol. 1988, 33, 225–230. [Google Scholar] [PubMed]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Prototropic Transformation and Rotational-Relaxation Dynamics of a Biological Photosensitizer Norharmane inside Nonionic Micellar Aggregates. J. Phys. Chem. B 2014, 118, 11209–11219. [Google Scholar] [CrossRef] [PubMed]
- Reyman, D.; Vinas, M.H.; Tardajos, G.; Mazario, E. The Impact of Dihydrogen Phosphate anions on the Excited-State Proton Transfer of Harmane. Effect of β-Cyclodextrin on These Photoreactions. J. Phys. Chem. A 2012, 116, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Varela, A.P.; da Miguel, M.G.; Macanita, A.L.; Becker, R.S. β-Carboline Photosensitizers. 1. Photophysics, Kinetics and Excited–State Equilibria in Organic Solvents, and Theoretical Calculations. J. Phys. Chem. 1992, 96, 10290–10296. [Google Scholar] [CrossRef]
- Connelly, P.R.; Thomson, J.A. Heat capacity changes and hydrophobic interactions in the binding of FK506 and rapamycin to the FK506 binding protein. Proc. Natl. Acad. Sci. USA 1992, 89, 4781–4785. [Google Scholar] [CrossRef]
- Record, M.T., Jr.; Anderson, C.F.; Lohman, T.M. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: The roles of ion association or release, screening and ion effects on water activity. Q. Rev. Biophys. 1978, 11, 103–178. [Google Scholar] [CrossRef]
- Homans, S.W. Dynamics and Thermodynamics of Ligand–Protein Interactions. Top. Curr. Chem. 2007, 272, 51–82. [Google Scholar]
- Huang, H.M.; Lin, F.Y.; Chen, W.Y.; Ruaan, R.C. Isothermal Titration Microcalorimetric Studies of the Effect of Temperature on Hydrophobic Interaction between Proteins and Hydrophobic Adsorbents. J. Colloid Interface Sci. 2000, 229, 600–606. [Google Scholar] [CrossRef]
- Joseph, D.; Sachar, S.; Kishore, N.; Chandra, S. Mechanistic insights into the interactions of magnetic nanoparticles with bovine serum albumin in presence of surfactants. Colloids Surf. B Biointerfaces 2015, 135, 596–603. [Google Scholar] [CrossRef]
- Paul, B.K. Classical vs nonclassical hydrophobic interactions underlying various interaction processes: Application of isothermal titration calorimetry. Chem. Phys. Impact 2022, 5, 100104–100122. [Google Scholar] [CrossRef]
- Lin, C.-M.; Li, C.-S.; Sheng, Y.-J.; Wu, D.T.; Tsao, H.-K. Size-dependent properties of small unilamellar vesicles formed by model lipids. Langmuir 2012, 28, 689–700. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of Univalent Ions across Lamellae of Swollen Phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Schreier, H.; Bouwstra, J. Liposomes and Niosomes as Topical Drug Carriers: Dermal and Transdermal Drug Delivery. J. Control. Release 1994, 30, 1–15. [Google Scholar] [CrossRef]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles–From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]
- Crini, G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- Szente, L.; Szeman, J.; Sohajda, T. Analytical characterization of cyclodextrins: History, official methods and recommended new techniques. J. Pharm. Biomed. Anal. 2016, 130, 347–365. [Google Scholar] [CrossRef]
- Martins, J.N.; Lima, J.C.; Basílio, N. Selective Recognition of Amino Acids and Peptides by Small Supramolecular Receptors. Molecules 2021, 26, 106. [Google Scholar] [CrossRef]
- Murray, J.; Kim, K.; Ogoshi, T.; Yao, W.; Gibb, B.C. The aqueous supramolecular chemistry of cucurbit[n]urils, pillar[n]arenes and deep-cavity cavitands. Chem. Soc. Rev. 2017, 46, 2479–2496. [Google Scholar] [CrossRef] [Green Version]
- Hazra, S.; Kumar, G.S. Physicochemical properties of inclusion complexes of sanguinarine with natural cyclodextrins: Spectroscopy, calorimetry and NMR studies. RSC Adv. 2015, 5, 1873–1882. [Google Scholar] [CrossRef]
- Lenfeld, J.; Kroutil, M.; Marsálek, E.; Slavík, J.; Preininger, V.; Simánek, V. Antiinflammatory activity of quaternary benzophenanthridine alkaloids from Chelidonium majus. Planta Med. 1981, 43, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Godowski, K.C. Antimicrobial action of sanguinarine. J. Clin. Dent. 1989, 1, 96–101. [Google Scholar] [PubMed]
- Southard, G.L.; Boulware, R.T.; Walborn, D.R.; Groznik, W.J.; Thorne, E.E.; Yankell, S.L. Sanguinarine, a new antiplaque agent: Retention and plaque specificity. J. Am. Dent. Assoc. 1984, 108, 338–341. [Google Scholar] [CrossRef]
- Paul, B.K.; Ghosh, N.; Mondal, R.; Mukherjee, S. Contrasting Effects of Salt and Temperature on Niosome-Bound Norharmane: Direct Evidence for Positive Heat Capacity Change in the Niosome: β-Cyclodextrin Interaction. J. Phys. Chem. B 2016, 120, 4091–4101. [Google Scholar] [CrossRef]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Direct Insight into the Nonclassical Hydrophobic Effect in Bile Salt: β-Cyclodextrin Interaction: Role of Hydrophobicity in Governing the Prototropism of a Biological Photosensitizer. RSC Adv. 2016, 6, 9984–9993. [Google Scholar] [CrossRef]
- Spolar, R.S.; Record, M.T., Jr. Coupling of Local Folding to Site-Specific Binding of Proteins to DNA. Science 1994, 263, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.P.; Privalov, P.L.; Gill, S.J. Common features of protein unfolding and dissolution of hydrophobic compounds. Science 1990, 247, 559–561. [Google Scholar] [CrossRef]
- Ondo, D.; Costas, M. Complexation Thermodynamics of α-Cyclodextrin with Ionic Surfactants in Water. J. Colloid Interface Sci. 2017, 505, 445–453. [Google Scholar] [CrossRef]
- Kresheck, G.C. The Temperature dependence of the Heat Capacity Change for Micellization of Nonionic Surfactants. J. Colloid Interface Sci. 2006, 298, 432–440. [Google Scholar] [CrossRef]
- Niedzwiecka, A.; Stepinski, J.; Darzynkiewicz, E.; Sonenberg, N.; Stolarski, R. Positive Heat Capacity Change upon Specific Binding of Translation Initiation Factor eIF4E to mRNA 5′ Cap. Biochemistry 2002, 41, 12140–12148. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Ghosh, S.; Adhikari, A.; Sen Mojumdar, S.; Bhattacharyya, K. A Fluorescence Correlation Spectroscopy Study of the Diffusion of an Organic Dye in the Gel Phase and Fluid Phase of a Single Lipid Vesicle. J. Phys. Chem. B 2010, 114, 5736–5741. [Google Scholar] [CrossRef]
- De, D.; Sajjan, M.; Datta, A. Anisotropic Dynamics of Guest Molecules in Aerosol OT Lamellar Structures. Phys. Chem. Chem. Phys. 2013, 15, 19724–19729. [Google Scholar] [CrossRef]
- Garcia, A.C.; Zakharov, L.N.; Pluth, M.D. Supramolecular Activation of S8 by Cucurbiturils in Water and Mechanism of Reduction to H2S by Thiols: Insights into Biological Sulfane Sulfur Trafficking. J. Am. Chem. Soc. 2022, 144, 15324–15332. [Google Scholar] [CrossRef]
- Das Saha, N.; Pradhan, S.; Sasmal, R.; Sarkar, A.; Berač, C.M.; Kölsch, J.C.; Pahwa, M.; Show, S.; Rozenholc, Y.; Topçu, Z.; et al. Cucurbit[7]Uril Macrocyclic Sensors for Optical Fingerprinting: Predicting Protein Structural Changes to Identifying Disease-Specific Amyloid Assemblies. J. Am. Chem. Soc. 2022, 144, 14363–14379. [Google Scholar] [CrossRef]
- Chakraborty, G.; Choudhary, M.K.; Sundararajan, M.; Ray, A.K.; Mula, S.; Pal, H. Stimuli Responsive Confinement of a Molecular Rotor Based BODIPY Dye inside a Cucurbit[7]uril Nanocavity. J. Phys. Chem. B 2021, 125, 7946–7957. [Google Scholar] [CrossRef]
- Deng, C.-L.; Murkli, S.L.; Isaacs, L.D. Supramolecular hosts as in vivo sequestration agents for pharmaceuticals and toxins. Chem. Soc. Rev. 2020, 49, 7516–7532. [Google Scholar] [CrossRef]
- Barrow, S.J.; Kasera, S.; Rowland, M.J.; del Barrio, J.; Scherman, O.A. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115, 12320–12406. [Google Scholar] [CrossRef] [Green Version]
- Masson, E.; Ling, X.; Joseph, R.; Kyeremeh-Mensah, L.; Lu, X. Cucurbituril chemistry: A tale of supramolecular success. RSC Adv. 2012, 2, 1213–1247. [Google Scholar] [CrossRef]
- Jeon, W.S.; Moon, K.; Park, S.H.; Chun, H.; Ko, Y.H.; Lee, J.Y.; Lee, E.S.; Samal, S.; Selvapalam, N.; Rekharsky, M.V.; et al. Complexation of Ferrocene Derivatives by the Cucurbit[7]uril Host: A Comparative Study of the Cucurbituril and Cyclodextrin Host Families. J. Am. Chem. Soc. 2005, 127, 12984–12989. [Google Scholar] [CrossRef]
- Lazar, A.I.; Biedermann, F.; Mustafina, K.R.; Assaf, K.I.; Hennig, A.; Nau, W.M. Nanomolar Binding of Steroids to Cucurbit[n]urils: Selectivity and Applications. J. Am. Chem. Soc. 2016, 138, 13022–13029. [Google Scholar] [CrossRef] [PubMed]
- Koninti, R.K.; Sappati, S.; Satpathi, S.; Gavvala, K.; Hazra, P. Spectroscopy and Dynamics of Cryptolepine in the Nanocavity of Cucurbit[7]uril and DNA. ChemPhysChem 2016, 17, 506–515. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial Number | Biological/Biomimicking Environment | Small Molecule/Drug | Sequestrating Agent | Reference(s) |
|---|---|---|---|---|
| 1. | Herring Sperm DNA | Harmane | CTAB | [29] |
| 2. | Calf Thymus DNA | Phenosafranin | SDS | [30] |
| 3. | Calf Thymus DNA | 3-Acetyl-4-oxo-6,7-dihydro-12H-indolo-[2,3-a]-quinolizine | CTAB | [31] |
| 4. | Herring Sperm DNA | Safranin O | Triton X-114 | [32] |
| 5. | DNA | Mitoxantrone | SDS | [33] |
| 6. | Heparin | Mallard Blue | SDS | [34] |
| 7. | Herring Sperm DNA | Norharmane | DTAB, CTAB, TTAB | [35] |
| 8. | DNA | Ethidium Bromide | P105, F127, and P123 with SDS | [36,37] |
| 9. | DNA | Epirubicin | P123-SDS Mixed Micelles | [38] |
| 10. | BSA Protein | 9-(2-Caboxy-2-cyano)vinyl-julolidine | CTAB-P123 Mixed Micelles | [39] |
| 11. | Calf Thymus DNA | Piperine | Surface Active Ionic Liquid | [40] |
| 12. | Calf Thymus DNA | Doxorubicin | Liposomes | [41,42,43] |
| 13. | DMPC lipids | Phenosafranin | β-Cyclodextrin | [44] |
| 14. | DMPG Lipids | Nile Red | β-Cyclodextrin | [45] |
| 15. | EYPC Lipids | Phenosafranin, ANS, Nile Red | β-Cyclodextrin | [46] |
| 16. | Liposomes | Sanguinarine | β-Cyclodextrin | [47] |
| 17. | Torula Yeast RNA | Cryptolepine | Cucurbit[7]uril | [48] |
| Surfactant | n a | kd (s−1) |
|---|---|---|
| CTAB | 16 | 65 × 10−3 |
| TTAB | 14 | 12 × 10−3 |
| DTAB | 12 | 7.96 × 10−3 |
| Surfactant a | Ka (M−1) | ∆H (kJ mol−1) | T∆S (kJ mol−1) | ∆G (kJ mol−1) |
|---|---|---|---|---|
| CTAB (n = 16) | (3.5 ± 0.12) × 105 | −3.85 ± 0.19 | 27.79 ± 0.08 | −31.63 |
| TTAB (n = 14) | (2.6 ± 0.10) × 105 | −1.56 ± 0.10 | 29.36 ± 0.10 | −30.93 |
| DTAB (n = 12) | (1.84 ± 0.11) × 104 | 0.59 ± 0.10 | 24.92 ± 0.11 | −24.33 |
| Temperature (K) | Ka (M−1) | ΔH (kJ mol−1) | TΔS (kJ mol−1) | ΔG (kJ mol−1) | ΔCp (J mol−1 K−1) |
|---|---|---|---|---|---|
| 303 | (1.08 ± 0.04) × 103 | −31.56 ± 1.2 | −13.95 ± 0.6 | −17.61 | 514 ± 20 |
| 308 | (1.27 ± 0.05) × 103 | −28.78 ± 1.2 | −10.74 ± 0.43 | −18.04 | |
| 313 | (1.47 ± 0.06) × 103 | −26.41 ± 1.1 | −6.91 ± 0.28 | −19.5 |
| System | Dt (µm2 s−1) |
|---|---|
| Aqueous buffer | 63.8 ± 3 |
| 1.0 mM DMPG | 9.07 ± 0.42 |
| 1.0 mM DMPG + 4.0 mM βCD | 14.33 ± 0.67 |
| 1.0 mM DMPG + 6.0 mM βCD | 25.76 ± 1.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, R.; Paul, B.K.; Mukherjee, S. Sequestration of Drugs from Biomolecular and Biomimicking Environments: Spectroscopic and Calorimetric Studies. Colloids Interfaces 2023, 7, 51. https://doi.org/10.3390/colloids7030051
Yadav R, Paul BK, Mukherjee S. Sequestration of Drugs from Biomolecular and Biomimicking Environments: Spectroscopic and Calorimetric Studies. Colloids and Interfaces. 2023; 7(3):51. https://doi.org/10.3390/colloids7030051
Chicago/Turabian StyleYadav, Rahul, Bijan Kumar Paul, and Saptarshi Mukherjee. 2023. "Sequestration of Drugs from Biomolecular and Biomimicking Environments: Spectroscopic and Calorimetric Studies" Colloids and Interfaces 7, no. 3: 51. https://doi.org/10.3390/colloids7030051